
Abstract
Although chirality is an ever-present characteristic in biology and some artificial molecules, controlling the chirality and demystifying the chirality origin of complex assemblies remain challenging. Herein, we report two homochiral Ag14 nanoclusters with inherent chirality originated from identical rotation of six square faces on a Ag8 cube driven by intra-cluster π···π stacking interaction between pntp− (Hpntp = p-nitrothiophenol) ligands. The spontaneous resolution of the racemic (SD/rac-Ag14a) to homochiral nanoclusters (SD/L-Ag14 and SD/R-Ag14) can be realized by re-crystallizing SD/rac-Ag14a in acetonitrile, which promotes the homochiral crystallization in solid state by forming C–H···O/N hydrogen bonds with nitro oxygen atoms in pntp− or aromatic hydrogen atoms in dpph (dpph = 1,6-bis(diphenylphosphino)hexane) on Ag14 nanocluster. This work not only provides strategic guidance for the syntheses of chiral silver nanoclusters in an all-achiral environment, but also deciphers the origin of chirality at molecular level by identifying the special effects of intra- and inter-cluster supramolecular interactions.
Similar content being viewed by others

Introduction
Chirality is ubiquitous in (supra)molecular structures, such as DNA in living organisms and natural products in plants, which plays a pivotal role in biological activity, catalysis, medicines, and a variety of other applications1,2. Although the enantioselective preparation and separation of chiral small molecules induced by chiral reagents have been well established, the realization of this process with achiral reagents still faces great challenges, especially for these higher-order motifs such as nanoclusters, nanoparticles, and supramolecules3,4,5,6,7,8,9,10,11,12,13,14. Recent research on the origin of chirality formed in achiral systems has led to great advances in chiral metal nanoclusters protected by achiral ligands; the origin of chirality of them has been typically classified into three types: (1) chiral arrangement of inner metal core atoms;15,16 (2) asymmetric arrangement of surface structure to form a chiral shell;17,18,19,20,21 (3) distortion or rotation induced structure chirality22,23,24. The majority of chirality typically belongs to the second type in nanoclusters because inter-ligand non-covalent interactions such as π···π stacking, C–H···π, hydrogen bond interactions etc. can effectively drive the asymmetric arrangement of surface structures25. While the last type is rather difficult to access in experiments due to the more subtle influencing factors. As we know, while the silver nanoclusters are more likely to adopt polyhedral structures with high symmetry, such as Ag180, Ag100, Ag90, Ag63, Ag48, Ag46, Ag40, and Ag3826,27,28,29,30,31, those with chiral polyhedral cores, however, are rarely reported32.
For achiral ligands protected chiral nanoclusters, a long-standing challenge lies in the fact that most of them will crystallize as racemates, and highly efficient enantioseparation technologies are thus urgently needed. To date, some enantioseparation technologies such as high-performance liquid chromatography, capillary electrophoresis and chiral ion pairs have been developed to separate enantiomers, but there are still some limitations to enantioseparation for most racemates33,34,35,36,37,38. For example, for some chiral molecules that cannot maintain a stable chiral conformation in solution, above enantioseparation technologies operated in solution phase will not be efficient enough. Thus, the solvent-induced crystallization resolution may serve as an upgrade alternative to achieve enantiomer separation.
In this work, we firstly isolate a racemic Ag14 nanocluster ([Ag14(pntp)10(dpph)4Cl2], SD/rac-Ag14a, Hpntp = p-nitrothiophenol and dpph = 1,6-bis(diphenylphosphino)hexane) in acetone/CH2Cl2, which is further subjected to a separation into their component enantiomers, SD/L-Ag14 and SD/R-Ag14, by re-crystallizing SD/rac-Ag14a in acetonitrile. As revealed by single-crystal X-ray diffraction (SCXRD), the inherent chirality of Ag14 nanocluster originates from identical rotational directionality of six square faces on Ag8 cube, which is mainly driven by intra-cluster π···π stacking interaction between pntp− ligands, while homochiral crystallization is promoted by C–H···O/N hydrogen bonds formed between lattice acetonitrile and the nitro oxygen atoms in pntp− or aromatic hydrogen atoms in dpph on Ag14 nanocluster. The enantiomeric conglomerates show mirror-imaged circular dichroism (CD) and circularly polarized luminescence (CPL) responses. These results give us precise answers at molecular-level to (i) How to desymmetrize the highly symmetrical polyhedral silver nanocluster to become a chiral one; (ii) what is responsible for the chirality of the overall molecule; (iii) what is the driving force to enantioseparate racemates during the crystallization.
Results
Syntheses and characterizations of SD/rac-Ag14a and SD/L/R-Ag14
The chiral Ag14 nanocluster was one-pot synthesized as racemates (SD/rac-Ag14a) using phosphine (dpph or PPh3), Hpntp, and CF3COOAg with a ratio of 1:2:2 in acetone/CH2Cl2 at room temperature. The filtrate was evaporated slowly to produce orange rod-like crystals of SD/rac-Ag14a. NaBH4 was chosen as the reducing agent to obtain the subvalent kernel Ag64+. The amount of triethylamine has a nontrivial influence on the dimensions of single crystals. The 0–100 μL triethylamine has been attempted in this reaction system with 10 μL as an interval and larger single crystals can be formed in the case of 40 μL triethylamine. Furthermore, the other two bases such as NaOH and N,N,N′,N′-Tetramethylethylenediamine (TMEDA) were also tried in the synthesis of Ag14 nanocluster. The TMEDA can also work as triethylamine, whereas the NaOH is not the case; this is probably due to the slower reduction kinetics of NaBH4 in NaOH, which impedes the formation of Ag14 nanocluster. The growth of large-sized single crystals of the enantiopure SD/L/R-Ag14 is difficult but necessary for collecting their more reliable solid-state CD and CPL signals. The detailed synthetic route to SD/Ag14 is shown in the Supplementary Fig. 1.
Separation of the racemates into single enantiomer by spontaneous resolution is very hard to achieve for metal nanoclusters because of inherent difficulty during crystallization. Therefore we try to enantioseparate SD/rac-Ag14a by tuning the solvent system. Initially, SD/rac-Ag14a crystallized in the C2/c space group in acetone/CH2Cl2. In comparison, the enantiomeric SD/L-Ag14 and SD/R-Ag14 can be directly obtained in MeCN/CH2Cl2 with a crystallographic space group of P212121. They possess completely different morphology (block) with respect to SD/rac-Ag14a (rod) (Supplementary Fig. 2). We also found that re-crystallization of SD/rac-Ag14a in MeCN can also produce enantiomeric SD/L-Ag14 and SD/R-Ag14, which can convert back to SD/rac-Ag14a through re-crystallization in acetone (Fig. 1). All these results suggest the important role of solvent in regulating the crystallization of chiral silver nanoclusters.

Solvent-induced spontaneous resolution of SD/rac-Ag14a to SD/L-Ag14 and SD/R-Ag14. Color labels: cyan and pink, Ag; yellow, S; orange, P; green, Cl; gray, C; blue, N; red, O. All H atoms are omitted for clarity.
Apart from SCXRD, SD/rac-Ag14a, SD/L-Ag14, and SD/R-Ag14 were thoroughly characterized by energy dispersive spectroscopy (EDS), nuclear magnetic resonance spectroscopy (NMR), electrospray ionization mass spectrometry (ESI-MS), ultraviolet-visible (UV-Vis), powder X-ray diffraction (PXRD) and infrared spectroscopy (IR). These characterizations, additional structural graphics, as well as crystallography-related information are collected in the Supplementary Figs. 3–23, Supplementary Tables 1–6. Moreover, density functional theory (DFT) calculations were also performed to understand the electronic structure and optical properties.
Crystal structure of SD/rac-Ag14a
SCXRD analysis determined the molecular formula of Ag14 nanocluster to be [Ag14(pntp)10(dpph)4Cl2]. SD/rac-Ag14a crystallizes in the monoclinic space group C2/c with a complete cluster in the asymmetric unit (Supplementary Table 1). The symmetry breaking of Ag14 nanocluster with alternating packing of opposite handedness ones finally results in racemization, therefore the unit cell of SD/rac-Ag14a contains two equivalent enantiomeric nanoclusters, SD/L-Ag14 and SD/R-Ag14. The Ag14 nanocluster is composed of 14 silver atoms, 10 pntp− ligands, 4 dpph ligands, and two chlorides (Fig. 2a). The EDS mapping clearly demonstrate the uniform distribution of elements in the nanoclusters and the presence of Cl (Supplementary Fig. 3). The Ag14 nanocluster can be regarded as a core-shell structure, with the octahedral Ag6 as the core surrounded by a twisted Ag8 cube as the shell. SD/rac-Ag14a is a neutral cluster as indicated by ESI-MS discussed below, thus two additional electrons should be injected into it, making the central Ag6 octahedron +4 charged39. The distorted Ag8 cube breaks the overall Oh symmetry, and therefore endows the nanocluster with chirality (Fig. 2a). In addition, the weak π···π interactions between pntp− ligands with the centroid-centroid distance ranging from 3.55 to 3.77 Å are clearly observed (Fig. 2b)40,41.

a Mirror symmetry of the enantiomers with Ag6 core and Ag8 shell depicted as colored polyhedra. b The coordination mode of pntp− (Hpntp = p-nitrothiophenol) and the π···π interaction (black dashed lines) between pntp− ligands in SD/rac-Ag14a. c The coordination mode of dpph (dpph = 1,6-bis(diphenylphosphino)hexane). Color labels: cyan and pink, Ag; yellow, S; orange, P; green, Cl; gray, C; blue, N; red, O. All H atoms are omitted for clarity.
SD/L-Ag14 was chosen as the representative for more detailed structural analysis. The Ag···Ag distance in Ag6 fall in the range of 2.79–2.89 Å (Supplementary Table 2), indicating the presence of argentophilic interactions42,43,44. The coordination modes of pntp− and dpph are shown in Figs. 2b, c. The ten pntp− ligands ride on ten edges of Ag8 cube, whereas the remaining two edges are ridden by two Cl− ions. Each pntp− ligand adopts a μ3 bridging mode to bind two silver atoms on an edge and one apical silver atom from Ag6 octahedron (Fig. 2b). The Agoctahedron–S bond lengths lie in the range of 2.48–2.55 Å and the Agcube–S bond lengths in the range of 2.53–2.70 Å (Supplementary Table 2). The S–Ag–S bond angles lie in the range of 86.20–120.04° (Supplementary Table 2). Four of twelve edges of Ag8 cube are additionally bridged by four μ2-dpph ligands (Ag–P: 2.40–2.47 Å, Supplementary Table 2). The steric hindrance of dpph impedes the full coordination of large pntp− ligand at all edges of Ag8 cube and two of them are bound by small Cl− ion instead. The coordination behavior of Cl− ion is quite similar to that of S atom of pntp− ligand (Ag-Cl: 2.61–2.83 Å). Of note, the 31P NMR spectrum shows eight different 31P chemical shifts with nearly identical integration values ranging from −1.79 to 2.02 ppm in the CD2Cl2, which suggests all four dpph ligands in SD/L-Ag14 and SD/R-Ag14 locate in completely different chemical environments, ruling out any symmetry for the overall structure in solution (Supplementary Fig. 4).
Based on the above crystallographic analyses, we make two hypotheses responsible for the origin of chirality: (i) the dpph immobilizes the asymmetric arrangement of vertex Ag atoms of Ag8 cubic shell; (ii) the locking of identical rotational directionality (Supplementary Fig. 6) of six square faces on Ag8 cube by the π···π interaction between the pntp− ligands. To verify the above hypotheses, the dpph ligand was replaced by PPh3 under similar synthetic conditions and another similar racemic nanocluster SD/rac-Ag14b was isolated. Some structural diagrams and crystallographic tables for it are shown in Supplementary Information. A detailed analysis of SD/rac-Ag14b revealed that the π···π interaction between the pntp− ligands and the identical rotational directionality of six square faces on Ag8 cube still exist (Supplementary Fig. 7), even in the absence of the immobilization of bridging dpph ligands, which means the factor (ii) may dominate the symmetry breaking of Ag8 cube.
In the 2 × 2 × 2 cell of SD/rac-Ag14a, the Ag14 nanoclusters with opposite chirality alternatively align in the planes parallel to crystallographic ab plane (Fig. 3a, b), causing the final racemic crystals. The inter-cluster Cdpph–H···Onitro hydrogen bonds (2.53–2.59 Å; Fig. 3c) consolidate the packing of homochiral Ag14 nanoclusters in the same layer. The homochiral layers composed of SD/L-Ag14 or SD/R-Ag14 alternate in an AB-type packing fashion along the c axis through similar hydrogen bonds (2.35–2.84 Å; Fig. 3d).

a Packing structure of enantiomers in a 2 × 2 × 2 cell viewed along the c axis. b Packing structure of enantiomers in a 2 × 2 × 2 cell viewed along the a axis. c Zoom-in inter-cluster weak interactions in the same layer. d Zoom-in inter-cluster weak interactions between neighboring layers. Color labels: cyan and pink, Ag; yellow, S; orange, P; gray, C; green, Cl; blue, N; red, O. white, H. Black dashed lines indicate the weak interactions.
Crystal structures of SD/L-Ag14 and SD/R-Ag14
To realize the homochiral crystallization of SD/rac-Ag14a for the following chiroptical studies, we changed the acetone/CH2Cl2 to MeCN/CH2Cl2 to get enantiomeric SD/L-Ag14 and SD/R-Ag14. The bulk samples of them were collected by manually picking single crystals then checking their configurations one by one by SCXRD. Crystallographic analysis showed that the unit cell indeed contains homochiral nanoclusters, and additionally co-crystallized MeCN was identified, thus the molecular formula of enantiomeric Ag14 nanoclusters is [Ag14(pntp)10(dpph)4Cl2]·MeCN (SD/L-Ag14 or SD/R-Ag14). SD/L-Ag14 and SD/R-Ag14 both crystallized in an orthorhombic space group of P212121 (Supplementary Table 1). All 40 crystals grown in one beaker from one-batch synthesis were analyzed by SCXRD and the ratio of SD/L-Ag14 and SD/R-Ag14 is 21: 19, indicating the spontaneous enantiomer resolution occurs (Supplementary Table 3).
To obtain deep insights into the chiral origin of SD/L-Ag14 and SD/R-Ag14, the rotation direction of six square faces of Ag8 cube are scrutinized by unfolding them in a two-dimensional plane (Fig. 4). It can be clearly seen that every square face of the Ag8 cube shows the same rotation direction in SD/L-Ag14 or SD/R-Ag14. Such rotational configuration is locked by the π···π interaction between the pntp− ligands to maintain such chirality in solid state. In both, codirectional face-rotation of polyhedron generates the inherent chirality, which is the origin of chirality of Ag14 nanocluster.

The rotational direction of six square faces of Ag8 cube in SD/L-Ag14 (left) and SD/R-Ag14 (right) nanoclusters. Color labels: pink, Ag; yellow, S; orange, P; green, Cl. All C, N, O, and H atoms are omitted for clarity.
Why does the CH3CN has such magic to homochiral crystallization of Ag14 nanocluster? We chose the packing structure of SD/L-Ag14 as a representative for the following analysis. In a 2 × 2 × 2 cell of SD/L-Ag14, the homochiral Ag14 nanoclusters are self-assembled into one-dimensional columnar arrangements along b axis (Fig. 5a, c). Such assembly is mainly driven by hydrogen bonds which contain non-classic but important Cacetonitrile–H···Onitro and Cdpph–H···Nacetonitrile hydrogen bonds (Cacetonitrile–H···Onitro and Cdpph–H···Nacetonitrile: 2.44–2.67 Å; Fig. 5b).

a Packing structure of SD/L-Ag14 in a 2 × 2 × 2 cell viewed along the c axis. b Zoom-in inter-cluster Cacetonitrile–H···Onitro and Cdpph–H···Nacetonitrile hydrogen bonds in the same layer (dpph = 1,6-bis(diphenylphosphino)hexane). c Packing structure of SD/L-Ag14 in a 2 × 2 × 2 cell viewed along the a axis. Color labels: cyan, Ag; yellow, S; orange, P; gray, C; green, Cl; blue, N; red, O; white, H. Black dashed lines indicate the weak interactions.
Mass spectrometry of SD/L-Ag14 and SD/R-Ag14
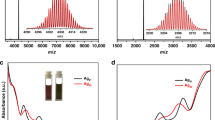
As we know, ESI-MS is a powerful analyzing tool to verify the chemical composition, charge state and the stability of metal nanoclusters45,46,47,48. The positive-ion ESI-MS of SD/L-Ag14 and SD/R-Ag14 was measured by dissolving them in methanol/CH2Cl2. As shown in Supplementary Fig. 8, there are three most prominent peaks in the mass-to-charge ratio (m/z) ranges of 4415–4435 (A), 4955–4972 (B), and 5368–5388 (C), respectively. Each group of isotopic peaks of them contains monovalent species, where the most dominant peak (B) is intact molecular ion species but with one sodium attachment. The peak B is located at 4964.3536 and can be assigned to [NaAg14(pntp)10(dpph)4Cl2]+ (SD/L-Ag14 + Na+; Calcd. 4964.3537), therefore, the Ag14 nanocluster is a 2-electron superatom system. The three species were identified based on the perfect matching of observed and simulated isotopic distributions. All these assigned formulas are listed in Supplementary Table 4.
The solid-state UV-Vis and CD spectra of SD/L-Ag14 and SD/R-Ag14
The UV-Vis diffuse reflection spectra of SD/rac-Ag14a, SD/L-Ag14, and SD/R-Ag14 show the same absorption peaks in solid state at room temperature, indicating that the cluster packing of nanoclusters does not essentially contribute to the optical absorbance. The broad absorption at 300–600 nm (Fig. 6a), consistent with its orange color of crystals, probably involve the ligand-to-ligand charge transfer (LLCT) mixed with small proportion of metal-to-ligand charge transfer (MLCT) transitions (vide infra). The CD and corresponding absorption spectra of SD/L-Ag14 and SD/R-Ag14 were also collected at 300–600 nm in solid state (Fig. 6b and Supplementary Fig. 9). The CD spectra of SD/L-Ag14 and SD/R-Ag14 are nearly perfect mirror images of each other, confirming their enantiomeric relationship; the broad absorption peaks are split into several peaks by the Cotton effect, including a major peak at around 355 nm and other less resolved peaks ranging from 400 to 500 nm.

a UV-Vis absorption spectrum of racemic conglomerates SD/L-Ag14 and SD/R-Ag14 in solid state. b Circular dichroism spectra of the SD/L-Ag14 and SD/R-Ag14 in solid state.
To further understand the origin of chirality, taking SD/L-Ag14 as an example, its metal core after DFT optimization in gas phase remains almost intact and thus its calculated CD spectrum can be used to help interpret the origin of its chirality. The calculated CD spectrum of SD/L-Ag14 matches fairly well with its experimental spectrum in the region below 500 nm, given the blue-shift in its calculated UV-Vis spectrum (Supplementary Fig. 10). The broad calculated absorption peak was also split into three peaks by Cotton effect, including two positive peaks at 355 nm and 457, respectively, and one broad negative peak at 400 nm. All the CD responses are mainly associated with the LLCT transitions, despite the small proportion of metal-to-metal charge transfer (MMCT) and MLCT transitions, as illustrated in its UV-Vis absorption analysis (Supplementary Fig. 14 and Supplementary Table 6). This suggests that the chiral response in the Ag14 nanocluster mainly originates from the asymmetrical arrangement of the surface ligands, which is induced by non-covalent interactions (mainly the π···π stacking interaction) between pntp− ligands.
Photoluminescent and CPL properties
The photoluminescent properties of SD/L-Ag14 and SD/R-Ag14 were examined in solid state and exhibited maximum emission around at 680 nm under the excitation of 365 nm at room temperature (Fig. 7a). The variable-temperature fluorescence spectrum of SD/rac-Ag14a (Supplementary Fig. 11) is almost identical to those of SD/L-Ag14 and SD/R-Ag14. The luminescence lifetimes are also determined in microsecond regime at 83 and 293 K (Supplementary Fig. 12), indicating that excited states exist in a triplet state. Upon cooling from 293 to 83 K, the emission maximum slightly blue-shifted from 680 to 668 nm (Δλ = 12 nm) with a 9-fold enhancement in intensity, which is mainly ascribed to the reduced nonradiative decay by limiting the intramolecular rotations and vibrations at low-temperature. Furthermore, the long-wavelength emissions with microsecond lifetime are predominantly originated from 3MLCT transitions5,6. The CPL properties of SD/L-Ag14 and SD/R-Ag14 were also explored. As shown in Fig. 7b, c, enantiomers display highly symmetric CPL patterns in the 550–750 nm range under the excitation of 365 nm at room temperature. The dissymmetry g-factor (glum) was calculated to be and its value 2.97 × 10−3 at 650 nm by glum = 2 × [CPL/(32 980/ln10)]/DC, which is comparable to those of the reported metal nanoclusters49,50.

a Variable-temperature emission spectra of racemic conglomerates SD/L-Ag14 and SD/R-Ag14 in the solid state under the excitation of 365 nm. b Circularly polarized luminescence spectra of SD/L-Ag14 or SD/R-Ag14 in the solid state under the excitation of 365 nm. c Total fluorescence intensity spectra of SD/L-Ag14 (blue solid line) or SD/R-Ag14 (orange solid line) in the solid state under the excitation of 365 nm.
Density functional theory calculation
The UV-Vis spectrum of SD/Ag14 in CH2Cl2 shows a strong and broad peak at 398 nm (Supplementary Fig. 13). Time-dependent DFT calculations on SD/L-Ag14 were performed to better understand its electronic structure (Supplementary Data 1). As shown in Fig. 8, the calculated electronic spectrum also exhibits one broad peak centered at 358 nm, which most probably corresponds to the experimental peak at 398 nm although it is blue-shifted by 40 nm. The HOMO orbital of SD/L-Ag14 has an obvious superatom S character, consistent with its identity as a 2-electron superatom. The other occupied orbitals from HOMO-1 to HOMO-8, and even those with lower energy such as HOMO-12 and HOMO-14, mainly comprises of the p orbitals of S and C atoms in pntp− ligands. The unoccupied orbitals from LUMO to LUMO + 9 are solely contributed by the π* orbitals of C = C and N = O bonds in the pntp− ligands. The LUMO + 10, LUMO + 11, LUMO + 12 orbitals are essentially P superatom orbitals (Supplementary Fig. 14). The calculated absorption at 358 nm was mainly contributed by the transitions from the occupied orbitals (HOMO to HOMO-8) to unoccupied orbitals (LUMO to LUMO + 12). Consequently, the electronic transitions contributed to the calculated absorption peak are mainly of LLCT character, such as HOMO-2→LUMO + 2/LUMO + 3/LUMO + 7 and HOMO-3/HOMO-4/HOMO-6→LUMO; apart from these, some MLCT (metal-to-ligand charge transfer) and MMCT (metal-to-metal charge transfer) transitions also contributed, as represented by HOMO → LUMO/LUMO + 1/LUMO + 2 and HOMO → LUMO + 10/LUMO + 11/LUMO + 12, respectively.

a Experimental and time-dependent density functional theory calculated UV-Vis absorption spectra of SD/Ag14. b The calculated HOMO, LUMO, HOMO-2, and LUMO + 10 orbitals of SD/L-Ag14.
Discussion
In conclusion, we isolated a racemic Ag14 nanocluster (SD/rac-Ag14a) protected by achiral ligands of pntp− and dpph in acetone/CH2Cl2, and further separated it into their component enantiomers by re-crystallizing SD/rac-Ag14a in acetonitrile. The inherent chirality of Ag14 nanocluster is induced by identical rotational directionality of six square faces of Ag8 cube, which is locked by π···π interaction between pntp− ligands, maintaining the chirality in solid state. Homochiral crystallization is promoted by C–H···O/N hydrogen bonds formed between lattice acetonitrile and the nitro oxygen atoms in pntp− or aromatic hydrogen atoms in dpph on Ag14 nanocluster. The enantiomeric conglomerates show mirror-imaged CD and CPL responses. This work not only presents an approach for the synthesis and enantioseparation of face-rotating induced chiral silver nanoclusters protected by achiral ligands, but also provides a deeper insight into the origin and spontaneous resolution of chirality at molecular level.
Methods
Synthesis of SD/rac-Ag14a
CF3COOAg (11.0 mg, 0.05 mmol), and 1,6-bis(diphenylphosphino)hexane (11.4 mg, 0.025 mmol) were mixed in 6 mL of acetone/CH2Cl2 (v:v = 4:2) followed by addition of Hpntp (7.8 mg, 0.05 mmol). After stirring (1000 rpm) for 5 min, 40 μL trimethylamine was added to the above solution. After stirring for 20 min, 0.5 mL of EtOH solution of NaBH4 (4 mg/mL) was added and the resulting mixture was further stirred (1000 rpm) at room temperature for 6 h, during which the color rapidly changed from yellow to black. The yellow filtrate was left to stand in the dark at room temperature. After slow evaporation for 14 days, yellow block crystals of SD/rac-Ag14a were collected and washed with ethanol (EtOH). Yield: 5.0 mg (34 %). Selected IR peaks (cm−1) of SD/rac-Ag14a: 1568 (s), 1500 (s), 1473 (m), 1432 (m), 1328 (s) 1178 (m), 1083 (s), 848 (s), 734 (s), 689 (s), 508 (s). Yield: 5.0 mg (34 %).
Synthesis of racemic conglomerates SD/L-Ag14 and SD/R-Ag14
CF3COOAg (11.0 mg, 0.05 mmol), and 1,6-bis(diphenylphosphino)hexane (11.4 mg, 0.025 mmol) were mixed in 6 mL of acetonitrile/CH2Cl2 (v:v = 4:2) followed by addition of Hpntp (7.8 mg, 0.05 mmol). After stirring (1000 rpm) for 5 min, 40 μL trimethylamine was added to the above solution. After stirring for 20 min, 0.5 mL of EtOH solution of NaBH4 (4 mg/mL) was added and the resulting mixture was further stirred (1000 rpm) at room temperature for 6 h, during which the color rapidly changed from yellow to black. The orange filtrate was left to stand in the dark at room temperature. After slow evaporation for 14 days, orange block crystals of racemic conglomerates SD/L-Ag14 and SD/R-Ag14 were collected and washed with ethanol (EtOH). Yield: 4.5 mg (30 %). In addition, we also found the re-crystallization of SD/rac-Ag14a in acetonitrile can also obtain racemic conglomerates SD/L-Ag14 and SD/R-Ag14, which can return back to SD/rac-Ag14a through re-crystallizing in acetone.
Synthesis of SD/rac-Ag14b
CF3COOAg (11.0 mg, 0.05 mmol), and PPh3 (6.5 mg, 0.025 mmol) were mixed in 6 mL of MeOH/CH2Cl2 (v:v = 4:2) followed by addition of Hpntp (7.8 mg, 0.05 mmol). After stirring (1000 rpm) for 5 min, 40 μL trimethylamine was added to the above solution. After stirring for 20 min, 0.5 mL of EtOH solution of NaBH4 (4 mg/mL) was added and the resulting mixture was further stirred (1000 rpm) at room temperature for 6 h, during which the color rapidly changed from yellow to black. The yellow filtrate was left to stand in the dark at room temperature. After slow evaporation for 14 days, yellow block crystals of SD/rac-Ag14b were collected and washed with ethanol (EtOH). Yield: 2.0 mg (13 %).
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. Other relevant data are available from the corresponding author upon reasonable request. The raw data for the computational calculations are provided with Supplementary Data 1. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC: 2070983-2070986 for SD/rac-Ag14a, SD/L-Ag14, SD/R-Ag14, and SD/rac-Ag14b. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Ryssy, J. et al. Light-responsive dynamic DNA-origami-based plasmonic assemblies. Angew. Chem. Int. Ed. 60, 5859–5863 (2021).
Morrow, S. M., Bissette, A. J. & Fletcher, S. P. Transmission of chirality through space and across length scales. Nat. Nanotechnol. 12, 410–419 (2017).
Zhang, D. et al. Diversified transformations of tetrahydroindolizines to construct chiral 3-arylindolizines and dicarbofunctionalized 1,5-diketones. J. Am. Chem. Soc. 142, 15975–15985 (2020).
Li, S. et al. Stepwise achievement of circularly polarized luminescence on atomically precise silver clusters. Adv. Sci. 7, 2000738 (2020).
Kong, Y. J. et al. Photoresponsive propeller-like chiral AIE copper(I) clusters. Angew. Chem. Int. Ed. 59, 5336–5340 (2020).
Zhang, M. M. et al. AIE triggers the circularly polarized luminescence of atomically precise enantiomeric copper(I) alkynyl clusters. Angew. Chem. Int. Ed. 59, 10052–10058 (2020).
Wang, J. Q., Guan, Z. J., Liu, W. D., Yang, Y. & Wang, Q. M. Chiroptical activity enhancement via structural control: the chiral synthesis and reversible interconversion of two intrinsically chiral gold nanoclusters. J. Am. Chem. Soc. 141, 2384–2390 (2019).
Yang, H. Y. et al. From racemic metal nanoparticles to optically pure enantiomers in one pot. J. Am. Chem. Soc. 139, 16113–16116 (2017).
Deng, G. C. et al. From symmetry breaking to unraveling the origin of the chirality of ligated Au13Cu2 nanoclusters. Angew. Chem. Int. Ed. 57, 3421–3425 (2018).
Yang, Y., Pei, X. L. & Wang, Q. M. Postclustering dynamic covalent modification for chirality control and chiral sensing. J. Am. Chem. Soc. 135, 16184–16191 (2013).
Kong, F. K. W. et al. Strategy for the realization of efficient solution-processable phosphorescent organic light-emitting devices: design and synthesis of bipolar alkynylplatinum(II) complexes. J. Am. Chem. Soc. 139, 6351–6362 (2017).
Tan, Y. B. et al. Visible circularly polarized luminescence of octanuclear circular Eu(III) helicate. J. Am. Chem. Soc. 142, 17653–17661 (2020).
Deng, G. C. et al. Enhanced surface ligands reactivity of metal clusters by bulky ligands for controlling optical and chiral properties. Angew. Chem. Int. Ed. 60, 12897–12903 (2021).
Sugiuchi, M., Shichibu, Y. & Konishi, K. An inherently chiral Au24 framework with double-helical hexagold strands. Angew. Chem. Int. Ed. 57, 7855–7859 (2018).
Wan, X. K., Yuan, S. F., Lin, Z. W. & Wang, Q. M. A chiral gold nanocluster Au20 protected by tetradentate phosphine ligands. Angew. Chem. Int. Ed. 53, 2923–2926 (2014).
Tian, F. & Chen, R. Ag18(μ8-S)(p-TBBT)16(PPh3)8: symmetry breaking induced by the core to generate chirality. Chem. Commun. 56, 2719–2722 (2020).
Jadzinsky, P. D., Calero, G., Ackerson, C. J., Bushnell, D. A. & Kornberg, R. D. Structure of A thiol monolayer-protected gold nanoparticle at 1.1 angstrom resolution. Science 318, 430–433 (2007).
Knoppe, S. & Burgi, T. Chirality in thiolate-protected gold clusters. Acc. Chem. Res. 47, 1318–1326 (2014).
Liao, L. W. et al. Structure of chiral Au44(2,4-DMBT)26 nanocluster with an 18-electron shell closure. J. Am. Chem. Soc. 138, 10425–10428 (2016).
Yao, Q. F. & Xie, J. P. Pasteur-like separation of silver nanocluster racemates by conglomerate crystallization. ACS Cent. Sci. 6, 1862–1865 (2020).
Huang, J. H., Wang, Z. Y., Zang, S. Q. & Mak, T. C. W. Spontaneous resolution of chiral multi-thiolate-protected Ag30 nanoclusters. ACS Cent. Sci. 6, 1971–1976 (2020).
Liu, C. et al. Chiral Ag23 nanocluster with open shell electronic structure and helical face-centered cubic framework. Nat. Commun. 9, 744 (2018).
Wang, X. C. et al. Assembled molecular face-rotating polyhedra to transfer chirality from two to three dimensions. Nat. Commun. 7, 12469 (2016).
Qu, H. et al. Molecular face-rotating cube with emergent chiral and fluorescence properties. J. Am. Chem. Soc. 139, 18142–18145 (2017).
Jin, Y. et al. Cations controlling the chiral assembly of luminescent atomically precise copper(I) Clusters. Angew. Chem. Int. Ed. 58, 12143–12148 (2019).
Wang, Z. et al. Assembly of silver trigons into a buckyball-like Ag180 nanocage. Proc. Natl Acad. Sci. USA.114, 12132–12137 (2017).
Ma, X. Y. et al. Rhombicuboctahedral Ag100: four-layered octahedral silver nanocluster adopting the russian nesting doll model. Angew. Chem. Int. Ed. 59, 17234–17238 (2020).
Su, Y. M., Wang, Z., Schein, S., Tung, C. H. & Sun, D. A Keplerian Ag90 nest of platonic and archimedean polyhedra in different symmetry groups. Nat. Commun. 11, 3316 (2020).
Yang, H. Y. et al. Embryonic growth of face-center-cubic silver nanoclusters shaped in nearly perfect half-cubes and cubes. J. Am. Chem. Soc. 139, 31–34 (2017).
Chai, J. S. et al. A unique pair: Ag40 and Ag46 nanoclusters with the same surface but different cores for structure-property correlation. J. Am. Chem. Soc. 140, 15582–15585 (2018).
Xie, Y. P., Jin, J. L., Lu, X. & Mak, T. C. W. High-nuclearity silver thiolate clusters constructed with phosphonates. Angew. Chem. Int. Ed. 54, 15176–15180 (2015).
Wang, Z. et al. Chalcogens-induced Ag6Z4@Ag36 (Z = S or Se) core-shell nanoclusters: enlarged tetrahedral core and homochiral crystallization. J. Am. Chem. Soc. 141, 17884–17890 (2019).
Dolamic, I., Knoppe, S., Dass, A. & Burgi, T. First enantioseparation and circular dichroism spectra of Au38 clusters protected by achiral ligands. Nat. Commun. 3, 798 (2012).
Zeng, C. J., Li, T., Das, A., Rosi, N. L. & Jin, R. C. Chiral structure of thiolate-protected 28-gold-atom nanocluster determined by X-ray crystallography. J. Am. Chem. Soc. 135, 10011–10013 (2013).
Nagata, Y. et al. Solvent-dependent switch of helical main-chain chirality in sergeants-and-soldiers-type poly(quinoxaline-2,3-diyl)s: effect of the position and structures of the “sergeant” chiral units on the screw-sense induction. J. Am. Chem. Soc. 135, 10104–10113 (2013).
Knoppe, S. et al. Chiral phase transfer and enantioenrichment of thiolate-protected Au102 clusters. J. Am. Chem. Soc. 136, 4129–4132 (2014).
Zhu, Y. F. et al. Enantioseparation of Au20(PP3)4Cl4 clusters with intrinsically chiral cores. Angew. Chem. Int. Ed. 57, 9059–9063 (2018).
Yan, J. Z. et al. Asymmetric synthesis of chiral bimetallic Ag28Cu12(SR)244- nanoclusters via ion pairing. J. Am. Chem. Soc. 138, 12751–12754 (2016).
Chen, S., Fang, W. H., Zhang, L. & Zhang, J. Atomically precise multimetallic semiconductive nanoclusters with optical limiting effects. Angew. Chem. Int. Ed. 57, 11252–11256 (2018).
Hunter, C. A. & Sanders, J. K. M. The Nature of π···π interactions. J. Am. Chem. Soc. 112, 5525–5534 (1990).
Chen, Z. J., Lohr, A., Saha-Moller, C. R. & Wurthner, F. Self-assembled π-stacks of functional dyes in solution: structural and thermodynamic features. Chem. Soc. Rev. 38, 564–584 (2009).
Schmidbaur, H. & Schier, A. Argentophilic interactions. Angew. Chem. Int. Ed. 54, 746–784 (2015).
Hau, S. C. K. & Mak, T. C. W. Synthesis of unstable carbides Ag2C2n (n=3, 4) and characterization via crystallographic analysis of their double salts with silver(I) trifluoroacetate. J. Am. Chem. Soc. 136, 902–905 (2014).
Alhilaly, M. J. et al. [Ag67(SPhMe2)32(PPh3)8]3+: synthesis, total structure, and optical properties of a large box-shaped silver nanocluster. J. Am. Chem. Soc. 138, 14727–14732 (2016).
Chen, T. K., Yao, Q. F., Nasaruddin, R. R. & Xie, J. P. Electrospray ionization mass spectrometry: a powerful platform for noble-metal nanocluster analysis. Angew. Chem. Int. Ed. 58, 11967–11977 (2019).
Krishnadas, K. R. et al. Intercluster reactions between Au25(SR)18 and Ag44(SR)30. J. Am. Chem. Soc. 138, 140–148 (2016).
Nag, A. et al. Isomerism in supramolecular adducts of atomically precise nanoparticles. J. Am. Chem. Soc. 140, 13590–13593 (2018).
Xuan, W. M., Surman, A. J., Miras, H. N., Long, D. L. & Cronin, L. Controlling the ring curvature, solution assembly, and reactivity of gigantic molybdenum blue wheels. J. Am. Chem. Soc. 136, 14114–14120 (2014).
Shi, L. et al. Self-assembly of chiral gold clusters into crystalline nanocubes of exceptional optical activity. Angew. Chem. Int. Ed. 56, 15397–15401 (2017).
Sang, Y. T., Han, J. L., Zhao, T. H., Duan, P. F. & Liu, M. H. Circularly polarized luminescence in nanoassemblies: generation, amplification, and application. Adv. Mater. 32, 1900110 (2020).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 91961105, 21822107, 22001139, and 21827801), the Natural Science Foundation of Shandong Province (Nos. ZR2020ZD35, ZR2019ZD45, ZR2019BB058, JQ201803, and ZR2017MB061), the Taishan Scholar Project of Shandong Province of China (Nos. tsqn201812003 and ts20190908), the Qilu Youth Scholar Funding of Shandong University. Project for Scientific Research Innovation Team of Young Scholar in Colleges and Universities of Shandong Province (2019KJC028).
Author information
Authors and Affiliations
Contributions
Original idea was conceived by D.S., experiments and data analyses were performed by X.-Q.L., Y.-Z.L, Z.W., S.-S.Z., Y.-C.L., Z.-Y.G., and D.S., ESI-MS data were collected by L.F., circular dichroism data were collected by X.-Q.L., Z.W. and Z.-Z.C., luminescence data were collected by S.-S.Z. and Q.-W.X., DFT calculations data were collected by Y.-Z.L, structure characterizations were performed by X.-Q.L., Z.W., and D.S., manuscript was drafted by X.-Q.L., D.S., and C.-H.T. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, XQ., Li, YZ., Wang, Z. et al. Revealing the chirality origin and homochirality crystallization of Ag14 nanocluster at the molecular level. Nat Commun 12, 4966 (2021). https://doi.org/10.1038/s41467-021-25275-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25275-2
This article is cited by
-
Flow-induced periodic chiral structures in an achiral nematic liquid crystal
Nature Communications (2024)
-
Ligand effect on switching the rate-determining step of water oxidation in atomically precise metal nanoclusters
Nature Communications (2023)
-
Small symmetry-breaking triggering large chiroptical responses of Ag70 nanoclusters
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
