
Abstract
2-Fluoroindoles as an important structural scaffold are widely existing in many bioactive or therapeutic agents. Despite their potential usefulness, efficient constructions of 2-fluoroindole derivatives are very sparce. The development of straightforward synthetic approaches to access 2-fluoroindoles is highly desirable for studying their fundamental properties and applications. Herein, we report an efficient and general strategy for the construction of 2-fluoroindoles in which a wide variety of 2-fluoroindoles were accessed with high efficiency and chemoselectivity. Instead of starting from indole skeletons, our strategy constructs indole scaffolds alongside the incorporation of fluorine atom on C2 position in a formal [4+1] cyclization from readily accessible ortho-vinylanilines and difluorocarbene. In our protocol, commercially accessible halodifluoroalkylative reagents provide one carbon and one fluorine atom by cleaving one C-N tertiary bond and forming one C-N bond and one C-C double bond with ortho-vinylanilines. Downstream transformations on 2-fluoroindoles lead to various valuable bioactive molecules which demonstrated significant synthetic advantages over previous reports. And mechanistic studies suggest that the reaction undergoes a cascade difluorocarbene-trapping and intramolecular Michael addition reaction followed by Csp3-F bond cleavage.
Similar content being viewed by others

Introduction
The introduction of fluorinated groups (including fluorine atom) into a target molecule would have significant influence on the parent molecule’s reactivity, selectivity, physical and biological properties1,2,3,4,5,6,7. Therefore, it becomes a critical strategy in pharmaceuticals for finding therapeutical agents8,9,10,11,12,13,14,15,16, where fluorine atom is often employed as a bioisostere of hydrogen atom based on the above features.
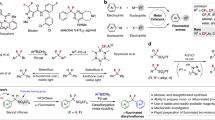
Indoles, as one of most important heterocyclic scaffolds, are widespread in a broad variety of natural products17,18,19,20 and bioactive compounds17,21,22 (Fig. 1A, Top). Meanwhile, 2-fluoroindoles are also key structural motifs which are widely existing in some biologically active compounds and demonstrate unique bioactivities23,24,25(Fig. 1A, Bottom). Because of the importance of C2-fluorine-containing indoles in medicinal chemistry, great attention has been devoted to the synthesis of 2-fluoroindoles26,27,28,29,30,31,32,33,34,35. However, efficient synthetic methods for the construction of this type of indole derivatives are very rare. Generally, there are three ways to access the target molecules (Fig. 1B): (a) copper-mediated aminoquinoline-directed fluorination of aromatic C–H bonds. However, there were only 3 examples about 2-fluoroindole synthesis with moderate yields26,27, and they are limited to 3-aminoquinolineindole derivatives, which seriously limits the application of indole skeleton. (b) pre-functionalization of indoles is mandatory to install some active functional groups, such as –COOH28 or –SnMe329, to C2 position of indoles for further fluorination, which require multi-step synthesis and need toxic organotin reagents, thus severely restrict the substrate scope. (c) silver or base-promoted C–N bond formation from ortho-amino-gem-difluorostyrenes to construct 2-fluororindoles30,31,32, this tactic requires complicated substrates with both gem-difluorostyrene and ortho-aromatic secondary amino functionalities, which are demanding and difficult to access, thus set a severe restriction on their applications in chemical field. Given the paucity of efficient synthetic methods and the huge challenges in the synthesis of 2-fluoroindole derivatives, the development of a convenient approach to synthesize 2-fluoroindole derivatives from simple and readily available starting materials is highly appealing and desirable.

A Importance of indoles and 2-fluoroindoles. B Approaches for the construction of 2-fluoroindoles. C Difluorocarbene enables to access 2-fluoroindoles from vinylanilines (this work).
Considering the difficulty in accessing the starting materials and limitations of substrates in the previous strategies, we wondered whether a strategy could be developed by using readily accessible starting materials in a one-step protocol. Retrosynthetic analysis of 2-fluoroindole is oriented to the o-alkenylanilines and a reagent which provides both C and F atoms (Fig. 1C). At this point, halodifluorinated reagents were considered due to their structural motif and their wide applications as fluorinated sources36,37. In addition, our recent research efforts culminated in discovery and development of several transformations featuring in situ generated difluorocarbene (:CF2) as C1 synthons to assemble various valuable N-containing compounds38,39,40,41,42,43,44. Moreover, the intramolecular capture of difluorocarbene by a thiolate and Frustrated Lewis Pairs was also developed45,46. Therefore, halodifluorinated reagent might be an ideal source to accomplish our goal. Inspired by our pioneering work38 and the previous research on interaction of tertiary amines with difluorocarbene47,48,49, and considering that difluoromethyl ammonium salt as the key intermediate from tertiary amine is readily decomposed to lead to a difluoromethyl anion in the reaction, we envisage that if a suitable electrophilic group is introduced into the substrate, difluorocarbene can be inserted into the targeted molecule via a nucleophilic attack with both carbon and fluorine atoms on, thus 2-fluoroindole framework might be resulted. There is a challenge in this hypothesis: maintenance of fluorine atom, since fluorine atoms from difluorocarbene are completely discarded and could not be reused in previous reactions, and it is an inevitable shortcoming for such transformations. To our delight, when we installed Michael reaction acceptors (MRAs)50,51 as electrophilic site on the substrates, a series of 2-fluoroindole derivatives were constructed smoothly (>90 examples). This strategy will open an avenue for difluorocarbene-involved transformations and will add significant synthetic values to fluorine chemistry.
Herein, we report the first general and highly efficient strategy for the synthesis of 2-fluoroindoles. This method would allow facile entry to construction of 2-fluoroindoles from simple and readily available starting materials under the mild conditions in the absence of transition-metal catalysis, and readily elaborate the late-stage modifications of pharmaceuticals and natural products with broad substrate scope and excellent functional group tolerance, and it will be a groundbreaking synthesis for 2-fluoroindole compounds, which will greatly promote the rapid development of fluorine chemistry and pharmaceutical chemistry52,53,54.
Results
Investigation of reaction conditions
To validate our conjecture, 3-(2-(dimethylamino)phenyl)-1-phenylprop-2-en-1-one (1a) and BrCF2COOEt (2a) were chosen as the model substrates using K2CO3 as base. To our delight, without any other additives in CH3CN, the desired 2-flurorindole product 3a was obtained in 76% isolated yield. Replacing K2CO3 with K3PO4 resulted in a superior result (Table 1, entry 7), and further base screening indicated that K3PO4 was the best one in comparison with KOH, Cs2CO3, Na2CO3, NaOH as well as Na3PO4 (entries 2–7). Encouraged by this promising result, we further screened other solvents (THF, 1,4-dioxane, DME and toluene), and CH3CN was found still to be the most effective one (entries 7–11). Difluorinated reagents were subsequently examined and the results suggested that among BrCF2COOEt (2a), BrCF2PO(OEt)2 (2b)55, TMSCF2Br (2c)56 ClCF2COONa (2d), BrCF2COOK (2e), BrCF2COONa (2f), the best reaction efficiency was endowed by 2a and 2b (entries 12–16). Given the price and the ready accessibility of raw materials, we finally chose 2a as the source of difluorocarbene (See Supplementary Tables 1–5 in Supplementary Information for details).
Synthetic scope
With the optimized conditions in hand, we systematically investigated the scope of the difluorocarbene-enabled access to 2-fluoroindoles from ortho-vinylanilines (Fig. 2). First, we explored the scope of α,β-unsaturated ketone moiety, which suggested that our reaction was compatible to both aryl and aliphatic α,β-unsaturated ketones. For aromatic ones, substrates bearing electron-neutral (1a–1b), electron-deficient (1c–1d), as well as electron-rich substituents (1e–1g) at the para-position of the aromatic rings all furnished the desired 2-fluoroindole products (3a–3g) smoothly. And with electronically neutral bis-methyl (1h) and ortho-methyl (1i) substrates, the corresponding products 3h–3i were procured in 81 and 93% yields respectively. The accommodation of iodine substituents (1j) signified the further potential structural elaborations as a handle. Fused ring reactant like 1-naphthaldehyde (1k) was also a suitable candidate for this transformation. This study was auspiciously and effortlessly extendable to a series of heteroaromatic ketone-containing furan (3l) and thiophene (3m) cores. And for cyclic aryl ketone (1o–1r), these targeted products could be obtained successfully under the standard conditions as well (3o–3r). Notably, this protocol also featured an admirable scope with respect to aliphatic ketone substrates. No matter that it was a chain ketone or a cyclic ketone, the corresponding desired products 3s–3ae were all smoothly delivered (52–80% yields), including four-membered (3ab), six-membered (3ac), eight-membered cyclic ketones (3ad) as well as bicyclic ketone (3ae). Interestingly, substrates tethered with two moieties of reaction sites (1n, 1af) underwent this transformation very well to deliver the products which contain two 2-fluoroindole skeletons (3n, 3af) by simple increasing the equivalent of BrCF2COOEt. We next surveyed the scope of R2 group on the aromatic ring of aniline skeleton. To our delight, a wide range of functionality was compatible under our standard conditions, and alkyl (1ag, 1aj), trifluoromethyl (1ah), alkoxyl (1ak) and halo (1ai, 1al–1an) substituted substrates were all converted into the corresponding products (3ag–3an) in moderate to excellent yields (39–81%). To assess the susceptibility of different C–N bonds towards scission, a panel of N-substituted tertiary amines was examined under the standard conditions. N-methylanilines with a different N-substituents (1ao, 1ap) were inspected, and when the R3 was ethyl group, N-ethylindole (3ao) was obtained in a satisfied yield. Interestingly, when the R3 is allyl group, contrary to expectations, it was cleaved prior to the Cmethyl–N bond and 3a was procured in 51% yield.

Reaction conditions: a1 (0.2 mmol), 2a (3 equiv., 0.6 mmol), K3PO4 (3 equiv.), H2O (0.1 mL), CH3CN (2 mL) under 90 °C for 12 h, N2. b1 (0.2 mmol), 2a (6 equiv., 1.2 mmol), K3PO4 (6 equiv.), H2O (0.15 mL), CH3CN (3 mL) under 90 °C for 12 h, N2. c24 h.
We next evaluated the scope of other Michael acceptors (Fig. 3), expectedly, these substrates were equipotent to afford the corresponding 2-fluoroindole products in moderate to excellent yields under marginally reoptimized conditions as follows: diethyl BrCF2PO(OEt)2 (2b) as the source of difluorocarbene, K2CO3 as the base and acetonitrile as the solvent at 90 °C for 12 h under N2 atmosphere (See Supplementary Tables 6–7 in Supplementary Information for details).

Reaction conditions: a4 (0.2 mmol), 2b (3 equiv., 0.6 mmol), K2CO3 (3 equiv.), H2O (0.1 mL), CH3CN (2 mL) under 90 °C for 12 h, N2. b6 (0.2 mmol), 2a (3 equiv., 0.6 mmol), K3PO4 (3 equiv.), CH3CN (2 mL) under 90 °C for 12 h, N2.
We then first explored α, β-unsaturated esters50,51. In addition to methyl acrylates, ethyl, tert-butyl and benzyl acrylates were also competent substrates to give the corresponding products (5a–5d) in moderate yields. Moreover, this system could also enable the synthesis of 2-fluoroindole scaffolds with other Michael acceptors such as acrylonitrile (4e) and para-quinone methides57 (6a–6e) with good efficiency, generating functional products (5e, 7a–7e) that are highly valuable structural motifs in medicinal chemistry. In order to further prove the universality of this reaction, we introduced other alkyl groups (like benzyl and allyl group) as substituents on N-atom, and also tried phosphonates (including phosphonic acid) as electron withdrawing groups. However, only trace amount of the corresponding products (7f–7i) was detected. We then tried several other difluoroalkylating reagents which could serve as the sources of difluorocarbene, but no good results were obtained.
We also paid our attention to the substrates with different cyclic tertiary amines, if successful, N-tethered long chain aliphatic halides would be rendered, which would add more values for further structural elaborations as a functional handle. The results were summarized in Fig. 4. It turned out that both four-membered (8a) and five-membered (8b–8u) cyclic tertiary amines were well-suited for this transformation, exhibiting insensitivity toward aryl substitutions (alkyl, alkoxyl, phenyl, halo, fused, thiophene and furan etc.). The corresponding target products (9a–9u) were procured in moderate yields. Remarkably, when KI as a nucleophile was added into the reaction system, the N-aliphatic-tethered-iodinated-2-fluoroindoles were obtained (9c, 9l, 9p) in decent yields accordingly.

Reaction conditions: a8 (0.2 mmol), 2a (3 equiv., 0.6 mmol), K3PO4 (3 equiv.), H2O (0.1 mL), CH3CN (2 mL) under 90 °C for 24 h, N2. b8 (0.2 mmol), 2a (3 equiv., 0.6 mmol), KI (3 equiv.), K3PO4 (3 equiv.), H2O (0.1 mL), CH3CN (2 mL) under 90 °C for 24 h, N2.
The above results clearly demonstrated that our method has a broad substrate scope and wide functional group compatibility, which further prompted our endeavors to extrapolate this strategy to late-stage modifications of bioactive molecules and therapeutic agents (Fig. 5). Gratifyingly, a series of bioactive molecules (Acetylferrocene, L-valine, DL-proline, Pregnenolone, Adamantane, Progesterone, Geraniol, (-)-Nopol, DL-Menthol, (-)-β-citronellol, Geraniol, Cholesterol) were all derivatized into the corresponding Michael acceptors and installed into our substrates (10a–o) which, upon treatment with BrCF2COOEt (2a) or BrCF2PO(OEt)2 (2b) under the established standard conditions, were all smoothly incorporated into the eventual 2-fluoroindole derivatives (11a–o). Moreover, drug compounds, such as (s)-Ibuprofen (10m, Antipyretic analgesics), Naproxen (10n, Anti-inflammatory), Probenecid (10o, Anti-gout agents) were also successfully introduced into the corresponding 2-fluoroindole derivatives (11m–o) without loss of efficiency. By combining the 2-fluoroindole skeleton and bioactive structural motifs together, it was thus envisioned that our system would simplify access to discover more potential bioactive molecules.

Reaction conditions: a10 (0.2 mmol), 2a (3 equiv., 0.6 mmol), K3PO4 (3 equiv.), H2O (0.1 mL), CH3CN (2 mL) under 90 °C for 12 h, N2. b10 (0.2 mmol), 2b (3 equiv., 0.6 mmol), K2CO3 (3 equiv.), CH3CN (2 mL) under 90 °C for 12 h, N2.
Synthetic application
To further demonstrate the synthetic utility of our strategy, we chose 5-chloro-N-methyl-2-fluoroindoles (13), the key intermediate for the synthesis of Syk (Spleen tyrosine kinase) inhibitors drug, as the target molecule (Fig. 6a). This reaction could be readily scaled-up from substrate 12a to 5 mmol without loss of efficiency (13a, 80% yield). Of note, according to the existing reports24, five-step synthesis was required for the construction of 5-chloro-N-methyl-2-fluoroindoles (13): (1) protection of N-H in indole with tosyl chloride, (2) trimethyltin was installed to activate indole by Sn(Me)3Cl, (3) fluorination with selectfluor, (4) removal of sufonamide by KOH, (5) dimethyl sulfate was introduced into the product as a methylation reagent. And for the starting material 12, it can be obtained by a sequential aromatic nucleophilic substitution between ortho-fluorobenzaldehyes and dimethylamines and aldol condensation between the aforementioned products and ketones. The final product (the starting material 12 for our reaction) could be obtained in the two-step synthesis with a total yield over 90%. And the price of the substrates is very cheap (ortho-fluorobenzaldehye: 1 $/g, dimethylamine: 0.03 $/g). Compared with the previous method, our route not only significantly decreases the step count (1 step vs. 5 steps), but also could avoid the use of strong base (KOH and NaH), toxic organotin reagents and dimethyl sulfate58,59. Meanwhile, our strategy could introduce very valuable functional groups in one step on C3 position of indoles as well. HYH42 is a highly specific inhibitor for human non-small cell lung cancer cells (NCI-H460, IC50 < 1 μM) which is commonly used in pharmaceutical research25. Remarkably, starting from readily accessible ortho-vinylaniline 15, our reaction enabled us to access the key intermediate 16 in a single step with a yield of 35% through the direct 2-fluoroindolylation reaction, then HYH42 can be obtained by coupling25,60 from the key intermediate 16 (Fig. 6b). Moreover, we utilized our methodology on the formal synthesis of 5a, which is the key intermediate of a potential plant growth hormone regulated transcription factor23, and the reaction could be scaled up to 5 mmol and the targeted product 5a was obtained in 64% yield (Fig. 6c). Of note, there is no other efficient report on its synthesis prior to our method.

Reaction conditions: a12 or 15 (0.2 mmol), 2a (3 equiv., 0.6 mmol), K3PO4 (3 equiv.), H2O (0.1 mL), CH3CN (2 mL) under 90 °C for 12 h, N2. b12a (5 mmol), 2a (3 equiv., 15 mmol), K3PO4 (3 equiv.), H2O (1.5 mL), CH3CN (25 mL) under 90 °C for 24 h, N2. c4a (5 mmol), 2b (3 equiv., 15 mmol), K2CO3 (3 equiv.), H2O (1.5 mL), CH3CN (25 mL) under 90 °C for 24 h, N2.
Mechanistic studies
Intrigued by the features of the presented methodology, we next conducted several control experiments to shed light on the reaction mechanism (Fig. 7). When water was replaced by deuterium oxide, one deuterium atom was incorporated at α-position of carbonyl group of the final product 3a-D with 95% deuterization rate in the crude mixture (Fig. 7, eq. a). When diflurocarbene trapping reagent, namely, benzimidazole (18), were added individually into this system, 1-(difluoromethyl)-1H-benzo[d]imidazole (19) was obtained in 78% yield alongside a trace amount of 3a, suggesting the existence of difluorocarbene species (Fig. 7, eq. b). We then considered whether other dihalocarbene species (:CCl2) are compatible with the reactions, of note, no desired products 20 were detected, which indicates that difluorocarbene species has some special characteristics, which isn’t possessed by other dihalocarbenes (Fig. 7, eq. c). In order to indicate the essence of tertiary amino group on the substrate, primary amine 21 was subjected to the reaction conditions, not surprisingly, no 2-fluoroindole formation was detected owing to the rapid cyclization to render 2-phenylquinoline 22 (Fig. 7, eq. d). Moreover, the HRMS (high resolution mass spectrometry) analysis of the mixture of the standard reaction for 30 min showed the two peaks at m/z = 303.1431 and m/z = 288.1199, which matched the ionic compound form possible intermediates 23 and 24 (calcd mass: 303.1429 and 288.1194) (Fig. 7, eq. e).

a Deuterium-labeling experiment. b Diflurocarbene trapping. c Control experiment of dichlorocarbene. d Experiments with non-substituted amino group. e Experiments with HRMS analysis.
Proposed mechanism
On the basis of the above results, a plausible mechanism is proposed as depicted in Fig. 8. Tertiary amine A reacts with the in situ generated difluorocarbene (:CF2) species which is unmasked from halodifluoroalkyl reagents (BrCF2COOEt (2a) or BrCF2PO(OEt)2 (2b)) in the presence of the base to deliver ammonium salt B. Internal nucleophile (X−) attacks the α carbon of ammonium salt B to break the C–N bond under mild and transition-metal free and oxidant-free conditions38,47. The newly formed difluoromethyl anion further attacks unsaturated double bond intramolecularly to render intermediate C via Michael addition and protonation. According to our previous work, the CF2 group on the nitrogen atom is unstable and readily to undertake C–F cleavage under the action of lone pair electrons of the nitrogen atom. Meanwhile, the hydrogen atom adjacent to CF2 is base-sensitive and easy to be deprotonated, thus a proposed E2cb mechanism is also possible. Based on the above discussion, there might be two possible paths to render products. One possible path is the Csp3–F bond adjacent to N atom is vulnerable in basic conditions to lead to intermediate D via a Csp3–F bond cleavage. Finally, with the participation of base, the final product is produced by the driving force to reconstruct the aromatic system (path a). Another way for the formation of the final product is caused by a E2cb pathway with the loss of H-F under basic conditions, once again, the driving force should be the reconstruction of the aromatic ring—indoles (path b).

Possible reaction mechanism of difluorocarbene enables to access 2-fluoroindoles from ortho-vinylanilines.
In summary, we have reported a straightforward synthesis of 2-fluoroindoles from readily accessible ortho-vinylanilines with halodifluoroalkylative reagents as difluorocarbene sources, which provide one carbon atom and one fluorine atom. This general and highly selective transformation provides operationally simple and robust access to versatile 2-fluoroindoles under a mild condition without any transition metals or oxidants. The products obtained can further engage in various derivatizations where the 2-fluoroindole motif functions as an ideal drug skeleton, enabling a fast and orthogonal transformation to many useful building blocks. In a broader context, these features emphasize the value of the presented methodology for the versatile synthesis of 2-fluoroindoles derivatives which are ubiquitously found in bioactive molecules. Further investigations to extend the reaction scope and applications of this process are currently in progress.
Methods
General procedure for synthesis of 2-fluoroindoles from chalcones or p-Quinone Methide
In air, chalcones or p-Quinone Methide (0.2 mmol) and K3PO4 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a stir bar. The vessel was evacuated and filled with N2 (three cycles). BrCF2COOEt (3 eq, 0.6 mmol), H2O (0.1 mL) and CH3CN (2 mL) added in turn by syringe under N2 atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 12 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography to give the desired products.
General procedure for synthesis of 2-fluoroindoles from α, β-unsaturated esters or acrylonitrile
In air, α, β-unsaturated esters (0.2 mmol) or acrylonitrile and K2CO3 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a stir bar. The vessel was evacuated and filled with N2 (three cycles). BrCF2PO(OEt)2 (3 eq, 0.6 mmol), H2O (0.1 mL) and CH3CN (2 mL) added in turn by syringe under N2 atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 12 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography to give the desired products.
General procedure for synthesis of N-tethered long chain aliphatic bromine
In air, cyclic tertiary amines (0.2 mmol) and K3PO4 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a stir bar. The vessel was evacuated and filled with N2 (three cycles). BrCF2COOEt (3 eq, 0.6 mmol), H2O (0.1 mL) and CH3CN (2 mL) added in turn by syringe under N2 atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 24 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography to give the desired products.
General procedure for synthesis of N-tethered long chain aliphatic iodine
In air, cyclic tertiary amines (0.2 mmol), KI (3 eq, 0.2 mmol) and K3PO4 (3 eq, 0.6 mmol) were added to a Schlenk tube equipped with a stir bar. The vessel was evacuated and filled with N2 (three cycles). BrCF2COOEt (3 eq, 0.6 mmol), H2O (0.1 mL) and CH3CN (2 mL) added in turn by syringe under N2 atmosphere. The resulting reaction mixture was stirred vigorously at 90 °C for 24 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure and the residue was purified by flash column chromatography to give the desired products.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2049500 (3c), 2049496 (3j), 2066928 (3af) and 2054216 (9k). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http:// www.ccdc.cam.ac.uk/data_request/cif.
References
“Fluoropolymers”: Topics in Applied Chemistry (Eds.: Hougham, G., Cassidy, P. E., Johns, K. & T. Davidson), Kluwer Academic Publishers, New York City, 2002.
Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Hagmann, W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 51, 4359–4369 (2008).
Wang, J. et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001-2011). Chem. Rev. 114, 2432–2506 (2014).
Gillis, E. P., Eastman, K. J., Hill, M. D., Donnelly, D. J. & Meanwell, N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 58, 8315–8359 (2015).
Aufiero, M. & Gilmour, R. Informing molecular design by stereoelectronic theory: the fluorine Gauche effect in catalysis. Acc. Chem. Res. 51, 1701–1710 (2018).
Li, X.-T., Gu, Q.-S., Dong, X.-Y., Meng, X. & Liu, X.-Y. Cinchona alkaloid-based sulfonamide/copper catalyst for asymmetric radical oxytrifluoromethylation of alkenyl oximes. Angew. Chem. Int. Ed. 57, 7668–7672 (2018).
Vogt, D. B., Seath, C. P., Wang, H. & Jui, N. T. Selective C-F functionalization of unactivated trifluoromethylarenes. J. Am. Chem. Soc. 141, 13203–13211 (2019).
Lin, J.-S. et al. A dual-catalytic strategy to direct asymmetric radical aminotrifluoromethylation of alkenes. J. Am. Chem. Soc. 138, 9357–9360 (2016).
Seath, C. P., Vogt, D. B., Xu, Z., Boyington, A. J. & Jui, N. T. Radical hydroarylation of functionalized olefins and mechanistic investigation of photocatalytic pyridyl radical reactions. J. Am. Chem. Soc. 140, 15525–15534 (2018).
Iwamoto, H., Imiya, H., Ohashi, M. & Ogoshi, S. Cleavage of C(sp3)-F bonds in trifluoromethylarenes using a Bis(NHC)nickel(0) complex. J. Am. Chem. Soc. 142, 19360–19367 (2020).
Doi, R., Ohashi, M. & Ogoshi, S. Copper‐catalyzed reaction of trifluoromethylketones with aldehydes via a copper difluoroenolate. Angew. Chem., Int. Ed. 55, 341–344 (2016).
Feng, Z., Min, Q.-Q., Fu, X.-P., An, L. & Zhang, X. Chlorodifluoromethane-triggered formation of difluoromethylated arenes catalysed by palladium. Nat. Chem. 9, 918–923 (2019).
Fu, X.-P. et al. Controllable catalytic difluorocarbene transfer enables access to diversified fluoroalkylated arenes. Nat. Chem. 11, 948–956 (2019).
Li, Z.-L., Li, X.-H., Wang, N., Yang, N.-Y. & Liu, X.-Y. Radical-mediated 1,2-formyl/carbonyl-functionalization of alkenes and application to construction of medium-sized-, fused-, and bridged-ring systems. Angew. Chem., Int. Ed. 55, 15100–15104 (2016).
Kochanowska-Karamyan, A. J. & Hamann, M. T. Marine indole alkaloids: potential new drug leads for the control of depression and anxiety. Chem. Rev. 110, 4489–4497 (2010).
Higuchi, K. & Kawasaki, T. Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat. Prod. Rep. 24, 843–868 (2007).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Stempel, E. & Gaich, T. Cyclohepta[b]indoles: a privileged structure motif in natural products and drug design. Acc. Chem. Res. 49, 2390–2402 (2016).
Tomoo, T. et al. Design, Synthesis, and biological evaluation of 3-(1-Aryl-1H-indol-5-yl)propanoic acids as new indole-based cytosolic phospholipase A2α inhibitors. J. Med. Chem. 57, 7244–7462 (2014).
Spencer, C. M., Gunasekara, N. S. & Hills, C. Zolmitriptan. Drugs 58, 347–374 (1999).
Schachtman, D. P. & Shin, R. Y. Plant growth hormone regulated transcription factors and promoters there of. WO2007127923A2. (2007).
Fujiwara, H. et al. Preparation of nicotinamide derivatives as Syk inhibitors. WO2012002577. (2012).
Hu, Y. et al. Preparation of aryl heterocyclic small-molecular compounds and derivatives as tubulin inhibitors for treating cancer. CN 2013-10312579, (2015).
Truong, T., Klimovica, K. & Daugulis, O. Copper-catalyzed, directing group-assisted fluorination of arene and heteroarene C-H bonds. J. Am. Chem. Soc. 135, 9342–9345 (2013).
Lee, S. J., Makaravage, K. J., Brooks, A. F., Scott, P. J. H. & Sanford, M. S. Copper-mediated aminoquinoline-directed radiofluorination of aromatic C-H bonds with K18F. Angew. Chem. Int. Ed. 59, 3119–3122 (2019).
Yuan, X., Yao, J.-F. & Tang, Z.-Y. Decarboxylative fluorination of electron-rich heteroaromatic carboxylic acids with selectfluor. Org. Lett. 19, 1410–1413 (2017).
Hodson, H. F., Madge, D. J. & Widdowson, D. A. Regioselective Electrophilic Fluorination of Alkenyl and Related Stannanes Using Caesium Fluoroxysulfate. Synlett 1992, 831–832 (1992).
Zhang, B., Zhang, X., Hao, J. & Yang, C. Direct approach to N‑substituted-2-fluoroindoles by sequential construction of C-N bonds from gem-difluorostyrenes. Org. Lett. 19, 1780–1783 (2017).
Fujita, T. et al. Silver-catalyzed vinylic C-F bond activation: synthesis of 2-fluoroindoles from β, β-difluoro-o-sulfonamidostyrenes. Chem. Lett. 45, 964–966 (2016).
Ichikawa, J., Wada, Y., Okauchi, T. & Minami, T. 5-endo-Trigonal cyclization of o-substituted gem-difluorostyrenes: syntheses of 2-fluorinated indoles, benzo[b]furans and benzo[b]thiophenes. Chem. Commun. 51, 1537–1538 (1997).
Fujita, T., Sakoda, K., Ikeda, M., Hattori, M. & Ichikawa, J. Nucleophilic 5-endo-trig cyclization of 3,3-difluoroallylic ketone enolates: synthesis of 5-fluorinated 2-alkylidene-2,3-dihydrofurans. Synlett 24, 57–60 (2013).
Fujita, T., Ikeda, M., Hattori, M., Sakoda, K. & Ichikawa, J. Nucleophilic 5-endo-trig cyclization of 3,3-difluoroallylic metal enolates and enamides: facile synthesis of ring-fluorinated dihydroheteroles. Synthesis 46, 1493–1505 (2014).
Nosova, E. V., Lipunova, G. N., Charushin, V. N. & Chupakhin, O. N. Fluorine-containing indoles: synthesis and biological activity. J. Fluor. Chem. 212, 51–106 (2018).
Ma, X. & Song, Q. Recent progress on selective deconstructive modes of halodifluoromethyl and trifluoromethyl-containing reagents. Chem. Soc. Rev. 49, 9197–9219 (2020).
Dilman, A. D. & Levin, V. V. Difluorocarbene as a building block for consecutive bond-forming reactions. Acc. Chem. Res. 51, 1272–1280 (2018).
Su, J., Ma, X., Ou, Z. & Song, Q. Deconstructive functionalizations of unstrained carbon-nitrogen cleavage enabled by difluorocarbene. ACS Cent. Sci. 6, 1819–1826 (2020).
Deng, S. et al. S8-Catalyzed triple cleavage of bromodifluoro compounds for the assembly of N-containing heterocycles. Chem. Sci. 10, 6828–6833 (2019).
Ma, X., Zhou, Y. & Song, Q. Synthesis of β-aminoenones via cross-coupling of in situ-generated isocyanides with 1,3-dicarbonyl compounds. Org. Lett. 20, 4777–4781 (2018).
Ma, X., Mai, S., Zhou, Y., Cheng, G.-J. & Song, Q. Dual role of ethyl bromodifluoroacetate in the formationof fluorine-containing heteroaromatic compounds. Chem. Commun. 54, 8960–8963 (2018).
Ma, X., Su, J., Zhang, X. & Song, Q. Chlorodifluoromethane as a C1 synthon in the assembly of N-containing compounds. iScience 19, 1–13 (2019).
Zhang, G. et al. Atom recombination of difluorocarbene enables 3-fluorinated oxindoles from 2-aminoarylketones. CCS Chem. 3, 1613–1621 (2021).
Ma, X., Xuan, Q. & Song, Q. N-H and O-H difluoromethylation of N-Heterocycles. Huaxue Xuebao 76, 972–976 (2018).
Liang, H. et al. Difluorocarbene-triggered cyclization: synthesis of (Hetero)arene-fused 2,2-difluoro-2,3-dihydrothiophenes. Org. Lett. 22, 7047–7051 (2020).
Smirnov, V. O., Volodin, A. D., Korlyukov, A. A. & Dilman, A. D. Trapping of difluorocarbene by frustrated Lewis Pairs. Angew. Chem. Int. Ed. 59, 12428–12431 (2020).
Kim, Y., Heo, J., Kim, D., Chang, S. & Seo, S. Ring-opening functionalizations of unstrained cyclic amines enabled by difluorocarbene transfer. Nat. Commun. 11, 4761–4771 (2020).
Mita, T., Harabuchi, Y. & Maeda, S. Discovery of a synthesis method for a difluoroglycine derivative based on a path generated by quantum chemical calculations. Chem. Sci. 11, 7569–7577 (2020).
Nawrot, E. & Jon˜czyk, A. Difluoromethyltrialkylammonium saltss their expeditious synthesis from chlorodifluoromethane and tertiary amines in the presence of concentrated aqueous sodium hydroxide. The Catalytic Process. J. Org. Chem. 72, 10258–10260 (2007).
Allgäuer, D. S. et al. Quantification and theoretical analysis of the electrophilicities of michael acceptors. J. Am. Chem. Soc. 139, 13318–13329 (2017).
Mayer, R. J. & Ofial, A. R. Nucleophilicity of glutathione: a link to michael acceptor reactivities. Angew. Chem. Int. Ed. 58, 7704–17708 (2020).
Gouverneur, V. & Seppelt, K. Introduction: fluorine chemistry. Chem. Rev. 115, 563–565 (2015).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Feng, Z., Min, Q. ‐Q., Xiao, Y. ‐L., Zhang, B. & Zhang, X. Palladium‐catalyzed difluoroalkylation of aryl boronic acids: a new method for the synthesis of aryldifluoromethylated phosphonates and carboxylic acid derivatives. Angew. Chem. Int. Ed. 53, 1669–1673 (2014).
Zafrani, Y., Sod-Moriah, G. & Segall, Y. Diethyl bromodifluoromethylphosphonate: a highly efficient and environmentally benign difluorocarbene precur. Tetrahedron 65, 5278–5283 (2009).
Krishnamoorthy, S. & Prakash, G. K. S. Silicon-based reagents for difluoromethylation and difluoromethylenation reactions. Synthesis 49, 3394–3406 (2017).
Alamsetti, S. K., Spanka, M. S. M. & Schneide, C. Synergistic rhodium/phosphoric acid catalysis for the enantioselective addition of oxonium ylides to ortho‐quinone methides. Angew. Chem., Int. Ed. 55, 2392–2396 (2016).
Vieira, F. T. et al. Synthesis, characterization and biocidal activity of new organotin complexes of 2-(3-oxocyclohex-1-enyl)benzoic acid. Eur. J. Med. Chem. 45, 883–889 (2010).
Selva, M. & Perosa, A. Green chemistry metrics: a comparative evaluation of dimethyl carbonate, methyl iodide, dimethyl sulfate and methanol as methylating agents. Green. Chem. 10, 457–464 (2008).
Mfu, A. M., Doyle, J. D., Chhetri, B., Arman, H. D. & Larionov, O. V. Scalable, metal- and additive-free, photoinduced borylation of haloarenes and quaternary arylammonium salts. J. Am. Chem. Soc. 138, 2985–2988 (2016).
Acknowledgements
Financial support from the National Natural Science Foundation (21772046 and 2193103) is gratefully acknowledged. The authors also thank the Instrumental Analysis Center of Huaqiao University for analysis support. J.S. thank the Subsidized Project for Cultivating Postgraduates’ Innovative Ability in Scientific Research of Huaqiao University.
Author information
Authors and Affiliations
Contributions
Q.S. designed and directed the project. J.S. performed the experiments and developed the reactions. X.H., H.H. and Y.G. helped collecting some experimental data. Q.S. and J.S. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Pingping Tang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, J., Hu, X., Huang, H. et al. Difluorocarbene enables to access 2-fluoroindoles from ortho-vinylanilines. Nat Commun 12, 4986 (2021). https://doi.org/10.1038/s41467-021-25313-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25313-z
This article is cited by
-
Single-atom skeletal editing of 2H-indazoles enabled by difluorocarbene
Science China Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
