
Abstract
Photon upconversion of near-infrared (NIR) irradiation into ultraviolet-C (UVC) emission offers many exciting opportunities for drug release in deep tissues, photodynamic therapy, solid-state lasing, energy storage, and photocatalysis. However, NIR-to-UVC upconversion remains a daunting challenge due to low quantum efficiency. Here, we report an unusual six-photon upconversion process in Gd3+/Tm3+-codoped nanoparticles following a heterogeneous core-multishell architecture. This design efficiently suppresses energy consumption induced by interior energy traps, maximizes cascade sensitizations of the NIR excitation, and promotes upconverted UVC emission from high-lying excited states. We realized the intense six-photon-upconverted UV emissions at 253 nm under 808 nm excitation. This work provides insight into mechanistic understanding of the upconversion process within the heterogeneous architecture, while offering exciting opportunities for developing nanoscale UVC emitters that can be remotely controlled through deep tissues upon NIR illumination.
Similar content being viewed by others
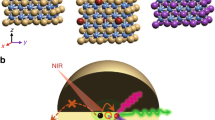

Introduction
Multiphoton upconversion processes that convert NIR excitation into visible emissions have attracted considerable attention owing to broad technical applications of anti-Stokes shifts1,2,3,4,5,6. UV upconversion luminescence can be a powerful tool for applications in biomedical7,8,9, environmental10,11, and industrial fields12,13, and converting NIR all the way upto UVC (100–290 nm) emissions holds promise in photocatalysis11, ultraviolet solid-state lasers12, and biomedical applications8,14,15,16,17. But, their practical implementations have been hindered by low emission intensities and difficulties in achieving large shifts into the UVC region. Apart from the intrinsic parity-forbidden nature of 4f−4f optical transitions in lanthanide systems, NIR-to-UVC upconversion can be significantly influenced by many deleterious factors, such as concentration quenching, surface quenching, cross-relaxation between lanthanide ions, and competitive energy harvesting from lower-lying energy levels. To minimize the unwanted energy consumption at high-lying emitting levels and reduce the chances for mitigating the upconverted UV emissions, attempts have been made to enhance the emission intensity in the UV range, for instance, by controlling the particle phase and size18, the pulse width of excitation beams19, dopant composition20, and nanoparticle core-shell structures12,21,22,23,24. To our best knowledge, little attention has been paid to the effect of interior defects on UVC upconversion luminescence25.
Compared with Yb3+-sensitized upconversion nanoparticles (UCNPs), Nd3+-sensitized UCNPs offer deep penetration depths and minimal over-heating effect, owing to low coefficients of water absorption under 800-nm excitation26. Nd3+-sensitized UCNPs are the promising candidates for photon-driven reactions in biosystems, such as biodetection27, photodynamic therapy28,29,30,31, light-triggered drug release32, and photocatalysis33. To enhance the brightness of Nd3+-sensitized UCNPs, core-shell nanostructural design has been typically utilized to prevent deleterious cross-relaxation34,35,36,37. By doping lanthanide ions and Nd3+ ions into the separated layer, the emission intensity can be notably enhanced while maintaining optical integrity38. Despite enticing prospects, UVC emission from Nd3+-sensitized UCNPs has been challenging because of the densely packed excited states of Nd3+ and dominant cross-relaxation within the nanoscale systems39.
Here we report the significantly enhanced UVC emission through Nd3+ sensitization by controlling upconverted excitation energy flux within Gd3+/Tm3+ codoped core and multishell nanostructures. Our mechanistic investigation reveals an upconverted excitation lock-in (UCEL) mode in which Gd3+-sensitized excitation energy can be retained by simply using an interlayer of the NaYF4 host lattice doped with Yb3+ that is optically inert to the excited Gd3+. This nanostructure preserves the upconverted UV energy within the core domain and effectively suppresses energy dissipation by interior traps, enabling six-photon-upconverted UV emission at 253 nm under 808 nm excitation.
Results
Heterogeneous nanostructural design
In our experiment, we designed a heterogeneous core-multishell structure to suppress surface quenching and achieve tunable emissions. In a conventional design35,40, under 808-nm excitation, Nd3+ sensitizers harvest excitation photons and subsequently pass them to Yb3+ ions with an excited state at ~10 000 cm−1. Energy migration through a network of high concentration Yb3+ ions promotes energy transfer of the NIR excitation to Tm3+ emitters with ladder-like metastable intermediate states, facilitating sequential upconversion processes from NIR to visible/UV. Subsequently, upconverted UV emission from high-lying states of Tm3+ can be further transferred to Gd3+ ions embedded in the nanoparticle core as the UVC energy reservoirs.
The key to our design is the use of a NaYF4 host lattice doped with the same amount of Yb3+ locating in the first shell layer of NaGdF4:49%Yb, 1%Tm@NaGdF4:20%Yb@NaGdF4:10%Yb, 50%Nd@NaGdF4 (Gd-CSGdS2S3) nanoparticle (Fig. 1). This layer of NaYF4:20%Yb is optically inert to the excited states (6DJ, 6IJ, and 6PJ) of Gd3+ ions and can lock-in the upconverted UVC and ultraviolet-B (UVB) energy of Gd3+ ions. The Gd3+ network can then reuse the upconverted excitation energy and prevent depopulation by deleterious energy traps within the nanoparticles, as well as absorb additional photon energy from the excited state Yb3+ ions. The NaYF4 layer plays a key role in interdicting detrimental energy transfer between Gd3+ and interior traps, enhancing five- and six-photon-upconverted UVB and UVC emissions.

The proposed UCEL scheme involving a heterogeneous, core-multishell nanostructure (Gd-CSYS2S3). A multistep cascade energy transfer (Nd3+→Yb3+→Tm3+→Gd3+) leads to populate the excited states of Gd3+. The layer of an optical inert NaYF4 host lattice doped with 20% Yb3+ locating in the first shell layer of nanoparticles can lock-in the upconverted excitation energy of Gd3+ ions and prevent depopulation by deleterious energy traps within the nanoparticles, resulting in intense UVC upconversion emission. S, M, E, R, and T denote sensitizer Nd3+, migrator Yb3+, emitter Tm3+, recycler Gd3+, and energy traps, respectively.
Upconverted excitation lock-in (UCEL) mode
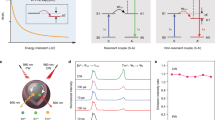
The UCEL mode requires both an interlayer of optical inert NaYF4 host lattice doped with Yb3+ and a network of Gd3+ ions to recycle upconversion energy for UVC emission amplification. Fig. 2 illustrates a typical upconversion process in the heterogeneous core-multishell nanoparticles upon 808-nm excitation. The 808 nm photons are first sensitized by Nd3+ sensitizer ions, being populated at the 4F5/2 energy state and quickly relaxed to the 4F3/2 energy state of Nd3+. The excited Yb3+ ions serve as an energy migrator to sensitize and pass on the energy from Nd3+ and to populate the 3P2 state of Tm3+ through a five-photon upconversion process. Subsequently, the energy at the 3P2 state, relax non-radiatively to populate 1I6 and give rise to UVB emissions at 290 nm. Besides, Gd3+ ions in the core domain extract the energy through an energy transfer process of 1I6 → 3H6 (Tm3+): 8S7/2 → 6PJ (Gd3+). The excitation energy of Gd3+ at 6PJ can resist nonradiative quenching due to its large energy gap (~32 000 cm−1 from 6PJ to 8S7/2). Thus, the lifetime of Gd3+ at 6PJ energy state is long enough for the sixth photon to be absorbed from the excited Yb3+. Therefore, the 6DJ state of Gd3+ is further populated by the appropriate energy matching of the following transitions of 2F5/2 → 2F7/2 (9750 cm−1, Yb3+): 6PJ → 6DJ (∼8750 cm−1, Gd3+)41,42,43. Thus, UVC and UVB upconversion emission peaked at 253, 273, 276, 279, 306, and 311 nm from 6DJ, 6IJ, and 6PJ of Gd3+ can be obtained. Noted that, the probability of nonradiative relaxation of 6DJ, 6IJ → 6PJ is larger than that of the radiative transition of 6DJ, 6IJ → 8S7/2, resulting in an efficient population of the 6P7/2 state, commonly observed in Gd-based homogeneous nanostructures22. In our design, the NaYF4-based first shell layer selectively blocks the energy transfer from Gd3+ to interior energy traps (e.g., lattice defects and impurities). It preserves and recycles the excitation energy within the core region, leading to increased populations in the 6DJ, 6IJ, and 6PJ states of Gd3+ and intense UVC and UVB emissions of Gd3+.

When the nanoparticles are excited under 808 nm, Nd3+ sensitizers first absorb the excitation energy and pass it onto Yb3+. Subsequently, the 3P2 state of Tm3+ is populated by a sequential five-photon energy transfer from the network of excited Yb3+ ions and relaxes to 1I6. The 6DJ state of Gd3+ is populated via a stepwise process of a five-photon energy transfer process from Tm3+ and a further energy transfer from Yb3+, giving rise to the sixth-photon upconversion luminescence. The inert NaYF4 host lattice layer can lock-in the Gd3+ excitation energy and reuse the energy that would otherwise be depopulated by deleterious energy traps within the nanoparticles, resulting in upconversion emissions in the UVB and UVC regions.
Controlled synthesis
We used a layer-by-layer epitaxial growth method24 to synthesize a batch of Gd-CSYS2S3 nanoparticles with optimized concentrations of co-dopants40 following the design of NaGdF4:49%Yb,1%Tm@NaYF4:20%Yb@NaGdF4:10%Yb,50%Nd@NaGdF4 (Fig. 3a). Transmission electron microscopy (TEM) images of obtained Gd-CSYS2S3 nanoparticles show the average size of ~29 nm with each layer ~2.5 nm in thickness (Supplementary Fig. 1). High-resolution TEM shows the single-crystalline structure of the as-synthesized core-multishell nanoparticles (Fig. 3b inset), and X-ray powder diffraction result (XRD, JCPDS file number 27-0699, Supplementary Fig. 2) confirms the hexagonal phase of the as-prepared nanoparticles. High-angle annular darkfield scanning TEM identified the formation of the heterogeneous core-multishell structures (Fig. 3b), in which the brighter regions correspond to heavier elements (Gd, Yb, and Nd) and the darker parts correspond to lighter ones (Y). Energy-dispersive X-ray mapping analysis further confirms the heterogeneous core-multishell structures (Fig. 3c and Supplementary Fig. 3).

a Schematic illustration of the as-synthesized Gd-CSYS2S3 nanoparticles. b High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and high-resolution TEM image of the corresponding nanoparticles (inset). c HAADF-STEM image and elemental mapping of a single Gd-CSYS2S3 nanoparticle, indicating the spatial distribution of the Gd, Y, Nd, and Yb elements in the core-multishell structure. d Room-temperature emission spectra of Gd-CSYS2S3 nanoparticles in cyclohexane under 808 nm excitation. e Emission spectra of Gd-CSYS2S3 and Gd-CSGdS2S3 under excitation of 808 nm CW diode laser. The excitation power density is 10 W cm−2. f Excitation-power-dependent UV upconversion emission spectra of Gd-CYS2S3 nanoparticles under 808 nm excitation. g Log intensity-pump power of the 253 nm upconversion emission of Gd-CYS2S3 nanoparticles under 808 nm excitation.
Remarkable UVC enhancement
To investigate the unusual UVC upconversion emission from Gd3+, we recorded the photoluminescence spectra of the as-synthesized nanoparticles at room temperature. Usually, in favor of the lower 6P7/2 (311 nm) energy level, the Gd3+ emission in the UVC range is quenched, and optical transitions of (6DJ, 6IJ, 6P5/2 → 8S7/2) could hardly be spectroscopically detected (Supplementary Figs. 4–7)12. In contrast, as shown in Fig. 3d and Supplementary Fig. 8, intense upconversion emissions from 6DJ and 6IJ of Gd3+ peaked at 253 nm (6D9/2 → 8S7/2), 273 nm (6IJ → 8S7/2), 276 nm (6IJ → 8S7/2), 279 nm (6IJ → 8S7/2), 306 nm (6P5/2 → 8S7/2) and 311 nm (6P7/2 → 8S7/2) in the UV region were observed either under 808 nm or 980 nm excitation. Moreover, we observed more than 50-fold and 30-fold enhancements in Gd3+ emission (311 nm) by our Gd-CSYS2S3 heterogeneous core-multishell design compared with the conventional Gd-CSGdS2S3 nanoparticles under 808 and 980 nm excitation, respectively (Supplementary Figs. 9 and 10), although the absorption profile of Gd-CSYS2S3 is not changed compared with that of Gd-CSGdS2S3 nanoparticles (Supplementary Fig. 11). The approximate absorption cross-section σ of Nd3+ at 808 nm was calculated to be σ = 1.5 × 10−19 cm2 (Gd-CSYS2S3), σ = 1.3 × 10−19 cm2 (Gd-CSGdS2S3) from the UV−Vis absorption spectra of the nanoparticles44. As verified by the emission spectra of as-prepared nanoparticles from different batches of (Supplementary Fig. 12), our protocol to enhance the UVC upconversion emissions is reproducible.
We further studied the excitation power dependence of luminescence intensity from higher-lying 6DJ, 6IJ and 6PJ excited states of Gd3+ (Fig. 3f). The number of photons (n) required to populate the upper emitting state can be calculated by the luminescence intensity If, and the pump power of laser P following the relation of If∝Pn45. The output slope for 253 nm emission band was calculated as 6.29, indicating that six 808 nm photons were needed to populate the 6DJ level, following a six photon upconversion process (Fig. 3g), while n values obtained for 276 and 311 nm emissions were 5.27 and 4.94, indicating five-photon processes (Supplementary Fig. 13).
Quantitative study
The large energy gap of about 32 000 cm−1 of Gd3+ and intrinsic low phonon energy of NaGdF4 offer good possibilities to obtain 100% energy transfer efficiency from Gd3+-to-Gd3+46,47. The energy transfer efficiencies η of Nd3+-to-Yb3+, Yb3+-to-Tm3+, and Tm3+-to-Gd3+ energy transfer can be quantitatively estimated from the Eqs. 1 and 248,49
where τm is the mean lifetime of energy donor lanthanides (Ln) in the presence of energy acceptor, τLn is the intrinsic lifetime of energy donor, and α is the amplitude. To calculate the energy transfer efficiencies of Nd3+-to-Yb3+, Yb3+-to-Tm3+, and Tm3+-to-Gd3+, we designed and synthesized three pairs of heterogeneous nanoparticles (TEM results shown in Supplementary Fig. 14). In our experiment, to first determine the intrinsic lifetime of the corresponding energy donors, the energy acceptors of Yb3+, Tm3+, and Gd3+ were replaced by optically inert Y3+ ions.
In detail, to calculate the energy transfer efficiency of Nd3+-to-Yb3+, we produced a pair of samples of NaGdF4:49%Yb,1%Tm@NaYF4:20%Yb@NaGdF4:10%Yb,50%Nd@NaGdF4 (Gd-CSYSS in the presence of 20% Yb3+ energy acceptor) v.s. NaGdF4:49%Y,1%Tm@NaYF4@NaGdF4:10%Y,50%Nd@NaGdF4 (Gd-CSYSS in the absence of 20% Yb3+). The lifetimes of Nd3+ at 893 nm were measured under the 793 nm pulsed excitation, and the energy transfer efficiency of Nd3+-to-Yb3+ was calculated to be 79% (Fig. 4a). Similarly, to calculate the energy transfer efficiency of Yb3+-to-Tm3+, and to avoid the complex energy transfer pathways in the core-multishell structure, we produced a pair of simplified designs of NaGdF4:20%Yb,1%Tm,29%Y@NaYF4 (Gd-CSY in the presence of 1% Tm3+ energy acceptor) v.s. NaGdF4: 20%Yb,30%Y@NaYF4 (Gd-CSY in the absence of Tm3+). The 980 nm decay lifetimes of Yb3+ were measured under the 920 nm pulsed excitation, and the energy transfer efficiency of Yb3+-to-Tm3+ was estimated to be 62% (Fig. 4b). To calculate the energy transfer efficieny of Tm3+-to-Gd3+, we produced a pair of samples of NaGdF4:20%Yb,1%Tm,29%Y@NaYF4 (Gd-CSY in the presence of Gd3+) v.s. NaYF4: 20%Yb,1%Tm@NaYF4 (Gd-CSY in the absence of Gd3+). By exciting the samples at 980 nm, the lifetimes of Tm3+ at 290 nm were measured and the energy transfer efficiency of Tm3+-to-Gd3+ was estimated to be 1% (Fig. 4c).

a Luminescence decay curves of Nd3+ emissions measured at 893 nm for NaGdF4:49%Yb,1%Tm@NaYF4:20%Yb@NaGdF4:10%Yb,50%Nd@NaGdF4 (with Yb3+) and NaGdF4:49%Y,1%Tm@NaYF4@NaGdF4:10%Y,50%Nd@NaGdF4 (without Yb3+) by pulsed 793 nm excitation. b Luminescence decay curves of Yb3+ emissions measured at 980 nm for NaGdF4:20%Yb,1%Tm,29%Y@NaYF4 (with Tm3+) and NaGdF4: 20%Yb,30%Y@NaYF4 (without Tm3+) by pulsed 920 nm excitation. c Luminescence decay curves of Tm3+ emissions measured at 290 nm for NaGdF4:20%Yb,1%Tm,29%Y@NaYF4 (with Gd3+) and NaYF4: 20%Yb,1%Tm@NaYF4 (without Gd3+) by pulsed 980 nm excitation.
Furthermore, we conducted a quantitative study to compare the quantum yields of Gd-CSYSS and Gd-CSGdSS nanoparticles. The upconversion quantum yields from 240 to 750 nm of the as-prepared Gd-CSYS2S3 and Gd-CSGdS2S3 nanoparticles were estimated as 1.74 and 0.97%, respectively. To quantify the emission enhancement in the UV range from 240 to 325 nm, we also attempted to measure the upconversion quantum yields in the UV range, but without success due to the limited UVC emissions. Instead, we measured the quantum yields of upconversion emissions in the range from 240 to 400 nm, with the results being approximately 0.13 and 0.04%, respectively.
The role of the first layer of NaYF4 shell
To probe the role of NaYF4 layer in locking-in and recycling Gd3+ excitation energy, we have compared the excited state lifetime of Gd3+. As shown in Fig. 5 and Supplementary Fig. 15a significant prolonged (~4 times) lifetime of Gd3+ emission from the 6P7/2 level was achieved when the NaYF4 first layer was applied. In contrast, there were negligible changes in the Gd3+ lifetimes for emissions from 6DJ and 6IJ energy levels, indicating the energy loss from Gd3+ to interior energy traps was mainly through 6P7/2 energy level of Gd3+ due to small energy gap between 6DJ, 6IJ, and 6PJ (Supplementary Fig. 16). In addition, the emission intensities of Nd3+ at 893 nm (4F3/2 → 4I9/2), 1057 nm (4F3/2 → 4I11/2), and 1330 nm (4F3/2 → 4I13/2), and Tm3+ at ~1460 nm (3H4 → 3F4) in the near-infrared range were essentially unaltered (Supplementary Fig. 17). These results indicate that the NaYF4-assisted UCEL mechanism favors the upconversion emissions from high-lying energy levels.

Upconversion luminescence decay curves of Gd3+ emissions at 311 nm from Gd-CSYS2S3 v.s. Gd-CSGdS2S3 by pulsed 808 nm excitation.
To further verify the role of NaYF4 layer in enhancing the UVB and UVC emissions, we synthesized a group of Gd-CSYS2S3, NaGdF4:49%Yb,1%Tm@NaGdF4:20%Yb@NaYF4:10%Yb,50%Nd@NaGdF4 (Gd-CS1SYS3) and NaGdF4:49%Yb,1%Tm@NaGdF4:20%Yb@NaGdF4:10%Yb,50%Nd@NaYF4 (Gd-CS1S2SY) heterogeneous nanoparticles, in which NaGdF4 was selectively replaced by NaYF4 host lattice in the first, second and third layer, respectively (Fig. 6a). The intense UVB and UVC emission was only observed in Gd-CSYS2S3 nanoparticles. The emission profiles of Gd-CS1SYS3 and Gd-CS1S2SY were quite similar to Gd-CSGdS2S3 nanoparticles. Moreover, when the half of optically inert Y3+ ions in the first layer were replaced by the Gd3+ ions, a drastic reduction of the Gd3+ emission was observed, indicating that the NaYF4 with Yb3+ doping layer can effectively prevent the Gd3+ energy leakage (Supplementary Fig. 18).

a Room-temperature upconversion emission spectra of solutions containing Gd-CSYS2S3, Gd-CS1SYS3, Gd-CS1S2SY, and Gd-CSGdS2S3 nanoparticles under 808 nm excitation at a power density of 10 W cm−2. b, c Schematic illustration, TEM images and photoluminescence spectra of the as-synthesized Gd-CSY-15%TbS2S3 and Gd-CSY-15%EuS2S3 nanoparticles.
We further prepared a group of Gd-CSYS2S3 nanoparticles doped with Tb3+ or Eu3+ ions in the first layer NaGdF4:49%Yb,1%Tm@NaYF4:20%Yb,15%Tb@NaGdF4:10%Yb,50%Nd@NaGdF4 (Gd-CSY-15%TbS2S3) or NaGdF4:49%Yb,1%Tm@NaYF4:20%Yb,15%Eu@NaGdF4:10%Yb,50%Nd@NaGdF4 (Gd-CSY-15%EuS2S3), which can extract the excitation energy from Gd3+ to emit green and red upconversion emissions through the scheme of energy migration upconversion (EMU)22. Upon excitation at 808 nm, the characteristic emissions of Tb3+ and Eu3+ (highlighted in color) were observed (Fig. 6b, c and Supplementary Fig. 19), but no enhancement of UVB emissions observed. Doping with 15% Tb3+ or Eu3+ in the outmost layer only led to weak emission of Tb3+ or Eu3+ (Supplementary Fig. 20). The weak Tb3+ and Eu3+ emissions were attributed to the interior energy trapping of the excitation energy in the Gd3+ sublattice. Together, these results indicate that an efficient energy transfer pathway (Nd3+→Yb3+→Tm3+→Gd3+) occurs50, and the excitation energy of Gd3+ can be easily dissipated through the emission of Tb3+, Eu3+, or interior traps if without the first-shell layer of 20% Yb3+ doped NaYF4.
Determination of the interior traps and Gd3+ energy recycling above 6PJ
The interior energy flux leakage pathway through lattice vibration and multiphonon transitions can be neglected because of the large energy gap of about 32 000 cm−1 of Gd3+ compared with the intrinsic low phonon energy of host materials (∼350 cm−1)47. Besides, it was reported that an efficient energy transfer can occur between Gd3+ and Nd3+ ions51. However, in our design, the energy transfer between these two ions did not happen. To preclude the possibility of the interior Nd3+ energy trapping, we prepared a pair of Gd-CSGdS2S3 nanoparticles with and without Nd3+ dopant NaGdF4:49%Yb,1%Tm@NaGdF4:20%Yb@NaGdF4:10%Yb,50%Nd@NaGdF4 and NaGdF4:49%Yb,1%Tm @NaGdF4:20%Yb@NaGdF4:10%Yb,0%Nd@NaGdF4 (Gd-CSGdS50%NdS3 and Gd-CSGdS0%NdS3). The lifetimes of Gd3+ (6DJ, 6IJ, 6PJ) and Tm3+ (1I6, 1D2) were virtually unchanged after removing Nd3+ dopants in nanoparticles (Fig. 7a and Supplementary Fig. 21).

a Schematic illustration of the as-synthesized Gd-CSGdS0%NdS3 nanoparticles, and the upconversion luminescence decay curves of Gd3+ emission at 311 nm and Tm3+ emission at 450 nm of Gd-CSGdS50%NdS3 and Gd-CSGdS0%NdS3 nanoparticles under 980 nm excitation, respectively. b Schematic illustration of energy recycling in Gd3+ sublattice at the core domain of NaGdF4:Yb,Tm. c The upconversion luminescence decay curves of Gd3+ emission at 311 nm in Gd-CSYS2S3, Gd-CS1SYS3, and Gd-CS1S2SY nanoparticles by the pulsed 808 nm excitation. d The upconversion luminescence decay curves of Gd3+ emission at 311 nm in Gd-CSGdS2S3, Gd-CS1SYS3, and Gd-CS1S2SY nanoparticles by the pulsed 808 nm excitation.
Since the Gd3+_Gd3+ energy migration is efficient and it can travel long distances22, it is reasonable to assume that the excitation energy may be quenched by the interior lattice defects in the heterogenous structure with multi-layers of the shell. In our design, NaYF4 in the first layer, effectively blocks the energy transfer from Gd3+ to interior lattice defects in the outer shell layers, resulting in the migrating energy only recycling within the core domain of NaGdF4:Yb,Tm (Fig. 7b). To validate our hypothesis, we investigated the lifetimes of Gd-CSYS2S3, Gd-CS1SYS3, and Gd-CS1S2SY nanoparticles by changing the position of the NaYF4 layer. As shown in Fig. 7c, the lifetime (Gd3+: 311 nm) in Gd-CSYS2S3 is significantly longer than those in both Gd-CS1SYS3 and Gd-CS1S2SY nanoparticles. The prolonged lifetime of Gd3+ in Gd-CSYS2S3 nanoparticles is ascribed to the suppressed trapping of Gd3+ energy by interior lattice defects in the multi-shell regions. By contrast, a similar level of short lifetimes of Gd3+ in Gd-CSGdS2S3, Gd-CS1SYS3, and Gd-CS1S2SY was observed, indicating that surface quenching was not responsible for the weak UV upconversion in conventional nanoparticles (Fig. 7d). This result is also consistent with our luminescence analysis in that a significantly stronger UV luminescence of Gd-CSYS2S3 nanoparticles compared with those of Gd-CSGdS2S3, Gd-CS1SYS3, and Gd-CS1S2SY counterparts.
Furthermore, a Gd3+ content of 50 mol% produced an optimum energy-migration property with a Gd3+−Gd3+ separation of 5.32 Å, which can be approximately calculated using the Eq. 3:52
For the hexagonal NaGdF4 unit cell, a = 6.02 Å, c = 3.60 Å. The short distance between Gd3+ ions indicates that the Gd3+−Gd3+ energy migration is dominated by exchange interaction46. Moreover, it was reported by the Blasse group, Pem for Gd3+ is about 5 × 102 s−1, P(Gd3+→Gd3+) is about 107 s−1. Pem denotes the probability of emission, while P(Gd3+→Gd3+) denotes the probability for energy migration46. These results indicate that the excitation energy can be transferred more than 105 times for the excited Gd3+. After NaGdF4 was replaced with NaYF4 in the first layer, a significant increase in Gd3+ lifetime was observed (Fig. 7c), suggesting the probability of Gd3+-Gd3+ energy migration within the core domain was significantly increased. Taken these together, the results conclusively suggested that the NaYF4 shell can impede the fast migrating energy within the network of Gd3+ ions from being trapped by the interior lattice defects in the outer multi-layer shell, which promotes the occurrence of energy hopping in Gd3+ sublattice at the core domain of NaGdF4:Yb,Tm, thereby realizing the intense UVB and UVC upconversion emissions.
Enhancement in highly doped single nanoparticles
To further evaluate UCEL mode in enhancing the high-order upconversion emissions in the heterogenous core-multishell structures, we implemented the similar design in the highly doped UCNP core, e.g., NaGdF4:49%Yb,8%Tm@NaYF4:20%Yb @NaGdF4:10%Yb,50%Nd@NaYF4 and NaGdF4:49%Yb,8%Tm@NaGdF4:20%Yb@NaGdF4:10% Yb,50%Nd@NaGdF4 (Gd-C8%TmSYS2S3 and Gd-C8%TmSGdS2S3), and quantify the brightness of single UCNPs using a purpose-built confocal microscopy system (Supplementary Fig. 22). Due to the significant UV absorption by the optical components, including the objective lens and mirrors, instead of a direct quantification of the UVC emissions at a single nanoparticle level, we monitored the amount of the blue band emissions from a single nanoparticle. Under the same excitation power from both 808 nm and ~980 nm lasers, the emission intensities of Gd-C1%TmSYS2S3 and Gd-C1%TmSGdS2S3 nanoparticles under the 808 nm excitation were ~4 times and ~5 times higher than those under the ~980 nm excitation, respectively (Supplementary Fig. 23). In contrast, much higher enhancement factors of the highly doped Gd-C8%TmSYS2S3 (~25 times) and Gd-C8%TmSGdS2S3 (~15 times) nanoparticles were achieved under the 808 nm v.s. ~980 nm excitations. These results suggest UCEL mode could be broadly applied to a variety of UCNP core concentrations38 and under a large dynamic range of excitation power densities53, suitable for both ensemble and single nanoparticle applications54.
Potential in enhancing Reactive Oxygen Species (ROS) generation
Moreover, we prepared the titanium dioxide (TiO2)-coated UCNPs in which TiO2 serves as the photosensitizer55. TEM and X-ray powder diffraction (XRD) analysis confirmed the successful synthesis (Supplementary Fig. 24), and compositional analysis of these nanocomposites by energy-dispersive X-ray spectroscopy (EDX) confirms the presence of Ti4+, Nd3+, Gd3+, Yb3+, and Tm3+ (Supplementary Fig. 25). As shown in Supplementary Fig. 26, the emission Gd-CSYS2S3@TiO2 became weaker compared with Gd-CSYS2S3 nanoparticles due to the absorbance of UV emission by the TiO2 shell. The ability to generate singlet oxygen (1O2) of the as-synthesized nanocomposites was evaluated by the 1,3-diphenylisobenzofuran (DPBF) chemical probe under 808 nm laser irradiation. The characteristic absorbance of DPBF gradually decreased with the increase in irradiation time, the characteristic absorption decreased with time, indicating the successful generation of 1O2 (Supplementary Fig. 26b). These results indicate the enticing prospects of NIR light-mediated photosensitizing nanocomposites for ROS generation and their potential applications in photocatalysis and biomedical fields.
Discussion
In this study, we demonstrated a UCEL approach through the core-multishell heterogeneous structure design to regulate the energy transfer pathway in lanthanide-doped UCNPs for UVC and UVB generation by 808 nm excitation. The key to this design is the utilization of an optical inert NaYF4 host lattice with Yb3+ doping as an interlayer between the multiple cascade NIR photon sensitization shells and upconversion emitting core. Therefore, the sensitized NIR excitation energies can be transferred inbound and upconverted at the core domain of NaGdF4:Yb,Tm, where high-concentration Gd3+ ions can recycle among the higher-lying excited energy states above 6PJ to realize intense UVB and UVC upconversion emissions. We believe this approach will advance the design rationale for enhancing the NIR sensitized UV upconversion emissions towards the potential areas of biomedicine, information technology, photocatalysis, environmental science, and many other emerging fields.
Methods
Nanoparticles synthesis
We synthesized the core–multishell nanoparticles using the method described in ref. 24. and ref. 40. Additional experimental details are provided in the Supplementary Note.
Synthesis of UCNPs@TiO2 nanocomposites
Gd-CSYS2S3@TiO2 nanocomposites were synthesized according to a modified literature procedure55,56. Typically, 66 mg/mL (0.3 mL) as-prepared oleic acid nanoparticles Gd-CSYS2S3 were dispersed in a 0.2 M HCl solution followed by ultrasonication to remove the surface ligands. After that, ligand-free UCNPs were collected via centrifugation. The ligand-free nanoparticles were washed with deionized water and ethanol several times, and then dispersed in 4 mL of deionized water containing 0.8 g polyvinylpyrolidone (average Mw 40 000) with ultrasonication and stirring for 1 h. Then, 20 mL ethanol was added under magnetic stirring for 30 min. TiF4 aqueous solution (2.4 mL 0.025 M) was dropwise added into the solution under stirring. Then the whole solution was transferred into a 50 mL Teflon-lines autoclave and heated at 180 °C for 4 h. After cooling to the room temperature, the as-prepared products were collected by centrifugation, washed with deionized water and ethanol several times, and dried at 65 °C.
Single particle imaging
The emission intensities of single nanoparticles were recorded using a laboratory-built confocal microscopy system. Supplementary Fig. 25 shows the schematic drawing of the experimental setup, where UCNPs are excited by a polarization-maintaining single-mode fiber-coupled ~980 nm (BL976-PAG900, controller CLD1015, Thorlabs) or 808 nm (F280APC-808, Leoptics) diode lasers. The first half-wave plate (HWP, WPH05M-980, Thorlabs) and a polarized beam splitter (PBS, CCM1-PBS252/M, Thorlabs) are employed to control the excitation power by rotating HWP electronically. The purpose of the second HWP is to turn the polarization from horizontal to vertical. The same setup is used for the 808 nm laser excitation, combined to the ~980 nm excitation path by the first dichroic mirror (DM, T842lp, Chroma). After collimation, the excitation beam is reflected by the short-pass dichroic mirror (DM, T785spxrxt-UF1, Chroma), and focused through a high numerical aperture objective (UPlanSApo, 100×/1.40 oil, Olympus) to the sample slide. Photoluminescence is collected by the same objective and split from the excitation beams by a dichroic mirror DM. The emission signals are filtered by a short pass filter (SPF, FF01-750SP, Semrock), coupled into a multimode fiber (MMF, M42L02, Thorlabs), and detected by a single-photon counting avalanche photodiode (SPAD, SPCM-AQRH-14-FC, Excelitas). The MMF can also be switched to a monochromator (iHR550, Horiba) for upconversion emission spectrum measurement.
Evaluation of singlet oxygen generation
The chemical probe 1,3-diphenylisobenzofuran (DPBF) can be used to evaluate the amount of produced singlet oxygen (1O2) from the as-prepared Gd-CSYS2S3@TiO2 under 808 nm laser irradiation. DPBF can react with singlet oxygen (1O2) irreversibly and then cause the intensity decrease of its characteristic absorption at 417 nm55. Therefore, the amount of 1O2 produced under laser irradiation can be evaluated by the absorption signal of DPBF with a UV−Vis absorption spectrum.
Caculation of absorption cross-section σ of Nd3+
The approximate absorption cross-section σ of Nd3+ at 808 nm was calculated from the UV−Vis absorption spectra of the nanoparticles using the following equations44:
where A is the absorbance, ε is the absorption coefficient, M is the molar concentration of Nd3+, l is the path length, n is the atomic number density of Nd3+ ions. σ = 1.5 × 10−19 cm2 (808 nm for Gd-CSYS2S3), σ = 1.3 × 10−19 cm2 (808 nm for Gd-CSGdS2S3).
Data availability
All the relevant data are available from the correspondence authors upon reasonable request. Source data are provided with this paper.
References
Auzel, F. Upconversion and anti-stokes processes with f and d ions in solids. Chem. Rev. 104, 139–174 (2004).
Bünzli, J.-C. G. & Piguet, C. Taking advantage of luminescent lanthanide ions. Chem. Soc. Rev. 34, 1048–1077 (2005).
Dong, H., Sun, L. D. & Yan, C. H. Energy transfer in lanthanide upconversion studies for extended optical applications. Chem. Soc. Rev. 44, 1608–1634 (2015).
Wen, S. et al. Future and challenges for hybrid upconversion nanosystems. Nat. Photonics 12, 828–838 (2019).
Bettinelli, M., Carlos, L. D. & Liu, X. Lanthanide-doped upconversion nanoparticles. Phys. Today 68, 38–44 (2015).
Su, Q., Feng, W., Yang, D. & Li, F. Resonance energy transfer in upconversion nanoplatforms for selective biodetection. Acc. Chem. Res. 50, 32–40 (2017).
Jalani, G. et al. Photocleavable hydrogel-coated upconverting nanoparticles: a multifunctional theranostic platform for NIR imaging and on-demand macromolecular delivery. J. Am. Chem. Soc. 138, 1078–1083 (2016).
Yao, C. et al. Near-infrared-triggered azobenzene-liposome/upconversion nanoparticle hybrid vesicles for remotely controlled drug delivery to overcome cancer multidrug resistance. Adv. Mater. 28, 9341–9348 (2016).
Zhang, Z. et al. Upconversion superballs for programmable photoactivation of therapeutics. Nat. Commun. 10, 4586 (2019).
Gao, W., Zhang, W. & Lu, G. A two-pronged strategy to enhance visible-light-driven overall water splitting via visible-to-ultraviolet upconversion coupling with hydrogen-oxygen recombination inhibition. Appl. Catal. B-Environ. 212, 23–31 (2017).
Anwer, H. & Park, J. W. Near-infrared to visible photon transition by upconverting NaYF4: Yb3+, Gd3+, Tm3+@Bi2WO6 core@shell composite for bisphenol a degradation in solar light. Appl. Catal. B-Environ. 243, 438–447 (2019).
Chen, X. et al. Confining energy migration in upconversion nanoparticles towards deep ultraviolet lasing. Nat. Commun. 7, 10304 (2016).
Zheng, K. et al. Rewritable optical memory through high-registry orthogonal upconversion. Adv. Mater. 30, 1801726 (2018).
Bansal, A. & Zhang, Y. Photocontrolled nanoparticle delivery systems for biomedical applications. Acc. Chem. Res. 47, 3052–3060 (2014).
Jayakumar, M. K. G., Idris, N. M. & Zhang, Y. Remote activation of biomolecules in deep tissues using near-infrared-to-UV upconversion nanotransducers. Proc. Natl Acad. Sci. USA 109, 8483–8488 (2012).
Dai, Y. et al. In vivo multimodality imaging and cancer therapy by near-infrared light-triggered trans-platinum pro-drug-conjugated upconversion nanoparticles. J. Am. Chem. Soc. 135, 18920–18929 (2013).
Zhao, J., Chu, H., Zhao, Y., Lu, Y. & Li, L. A NIR light gated DNA nanodevice for spatiotemporally controlled imaging of microRNA in cells and animals. J. Am. Chem. Soc. 141, 7056–7062 (2019).
Shi, F., Wang, J., Zhang, D., Qin, G. & Qin, W. Greatly enhanced size-tunable ultraviolet upconversion luminescence of monodisperse β-NaYF4:Yb,Tm nanocrystals. J. Mater. Chem. 21, 13413–13421 (2011).
Dawson, P. & Romanowsk, M. Excitation modulation of upconversion nanoparticles for switch-like control of ultraviolet luminescence. J. Am. Chem. Soc. 140, 5714–5718 (2018).
Zhao, C. et al. Li+ ion doping: an approach for improving the crystallinity and upconversion emissions of NaYF4:Yb3+,Tm3+ nanoparticles. Nanoscale 5, 8084–8089 (2013).
Wang, F., Wang, J. & Liu, X. Direct evidence of a surface quenching effect on size-dependent luminescence of upconversion nanoparticles. Angew. Chem. Int. Ed. 49, 7456–7460 (2010).
Wang, F. et al. Tuning upconversion through energy migration in core–shell nanoparticles. Nat. Mater. 10, 968–973 (2011).
Sun, T. et al. Integrating temporal and spatial control of electronic transitions for bright multiphoton upconversion. Nat. Commun. 10, 1811 (2019).
Su, Q. et al. The effect of surface coating on energy migration-mediated upconversion. J. Am. Chem. Soc. 134, 20849–20857 (2012).
Chen, Q. et al. Confining excitation energy in Er3+-sensitized upconversion nanocrystals through Tm3+-mediated transient energy trapping. Angew. Chem. Int. Ed. 56, 7605–7609 (2017).
Wang, Y. et al. Nd3+-sensitized upconversion nanophosphors: efficient in vivo bioimaging probes with minimized heating effect. ACS Nano 7, 7200–7206 (2013).
Wang, H., Li, Y., Yang, M., Wang, P. & Gu, Y. FRET-based upconversion nanoprobe sensitized by Nd3+ for the ratiometric detection of hydrogen peroxide in vivo. ACS Appl. Mater. Interfaces 11, 7441–7449 (2019).
Xu, J. et al. Highly emissive dye-sensitized upconversion nanostructure for dual-photosensitizer photodynamic therapy and bioimaging. ACS Nano 11, 4133–4144 (2017).
Zuo, J. et al. Near infrared light sensitive ultraviolet−blue nanophotoswitch for imaging-guided “off−on” therapy. ACS Nano 12, 3217–3225 (2018).
Chan, M.-H. et al. Nanobubble-embedded inorganic 808 nm excited upconversion nanocomposites for tumor multiple imaging and treatment. Chem. Sci. 9, 3141–3151 (2018).
Liu, C. et al. sensitized upconversion metal–organic Nd3+-frameworks for mitochondria-targeted amplified photodynamic therapy. Angew. Chem. Int. Ed. 59, 2634–Nd32638 (2020).
Zhang, Y. et al. Ultrasmall-superbright neodymium upconversion nanoparticles via energy migration manipulation and lattice modification: 808 nm-activated drug release. ACS Nano 11, 2846–2857 (2017).
Tan, L. et al. Preparation of multishell-structured NaYF4:Yb,Tm,Nd@NaYF4:Yb,Nd@SiO2@ZnO nanospheres with effective NIR-induced photocatalytic activity. J. Phys. Chem. C. 124, 18081–18090 (2020).
Xie, X. et al. Mechanistic investigation of photon upconversion in Nd3+-sensitized core−shell nanoparticles. J. Am. Chem. Soc. 135, 12608–12611 (2013).
Wen, H. et al. Upconverting near-infrared light through energy management in core–shell–shell nanoparticles. Angew. Chem. Int. Ed. 52, 13419–13423 (2013).
Shen, J. et al. Engineering the upconversion nanoparticle excitation wavelength: cascade sensitization of tri-doped upconversion colloidal nanoparticles at 800 nm. Adv. Opt. Mater. 1, 644–650 (2013).
Zhong, Y. et al. Elimination of photon quenching by a transition layer to fabricate a quenching-shield sandwich structure for 800 nm excited upconversion luminescence of Nd3+-sensitized nanoparticles. Adv. Mater. 26, 2831–2837 (2014).
Wen, S. et al. Advances in highly doped upconversion nanoparticles. Nat. Commun. 9, 2415 (2018).
Liao, J., Jin, D., Chen, C., Li, Y. & Zhou, J. Helix shape power-dependent properties of single upconversion nanoparticles. J. Phys. Chem. Lett. 11, 2883–2890 (2020).
Wang, S. et al. Comparative investigation of the optical spectroscopic and thermal effect in Nd3+-doped nanoparticles. Nanoscale 11, 10220–10228 (2019).
Qin, W. et al. Ultraviolet upconversion fluorescence from 6DJ of Gd3+ induced by 980 nm excitation. Opt. Lett. 33, 2167–2169 (2008).
Zheng, K., Qin, W., Cao, C., Zhao, D. & Wang, L. NIR to VUV: seven-photon upconversion emissions from Gd3+ ions in fluoride nanocrystals. J. Phys. Chem. Lett. 6, 556–560 (2015).
Qin, W. et al. Multi-ion cooperative processes in Yb3+ clusters. Light Sci. Appl. 3, e193 (2014).
Xu, Y. et al. Crystal growth and optical properties of YbAl3(BO3)4: a promising stoichiometric laser crystal. J. Cryst. Growth 252, 241–245 (2003).
Pollnau, M., Gamelin, D. R., Lüthi, S. R., Güdel, H. U. & Hehlen, M. P. Power dependence of upconversion luminescence in lanthanide and transition-metal-ion systems. Phys. Rev. B 61, 3337–3346 (2000).
Blasse, G. The physics of new luminescent materials. Mater. Chem. Phys. 16, 201–236 (1987).
Blasse, G., Grabmaier, B. C. in Energy Transfer. Luminescent Materials Springer, (Springer, 1994).
Weber, M. J. Optical properties of Yb3+ and Nd3+-Yb3+ energy transfer in YAlO3. Phys. Rev. B 4, 3153–3159 (1971).
Bartosiewicz, K., Babin, V., Kamada, K., Yoshikawa, A. & Nikl, M. Energy migration processes in undoped and Ce-doped multicomponent garnet single crystal scintillators. J. Lumin. 166, 177–122 (2015).
Pokhrel, M., Valdes, C. & Mao, Y. Ultraviolet upconversion enhancement in triply doped NaYF4:Tm3+,Yb3+ particles: the role of Nd3+ or Gd3+ co-doping. Opt. Mat. 58, 67e75 (2016).
Zhu, Q. et al. Yb3+-sensitized upconversion and downshifting luminescence in Nd3+ ions through energy migration. Dalton Trans. 47, 8581–8584 (2018).
Zhou, B. et al. NIR II-responsive photon upconversion through energy migration in an ytterbium sublattice. Nat. Photonics 14, 760–766 (2020).
Wang, F. et al. Microscopic inspection and tracking of single upconversion nanoparticles in living cells. Light Sci. Appl. 7, 18007 (2018).
Zhou, J., Chizhik, A. I., Chu, S. & Jin, D. Single-particle spectroscopy for functional nanomaterials. Nature 579, 41–50 (2020).
Hou, Z. et al. UV-emitting upconversion-based TiO2 photosensitizing nanoplatform: near-infrared light mediated in vivo photodynamic therapy via mitochondria-involved apoptosis pathway. ACS Nano 9, 2584–2599 (2015).
Zhang, Y. & Hong, Z. Synthesis of lanthanide-doped NaYF4@TiO2 core–shell composites with highly crystalline and tunable TiO2 shells under mild conditions and their upconversionbased photocatalysis. Nanoscale 5, 8930–8933 (2013).
Acknowledgements
The authors thank the National Basic Research Program of China (No. 2016YFA0201600), the National Natural Science Foundation of China (Nos. 21701109 and 31771105), Shanghai Shuguang Program (18SG29) and Natural Science Foundation of Shanghai (18ZR1401700) for financial supports. The authors acknowledge the assistance of SUSTech Core Research Facilities. The authors thank Prof. X. Liu, Prof. F. Li, Prof. F. Wang, Prof. K. Zheng, Prof. X. Zhu, Prof. A. Cao, Dr. S. Han and Dr. J. Zhou for helpful discussions. The authors thank Prof. W. Feng and Y. Cai for their help with lifetime measurements. The authors thank R. Liu for her help with quantum field measurements.
Author information
Authors and Affiliations
Contributions
Q. S. and D. J. conceived the project. Q. S., H. W., and D. J. designed the experiments and supervised the research. Q. S., H. L. W., Y. L., C. C., M. G., S. W., Y. S., and Z. C. were primarily responsible for the experiments of nanoparticles synthesis and characterization. Q. S., D. J., H. L. W., Y. L., C. C., Y. S., and S. W. contributed to the data analyses and discussion. Q. S. and H. L. W. prepared the figures. Q. S. and D. J. wrote the paper with input from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, Q., Wei, HL., Liu, Y. et al. Six-photon upconverted excitation energy lock-in for ultraviolet-C enhancement. Nat Commun 12, 4367 (2021). https://doi.org/10.1038/s41467-021-24664-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-24664-x
This article is cited by
-
Ultra-wideband-responsive photon conversion through co-sensitization in lanthanide nanocrystals
Nature Communications (2023)
-
Proof of crystal-field-perturbation-enhanced luminescence of lanthanide-doped nanocrystals through interstitial H+ doping
Nature Communications (2023)
-
Upconversion nanoparticles for super-resolution quantification of single small extracellular vesicles
eLight (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
