
Abstract
Contemporary asymmetric catalysis faces huge challenges when prochiral substrates bear electronically and sterically unbiased substituents and when substrates show low reactivities. One of the inherent limitations of chiral catalysts and ligands is their incapability in recognizing prochiral substrates bearing similar groups. This has rendered many enantiopure substances bearing several similar substituents inaccessible. Here we report the rationale, scope, and applications of the strategy of kinetic resolution of auxiliary adjacent alcohols (KRA*) that can be used to solve the above troubles. Using this method, a large variety of optically enriched tertiary alcohols, epoxides, esters, ketones, hydroxy ketones, epoxy ketones, β-ketoesters, and tetrasubstituted methane analogs with two, three, and four spatially and electronically similar groups can be readily obtained (totally 96 examples). At the current stage, the strategy serves as the optimal solution that can complement the inability caused by direct asymmetric catalysis in getting chiral molecules with challenging fully substituted stereocenters.
Similar content being viewed by others


Introduction
The last several decades have witnessed great achievements in the field of asymmetric catalysis, which have laid a profound impact on various fields including chemical engineering, pharmaceutical industry, material science, and agrochemistry. The success relies mainly on the discovery of various elegantly designed chiral catalysts/ligands, which enable the excellent enantiodiscrimination of prochiral substrates in the reaction1,2,3,4. On the other side, fully substituted carbon stereocenters are ubiquitous in naturally occurred compounds, bioactive substances, and drug molecules, and the rapid and efficient construction of substances containing such key moieties represents one of the most important topics in asymmetric synthesis5,6,7,8,9,10.
Although great progress has been made in addressing the above issue, a series of challenging targets still exist. For instance, Fig. 1a lists two general classes of functionalized compounds with tetrasubstituted carbon centers. Because all the stereogenic centers are connected by two, three, and even four electronically and sterically similar groups, most of them are hard to be rapidly produced via currently known direct catalytic methods. The origin arises from two innate limitations concerning asymmetric catalysis (Fig. 1b): (1) when prochiral substrates contain two similar substituents, low ee of the products will be expected owing to the poor facial enantiodiscrimination; (2) when substrates display low reactivities, the corresponding asymmetric catalysis will be more challenging and harsh conditions have to be employed, which is very probable to lead to low levels of enantioselectivity. For instance, enantiopure tertiary alcohols are important building blocks and key units in a large amount of naturally occurring and artificial bioactive molecules; asymmetric nucleophilic additions to ketones and kinetic resolution of racemic tertiary alcohols represent two mostly used strategies allowing access to tertiary alcohols with high enantiopurity11,12,13,14,15,16,17,18,19. However, for the former one, the frequently used prochiral ketones are aryl/alkyl ketones (e.g., acetophenone), α-ketoesters, trifluoromethyl ketones, and isatin derivatives20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35; ketones with electronically and sterically unbiased substituents such as Ar1COAr2 and R1CH2COCH2R2, and less reactive ketones such as tBuCOPh and tBuCOiPr are not suitable reaction partners for nucleophilic additions. Kinetic resolution methods producing enantiopure tertiary alcohols are mostly constrained to those with aryl/alkyl substituents and sterically different alkyl-alkyl combinations36,37,38,39,40,41,42,43,44,45,46,47,48. A similar situation can also be found in the access to enantioenriched epoxides, ketones, and esters, etc. (Fig. 1a). Therefore, to promote the diversity of the current molecule types containing tetrasubstituted stereocenters and provide opportunities for bioactive molecule preparation and pharmaceutical discovery, a general solution to obtain all the above fully substituted molecules is highly desirable but still absent to date.

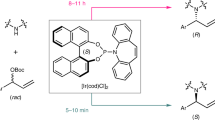
a Challenging targets in asymmetric synthesis. b Two formidable obstacles in asymmeric catalysis. Cat.* catalyst, O oxidants, Nu nucleophiles, E electrophiles. c Rationale of hydroxy addition-kinetic resolution. d Selected examples of diastereoselective synthesis of III. e Proof of concept. Ee values were determined via HPLC analysis on a chiral stationary phase; selectivity factors (s) were calculated according to the following equation: s = ln[(1 − conv)(1 − ee1a)]/ln[(1 − conv)(1 + ee1a)], conv = (ee1a)/(ee1a + ee2a).
In this work, we develop an alternative strategy of kinetic resolution of auxiliary adjacent alcohols (KRA*) to address the big challenge that is hard to be solved by designing chiral catalysts and ligands. The work features the combination of diastereoselective secondary alcohol synthesis and the following kinetic resolution, and in the process, the hydroxy group serves as a kinetic resolution auxiliary group.
Results
Design of the project
As illustrated in Fig. 1c, enantiopure targets (I) represent all types of challenging compounds shown in Fig. 1a, and are hard to be obtained using direct synthetic methods. However, inspired by Corey’s retrosynthetic analysis, we imagine that I can be readily derived from enantiopure medium compounds II via OH group transformations; enantiopure II then can be obtained from racemic substrates III through kinetic resolution, because now the two groups connected to the second alcohol moiety are with different steric and (or) electronic properties. If successful, this method of combining diastereoselective synthesis of secondary alcohols bearing adjacent full-substituted carbon centers and the following kinetic resolution will be a general, robust, and practical strategy for the access to many different types of challenging compounds bearing tetrasubstituted stereogenic centers. Noteworthy is that substrates III are relatively easier to be obtained as racemic form, but hard to be produced in their enantioenriched form using known methods; therefore, the kinetic resolution of III is still the optimal way to get the enantiopure ones. Selected examples of highly diastereoselective synthesis of III are shown in Fig. 1d (see the Supplementary Material for more details). Moreover, the conventional kinetic resolution has mainly focused on the separation of racemic compounds with a single stereocenter; resolution on racemic substances bearing two or more stereocenters has been underdeveloped and its great potential in organic synthesis remains to be demonstrated. Nevertheless, we believe that resolution on easily available racemic substrates with multiple stereocenters will be an inevitable tendency for the further development of the whole field of kinetic resolution.
Reaction optimization
To test the feasibility of our hypothesis, we commenced by selecting racemic diol 1a as the model substrate to check the possibility of achieving an efficient resolution of tertiary alcohol with two aryl groups. Acyl transfer catalysts49,50,51,52 BTM, HBTM, and HyperBTM were selected for the survey. As shown in Fig. 1e, among the three catalysts, both HBTM and HyperBTM showed good results, with the former one giving slightly higher selectivity factor (s) of 78. To our pleasure, lowering the temperature proved beneficial to the resolution when HBTM was used as the catalyst, and up to 127 of the s value was observed, which also proved the feasibility of our hypothesis. Noteworthy is that direct asymmetric dihydroxylation of 1,1-diaryl alkenes using Sharpless’ conditions can only afford the corresponding alcohols with poor enantioselectivity53, and such type of getting enantioenriched tertiary alcohols using the assistance of secondary alcohol resolution has been not disclosed.
Substrate scope
Having proved the conceptual feasibility of KRA*, we investigated a series of diols bearing diary tertiary alcohol units, and in all cases, excellent enantiopurity (91–99% ee) of the recovered diols were observed, with up to 340 of the s value obtained (Fig. 2a, 1j). It’s worthwhile to mention that although all racemic diols 1 can be readily made from the corresponding racemic α-hydroxy ketones, the state-of-art asymmetric synthesis of these α-hydroxy ketones has been not satisfied, and most of them cannot be obtained in a practically useful level (i.e., >60% yield and >95% ee, see the Supplementary Material for more details) for the further synthesis of chiral diols 1. This method also tolerated aryl groups with a minimal difference, such as 4-Br-C6H4/4-Cl-C6H4 (Fig. 2a, 1a and 1h), 4-Me-C6H4/4-OMe-C6H4 (Fig. 2a, 1b and 1j), 2-thienyl/2-furyl (Fig. 2a, 1f), 2-pyridinyl/Ph (Fig. 2a, 1g), and C6D5/C6H5 (Fig. 2a, 1l), and can be scaled up using gram scale of racemic 1h. Actually, the method shows no limitation with respect to the steric and electronic difference of the two aryl substituents, and furthermore, through simply changing the synthesis route of the racemic substrates, both enantiopure diastereomeric diols can be obtained, as witnessed by 1b and 1j (the absolute configurations are opposite at the tertiary alcohol units). In literature reports, Rh-catalyzed asymmetric addition of aryl boric acids to ketones has been developed to make diaryl tertiary alcohols, but the system is usually compatible with activated ketones such as trifluoromethyl ketones and α-ketocarbonyls21,54,55. Titanium-catalyzed asymmetric Grignard addition to aryl ketones has also been developed, but in most cases, methyl aryl ketones were tested to give moderate ee, and ethyl aryl ketone proved inert to the reaction20. Also noteworthy is that, different from the established secondary alcohol kinetic resolution mode using HBTM50, the π-π stacking in this process occurs between the Ar1 group connected to the tertiary alcohol unit and the catalyst (Fig. 2a, TS-1). To the best of our knowledge, such a resolution model has been not established in known reports49,50,51,52. This is further confirmed by the X-ray single-crystal analysis of 2j (Fig. 2a, and also see Supplementary Data 1).

a Scope of diaryl tertiary alcohols. b Scope of dialkyl tertiary alcohols and triaryl tertiary alcohols. Variations of the standard conditions: 1n: (S)-BTM (10 mol%), rt; 1r: anhydrous Na2SO4 (150 mg), toluene, 0 °C, 11 h; 1x: rt, 20 h; 1y: −40 °C; 1aa–1ad: (S)-BTM (10 mol%), Na2SO4 (150 mg), toluene; 1ad: −20 °C. c conversion.
Inspired by the success of accessing diaryl tertiary alcohols using KRA* method, we further surveyed tertiary alcohols with all aliphatic substituents (three sp3 carbons), which represents one of the most challenging topics in the field of asymmetric catalysis. Very limited methods have been developed to address the issue, and to date the most effective methods were reported by Hoveyda and co-workers, using asymmetric allylation and propargylation of methyl and mono, di, or trifluoromethyl ketones31,32,56,57,58 and the kinetic resolution of methyl tertiary alcohols39. Asymmetric desymmetrization has also been developed, but with specific types of substrates59,60,61,62. To our pleasure, under slightly modified conditions (using BTM instead of HBTM), a series of tertiary alcohols with Me/Et (Fig. 2b, 1m), Me/cyclohexyl (Fig. 2b, 1n), Me/iPr (Fig. 2b, 1o), Me/allyl (Fig. 2b, 1p), Me/4-Me-3-pentenyl (Fig. 2b, 1q), and Et/nBu (Fig. 2b, 1r) substituents were all successfully resolved, and the recovered diols were obtained with 94–97% ee. Synthetically more difficult tertiary alcohols with two methylene groups such as PhCH2/MeCH2 (Fig. 2b, 1s), BnCH2/PhCH2 (Fig. 2b, 1t), BnCH2/4-MeC6H4CH2 (Fig. 2b, 1u), and EtCH2CH2/PhCH2CH2 (Fig. 2b, 1v) can also be easily accessed via the KRA* method, and the s value can be up to >500 (Fig. 2b, 1u). Moreover, sterically bulky tBu-substituted diols can also be efficiently resolved (Fig. 2b, 1w and 1x). To the best of our knowledge, 1r-1x type tertiary alcohols are not accessible using currently known methods. Furthermore, tertiary alcohols bearing substituents with different electronic properties (Et/vinyl, nBu/phenylethynyl, cyclohexyl/Ph) were also tolerated under the standard conditions, leading to recovered 1y−1za with good to excellent enantiopurities (Figs. 2b, 1y−1za). Additionally, the state-of-art of asymmetric catalysis cannot produce enantiopure triaryl tertiary alcohols as far as we know. To test the robustness of KRA*, we found that enantioenriched triaryl alcohols 1aa−1ad can also be produced with up to 99% ee (Fig. 2b, 1aa−1ad). Noteworthy is that the resolution of 1aa was run on a 1.4 g scale. X-ray structure analysis of 1s (Supplementary Data 2) and 2ac (Supplementary Data 3) confirms the configuration of the enantioenriched alcohols.
Subsequently, we focused on the catalytic resolution of epoxides with geminal aryl or alkyl groups. The great importance of chiral epoxides has been well documented, and the asymmetric olefin epoxidation is the most direct and powerful approach to produce such molecules, as has been established by Sharpless, Jacobsen, Shi, and others63,64,65,66,67,68. However, alkenes having 1,1-diaryl or 1,1-dialkyl substituents with similar steric bulkiness are not suitable reaction partners for highly enantioselective epoxidation. Pleasingly, we found that the KRA* method can achieve the highly efficient resolution of epoxides containing diaryl substituents with minimal steric and electronic differences, producing 3a–3g with 91–99% ee (Fig. 3, 3a−3g). Similarly, the system showed no negative influence on epoxides with geminal aliphatic groups, delivering 3h–3k with 96–99% ee (Fig. 3, 3h−3k). The absolute configuration of 4d was confirmed by single-crystal X-ray structure analysis (Supplementary Data 4). It’s worthwhile to mention that enantioenriched epoxy alcohols (EEAs) with three stereogenic centers also hold great importance in modern organic synthesis, but the predominant catalytic method to make EEAs is still the kinetic resolution of racemic allylic alcohols using Sharpless epoxidation69,70. Therefore, this method provides a complementary approach for getting EEAs with germinal similar substituents, which are hard to be formed with high enantiopurity using Sharpless’ asymmetric epoxidation protocol. Also noteworthy is that, in principle, all the chiral epoxy alcohols shown in Fig. 3 can be made from the diastereoselective reduction of chiral epoxy ketones or epoxidation of chiral allylic alcohols, but a global literature survey shows that all epoxy alcohols 3 cannot be obtained with a useful level of enantiopurity starting from chiral chemicals (see the Supplementary Material for more details).

3a–3g, resolution of epoxides with two similar aromatic substituents. 3h–3k, resolution of epoxides with two similar aliphatic substituents. c conversion.
Asymmetric synthesis of acyclic esters with α-quaternary carbon centers has been a long-time challenge, owing to the limited activation methods towards esters, and the relatively low reactivity of α,α-disubstituted esters. Known methods have relied heavily on activated substrates such as α-branched β-ketoesters or α-cyano esters71,72,73,74,75,76. Catalytic approaches affording esters with α-quaternary carbons connected by three Csp3 groups are still rare. Therefore, our next goal is to test the capability of KRA* in accessing enantioenriched esters (especially unactivated esters) with α-quaternary stereocenters. As displayed in Fig. 4, racemic 5 can be readily made through the stereoselective reduction of the corresponding α,α-disubstituted β-ketoesters (see the Supplementary Material).

a Scope of esters with α-quaternary stereocenters. Variation of the standard conditions: 5f: 10 °C, 28 h; 5b/5j/5m: 20 °C; 5c/5d: (S)-BTM (10 mol%); 5g/5i/5l/5n/5o: (S)-BTM (10 mol%), −10 °C. b Scope of tetrasubstituted methane derivatives. Variations of the standard conditions: 7c/7g: −10 °C, 23 h; 7h: (S)-BTM (15 mol%), −20 °C, 40 h; 7i: (S)-BTM (4 mol%), iPr2NEt (1.0 equiv), toluene; 7j: iPr2NEt (1.0 equiv), toluene, 12 h; 7k/7m: (S)-BTM (5 mol%). c conversion.
Using the KRA* method, a variety of α-allyl-β-hydroxy esters can be successfully resolved, providing enantioenriched 5a–5f with 89–97% ee (Fig. 4a, 5a−5f). We were glad to find that esters with other functional groups such as propargyl group (5g), butyric ester group (5h), and vinyl group (5i) were also compatible under the resolution conditions (Fig. 4a, 5g−5i). Moreover, the substituents connected to the secondary alcohol moiety are not limited to the phenyl group, and other groups such as naphthyl, furyl, and styryl groups are also efficient in achieving the corresponding ester resolution (Fig. 4a, 5j−5o). Because racemic 5 were obtained via diastereoselective reduction of the corresponding disubstituted ketoesters, one will imagine the possibility of getting chiral 5 from chiral α,α-disubtituted β-ketoesters. However, the known methods cannot afford all the corresponding α,α-disubtituted β-ketoesters used in this work in acceptable results (see the Supplementary Material for more details).
All-carbon quaternary stereogenic centers are ubiquitous in naturally occurred substances and artificial pharmaceuticals10,77,78. However, the construction of enantiopure compounds with all-carbon quaternary centers also represents one of the most challenging topics in asymmetric catalysis, especially for those existing in acyclic molecules. Asymmetric nucleophilic addition, cascade annulation, and allylic alkylation are powerful methods to achieve the purpose, but in most cases, they deliver products with electronically distinct substituents5,6,7,8,9,10,79,80. Therefore, a remaining hard topic is the rapid asymmetric formation of all-sp3-carbon-substituted quaternary centers, and very limited reports have achieved this purpose. In this context, we tested the possibility of getting enantioenriched compounds with various different all-carbon quaternary centers using KRA* approach. To our delight, all of them can be easily resolved, and in all cases, excellent 91–99% ee values were detected (Fig. 4b, 7a–7n). The absolute configuration of 7k was confirmed by single-crystal X-ray structure analysis (Supplementary Data 5). Noteworthy is that molecules with quaternary carbon centers bearing three methylene groups were also efficiently resolved (Fig. 4b, 7c and 7d). The reaction using 1.8 g of racemic 7m also proved successful, again showing the potential of this method in big scale production.
Synthetic transformations
Having obtained a large number of enantioenriched molecules with quaternary and tetrasubstituted stereocenters and secondary alcohol units, we commenced completing the final purpose of getting the molecule types shown in Fig. 1a. To our pleasure, the readily transformable feature of the OH group makes further transformations simple. As shown in Fig. 5a, using easily operable Dess-Marin periodinane (DMP) oxidation conditions, α-hydroxy ketones with α,α-diaryl groups (9a), α,α-dimethylene substituents (9b), and bulky α-substituent (9c) were all concisely produced without any erosion of the enantiopurity. Then a variety of ketones with all-carbon α-quaternary centers were also released with 93–99% ee (Fig. 5b, 9d–9g), and 9f is a ketone with three different methylene groups. Furthermore, β-ketoesters with quaternary stereocenters are highly valuable building blocks in complex molecule synthesis, but in most cases, the α-functionalization of α-branched β-ketoesters cannot tolerate two bulky α-substituents (i.e., bulkier than a methyl group), which significantly limits the scope of this field81. In this study, ketoesters 9h and 9i bearing bulky substituents can also be easily obtained, thus complementing the drawbacks caused by the direct asymmetric catalysis. Additionally, the reaction could provide epoxy ketone bearing two aryl or two alkyl groups with minimal structural distinctions (Fig. 5b, 9j and 9k). Moreover, the secondary OH can be easily removed under reductive conditions, affording tertiary alcohols with three different alkyl groups (Fig. 5b, 10a and 10b). Similarly, enantioenriched molecules with all-carbon quaternary stereocenters such as 10c–10g were also gotten with excellent 90–95% ee (Fig. 5b, 10c–10g); noteworthy is that 10g has four distinct methylene substituents, which is almost inaccessible via conventional methods. Finally, ester 10h having three α-methylene substituents was also generated in quantitative yield with 90% ee, which is also a very challenging target using other protocols.

a Dess-Marin periodinane (DMP) oxidation. b OH group removal. Conditions I: Raney Ni (3 equiv), EtOH, reflux, 12 h. Conditions II: CF3CO2H (8 equiv), CH2Cl2, rt, 24 h; then H2, Pd/C (10 %), EtOAc, rt, 12 h. Conditions III: HBF4 (2 equiv), Et3SiH (2 equiv), CH2Cl2, 40 °C, 3 h. Conditions IV: Et3SiH (2.5 equiv), CF3CO2H (6 equiv), CH2Cl2, 0 °C −rt, 12 h. c Various other transformations.
Besides the OH-oxidation and removal reactions, the enantiopure substances obtained in this study can also participate in a series of useful further transformations. For instance, the ring-opening of 3h by NaN3 allowed access to 3-azido-1,2-diol 11a with excellent diastereo- and enantioselectivity (Fig. 5c, eq 1), and β-amino acid derivative 11b could be formed through a formal replacement of OH by NH2 group (Fig. 5c, eq 2). Moreover, 1aa was readily transferred to 1,3-dihydroisobenzofuran 11c in 94% yield with 92% ee (Fig. 5c, eq 3), and methyl ester 11d with a cyclopenta[a]naphthalene unit was delivered by a formal Friedel-Crafts alkylation of 5e (Fig. 5c, eq 4). The absolute configuration of 11c was confirmed by single-crystal X-ray structure analysis (Supplementary Data 6). Diaryl epoxide 11e was formed from 1b via a two-step method (Fig. 5c, eq 5), and the formal synthesis of serotonin antagonist 11g can be easily realized from 7h (Fig. 5c, eq 6)82.
Discussion
We have systematically demonstrated the rationale, scope, and applications of the strategy of kinetic resolution of auxiliary adjacent alcohol (KRA*). The combination of diastereoselective secondary alcohol synthesis and the following kinetic resolution has been exploited as a powerful strategy that can satisfactorily complement the limitations confronted by current direct asymmetric catalytic methods. The study shows a general scope in accessing enantioenriched molecules with various tetrasubstituted carbon stereocenters such as tertiary alcohols, epoxides, esters, quaternary methane derivatives, ketones, α-hydroxy ketones, epoxy ketones, β-ketoesters, etc., including those with two, three, and even four sterically and electronically unbiased substituents (96 examples). A detailed survey has shown that a vast majority of the products obtained via KRA* in this work cannot be gotten using either direct asymmetric catalysis or starting from enantiopure chiral chemicals. Moreover, besides the broad substrate scope and high resolution efficiency, the protocol is also featured by its very mild conditions, being able to scale up, and using readily available catalysts. These features guarantee the further application of KRA* in both academic and industrial communities. The study also enriches the toolbox during bioactive molecule synthesis and drug discovery, and represents a development tendency in the field of kinetic resolution. Studies on further applications of this strategy in accessing highly valuable enantioenriched molecules are ongoing in our group.
Methods
General
For 1H and 13C nuclear magnetic resonance (NMR) spectra of compounds in this manuscript and details of the synthetic procedures as well as more reaction conditions screening, see Supplementary Information.
General procedures for the kinetic resolution of diols 1a–1l, 1za
To a flask containing 1a (342 mg, 1.0 mmol), (S)-HBTM (10.65 mg, 0.04 mmol) and anhydrous Na2SO4 (200 mg) was added CHCl3 (5 mL) at 0 °C. After dissolvation of all compounds, (EtCO)2O (69.2 μL, 0.54 mmol) was added to the clear solution. After stirring for 12 h at 0 °C, solvent was removed under reduced pressure, the crude product was purified with a silica gel column chromatograghy using a mixture of hexane and ethyl acetate (10:1 v/v) as eluent gave 2a (171 mg, 43% yield) as a white solid and 1a (164 mg, 48% yield) as a white solid.
General procedures for the kinetic resolution of diols 1m–1z, 1aa–1ad, 3a–3k, 5a–5n, 7a–7l
Catalyst (S)-BTM (2.5 mg, 0.01 mmol) and 4 Å molecular sieve (50 mg) were added to a Schlenk tube at room temperature under argon atmosphere. A solution of rac-1a (49.6 mg, 0.2 mmol) and iPr2NEt (16.5 μL, 0.1 mmol) in distilled CHCl3 (1 mL) was added via syringe. After stirring for 10 min at 0 °C, (EtCO)2O (12.8 μL, 0.1 mmol) was added to the reaction via microsyringe. After stirred at 0 °C for 11 h, the reaction mixture was diluted with petroleum ether, and then purified by flash chromatography sing a mixture of hexane and ethyl acetate (20:1 v:v) as eluent to afford the products 2m (23.5 mg, 50% yield) and 1m (18 mg, 50% yield).
Data availability
X-ray structural data for 1s (CCDC 2026917), 2ac (2026918), 2j (CCDC 2026919), 4d (CCDC 2026920), 7k (CCDC 2026921), and 11c (CCDC 2026922) can be obtained free of charge from the Cambridge Crystallographic Data Centre (CCDC) via http://www.ccdc.cam.ac.uk/data_request/cif. All other experimental data are available in the main text or the supplementary materials, and also are available from the corresponding author upon reasonable request.
References
Ojima, I. Ed., Catalytic Asymmetric Synthesis, Second Edition (Wiley, 2000).
Jacobsen, E. N., Pfalz, A. & Yamamoto, H. Eds., Comprehensive Asymmetric Catalysis (Springer, 1999).
Lin, G.-Q., Li, Y.-M. & Chan, A. S. C. Eds., Principles and Applications of Asymmetric Synthesis (Wiley, 2001).
List, B. Ed., Asymmetric Organocatalysis (Springer, 2010).
Fuji, K. Asymmetric creation of quaternary carbon centers. Chem. Rev. 93, 2037–2066 (1993).
Corey, E. J. & Guzman-Perez, A. The catalytic enantioselective construction of molecules with quaternary carbon stereocenters. Angew. Chem. Int. Ed. 37, 388–401 (1998).
Quasdorf, K. W. & Overman, L. E. Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 516, 181–191 (2014).
Liu, Y., Han, S.-J., Liu, W.-B. & Stoltz, B. M. Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc. Chem. Res. 48, 740–751 (2015).
Zeng, X.-P., Cao, Z.-Y., Wang, Y.-H., Zhou, F. & Zhou, J. Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev. 116, 7330–7396 (2016).
Büschleb, M. et al. Synthetic strategies toward natural products containing contiguous stereogenic quaternary carbon atoms. Angew. Chem. Int. Ed. 55, 4156–4186 (2016).
Shibasaki, M. & Kanai, M. Asymmetric synthesis of tertiary alcohols and α-tertiary amines via Cu-catalyzed C-C bond formation to ketones and ketimines. Chem. Rev. 108, 2853–2873 (2008).
Trost, B. M. & Weiss, A. H. The enantioselective addition of alkyne nucleophiles to carbonyl groups. Adv. Synth. Catal. 351, 963–983 (2009).
Collados, J. F., Solà, R., Harutyunyan, S. R. & Maciá, B. Catalytic synthesis of enantiopure chiral alcohols via addition of Grignard reagents to carbonyl compounds. ACS Catal. 6, 1952–1970 (2016).
Liu, Y.-L. & Lin, X.-T. Recent advances in catalytic asymmetric synthesis of tertiary alcohols via nucleophilic addition to ketones. Adv. Synth. Catal. 361, 876–918 (2019).
France, S., Guerin, D. J., Miller, S. J. & Lectka, T. Nucleophilic chiral amines as catalysts in asymmetric synthesis. Chem. Rev. 103, 2985–3012 (2003).
Pellissier, H. Catalytic non-enzymatic kinetic resolution. Adv. Synth. Catal. 353, 1613–1666 (2003).
Gurubrahamam, R., Cheng, Y.-S., Huang, W.-Y. & Chen, K. Recent advances in organocatalytic kinetic resolution for the synthesis of functionalized products. ChemCatChem 8, 86–96 (2016).
Müller, C. E. & Schreiner, P. R. Organocatalytic enantioselective acyl transfer onto racemic as well as meso alcohols, amines, and thiols. Angew. Chem. Int. Ed. 50, 6012–6042 (2011).
Kourist, R., de María, P. D. & Bornscheuer, U. T. Enzymatic synthesis of optically active tertiary alcohols: expanding the biocatalysis toolbox. ChemBioChem 9, 491–498 (2008).
Fernández-Mateos, E., Maciá, B. & Yus, M. Catalytic enantioselective addition of aryl Grignard reagents to ketones. Eur. J. Org. Chem. 2014, 6519–6526 (2014).
Zhu, T.-S., Jin, S.-S. & Xu, M.-H. Rhodium-catalyzed, highly enantioselective 1,2-addition of aryl boronic acids to α-ketoesters and α-diketones using simple, chiral sulfur-olefin ligands. Angew. Chem. Int. Ed. 51, 780–783 (2012).
Bartlett, S. L., Keiter, K. M. & Johnson, J. S. Synthesis of complex tertiary glycolates by enantioconvergent arylation of stereochemically labile α-keto esters. J. Am. Chem. Soc. 139, 3911–3916 (2017).
Huang, L. et al. Highly enantioselective rhodium-catalyzed addition of arylboroxines to simple aryl ketones: efficient synthesis of escitalopram. Angew. Chem. Int. Ed. 55, 4527–4531 (2016).
Ramón, D. J. & Yus, M. Chiral tertiary alcohols made by catalytic enantioselective addition of unreactive zinc reagents to poorly electrophilic ketones? Angew. Chem. Int. Ed. 43, 284–287 (2004).
Murray, S. A., Green, J. C., Tailor, S. B. & Meek, S. J. Enantio- and diastereoselective 1,2-additions to α-ketoesters with diborylmethane and substituted 1,1-diborylalkanes. Angew. Chem. Int. Ed. 55, 9065–9069 (2016).
Huang, Y., Huang, R.-Z. & Zhao, Y. Cobalt-catalyzed enantioselective vinylation of activated ketones and imines. J. Am. Chem. Soc. 138, 6571–6576 (2016).
Zheng, Y. et al. Octahedral ruthenium complex with exclusive metal-centered chirality for highly effective asymmetric catalysis. J. Am. Chem. Soc. 139, 4322–4325 (2017).
Yang, Y., Perry, I. B., Lu, G., Liu, P. & Buchwald, S. L. Copper-catalyzed asymmetric addition of olefin-derived nucleophiles to ketones. Science 353, 144–150 (2016).
Silverio, D. L. et al. Simple organic molecules as catalysts for enantioselective synthesis of amines and alcohols. Nature 494, 216–221 (2013).
Lee, K. et al. Catalytic enantioselective addition of organoboron reagents to fluoroketones controlled by electrostatic interactions. Nat. Chem. 8, 768–777 (2016).
van der Mei, F. W., Qin, C., Morrison, R. J. & Hoveyda, A. H. Practical, broadly applicable, α-selective, Z-selective, diastereoselective, and enantioselective addition of allylboron compounds to mono-, di-, tri-, and polyfluoroalkyl ketones. J. Am. Chem. Soc. 139, 9053–9065 (2017).
Robbins, D. W. et al. Practical and broadly applicable catalytic enantioselective additions of allyl-B(pin) compounds to ketones and α-Ketoesters. Angew. Chem. Int. Ed. 55, 9610–9614 (2016).
Tsai, E. Y., Liu, R. Y., Yang, Y. & Buchwald, S. L. A regio- and enantioselective CuH-catalyzed ketone allylation with terminal allenes. J. Am. Chem. Soc. 140, 2007–2011 (2018).
Li, C. et al. Copper-catalyzed enantioselective Friedel–Crafts alkylation of pyrrole with isatins. Org. Lett. 16, 3192–3195 (2014).
Liu, Y.-L. et al. Organocatalytic asymmetric synthesis of substituted 3-hydroxy-2-oxindoles via Morita−Baylis−Hillman reaction. J. Am. Chem. Soc. 132, 15176–15178 (2010).
Jarvo, E. R., Evans, C. A., Copeland, G. T. & Miller, S. J. Fluorescence-based screening of asymmetric acylation catalysts through parallel enantiomer analysis. Identification of a catalyst for tertiary alcohol resolution. J. Org. Chem. 66, 5522–5527 (2001).
Lu, S., Poh, S. B., Siau, W.-Y. & Zhao, Y. Kinetic resolution of tertiary alcohols: highly enantioselective access to 3-hydroxy-3-substituted oxindoles. Angew. Chem. Int. Ed. 52, 1731–1734 (2013).
Čorić, I., Müller, S. & List, B. Kinetic resolution of homoaldols via catalytic asymmetric transacetalization. J. Am. Chem. Soc. 132, 17370–17373 (2010).
Zhao, Y., Mitra, A. W., Hoveyda, A. H. & Snapper, M. L. Kinetic resolution of 1,2-diols through highly site- and enantioselective catalytic silylation. Angew. Chem. Int. Ed. 46, 8471–8474 (2007).
Tosaki, S. et al. Mixed La-Li heterobimetallic complexes for tertiary nitroaldol resolution. J. Am. Chem. Soc. 128, 11776–11777 (2006).
Kim, J. H., Čorić, I., Palumbo, C. & List, B. Resolution of diols via catalytic asymmetric acetalization. J. Am. Chem. Soc. 137, 1778–1781 (2015).
Greenhalgh, M. D. et al. A C=O⋯isothiouronium interaction dictates enantiodiscrimination in acylative kinetic resolutions of tertiary heterocyclic alcohols. Angew. Chem. Int. Ed. 57, 3200–3206 (2018).
Pan, Y. et al. Kinetic resolution of 2‐N‐acylamido tertiary allylic alcohols: asymmetric synthesis of oxazolines. Adv. Synth. Catal. https://doi.org/10.1002/adsc.202001051 (2020).
Olivares-Romero, J. L., Li, Z. & Yamamoto, H. Catalytic enantioselective epoxidation of tertiary allylic and homoallylic alcohols. J. Am. Chem. Soc. 135, 3411–3413 (2013).
Seliger, J., Dong, X. & Oestreich, M. Kinetic resolution of tertiary propargylic alcohols by enantioselective Cu-H catalyzed Si-O coupling. Angew. Chem. Int. Ed. 58, 1970–1974 (2019).
Rajkumar, S., He, S. & Yang, X. Kinetic resolution of tertiary 2-alkoxycarboxamido-substituted allylic alcohols by chiral phosphoric acid catalyzed intramolecular transesterification. Angew. Chem. Int. Ed. 58, 10315–10319 (2019).
Rajkumar, S., Tang, M. & Yang, X. Chiral phosphoric acid catalyzed kinetic resolution of 2-amido benzyl alcohols: asymmetric synthesis of 4H-3,1-benzoxazines. Angew. Chem. Int. Ed. 59, 2333–2337 (2020).
Qu, S., et al. Isothiourea-catalyzed acylative kinetic resolution of tertiary α-hydroxy esters. Angew. Chem. Int. Ed. https://doi.org/10.1002/anie.202004354 (2020).
Birman, V. B. & Li, X. Benzotetramisole: a remarkably enantioselective acyl transfer catalyst. Org. Lett. 8, 1351–1354 (2006).
Birman, V. B. & Li, X. Homobenzotetramisole: an effective catalyst for kinetic resolution of aryl-cycloalkanols. Org. Lett. 10, 1115–1118 (2008).
Belmessieri, D. et al. Structure-enantioselectivity effects in 3,4-dihydropyrimido[2,1-b]benzothiazole-based isothioureas as enantioselective acylation catalysts. Org. Biomol. Chem. 9, 559–570 (2011).
Birman, V. B. Amidine-based catalysts (ABCs) design, development, and applications. Aldrichimica Acta 49, 23–33 (2016).
Han, P., Wang, R. & Wang, D. Z. Electronic polarizability-based stereochemical model for Sharpless AD reactions. Tetrahedron 67, 8873–8878 (2011).
Duan, H.-F., Xie, J.-H., Qiao, X.-C., Wang, L.-X. & Zhou, Q.-L. Enantioselective rhodium-catalyzed addition of arylboronic acids to α-ketoesters. Angew. Chem. Int. Ed. 47, 4351–4353 (2008).
Cai, F. et al. Chiral allene-containing phosphines in asymmetric catalysis. J. Am. Chem. Soc. 133, 18066–18069 (2011).
Fager, D. C., Lee, K. & Hoveyda, A. H. Catalytic enantioselective addition of an allyl group to ketones containing a tri-, a di-, or a monohalomethyl moiety. Stereochemical control based on distinctive electronic and steric attributes of C-Cl, C-Br, and C-F bonds. J. Am. Chem. Soc. 141, 16125–16138 (2019).
Lee, K. et al. Catalytic enantioselective addition of organoboron reagents to fluoroketones controlled by electrostatic interactions. Nat. Chem. 8, 768–777 (2016).
Mszar, N. W., Mikus, M. S., Torker, S., Haeffner, F. & Hoveyda, A. H. Electronically sctivated organoboron vatalysts for enantioselective propargyl addition to trifluoromethyl ketones. Angew. Chem. Int. Ed. 56, 8736–8741 (2017).
Jung, B., Hong, M. S. & Kang, S. H. Enantioselective synthesis of tertiary alcohols by the desymmetrizing benzoylation of 2-substituted glycerols. Angew. Chem. Int. Ed. 46, 2616–2618 (2007).
Wang, C., Zhu, R.-Y., Liao, K., Zhou, F. & Zhou, J. Enantioselective Cu(I)-catalyzed cycloaddition of prochiral siazides with terminal or 1-iodoalkynes. Org. Lett. 22, 1270–1274 (2020).
Jung, B. & Kang, S. H. Chiral imine copper chloride-catalyzed enantioselective desymmetrization of 2-substituted 1,2,3-propanetriols. Proc. Natl Acad. Sci. USA. 104, 1471–1475 (2007).
You, Z., Hoveyda, A. H. & Snapper, M. L. Catalytic enantioselective silylation of acyclic and cyclic triols: application to total syntheses of cleroindicins D, F, and C. Angew. Chem. Int. Ed. 48, 547–550 (2009).
Xia, Q.-H. et al. Advances in homogeneous and geterogeneous catalytic asymmetric epoxidation. Chem. Rev. 105, 1603–1662 (2005).
Johnson, R. A. & Sharpless, K. B. Catalytic asymmetric epoxidation of allylic alcohols. In Catalytic Asymmetric Synthesis, (ed. Ojima, I.) pp. 231–280 (Wiley, ed. 2, 2000).
Johnson, R. A. & Sharpless, K. B. Addition reactions with formation of carbon–oxygen bonds: asymmetric methods of epoxidation. In Comprehensive Organic Synthesis, (eds Trost, B. M. & Fleming, I.) vol. 7, pp. 389–436 (Pergamon Press, ed. 1, 1991).
Jacobsen, E. N. Transition metal-catalyzed oxidations: asymmetric epoxidation. In Comprehensive Organometallic Chemistry II, (eds Abel, E. W., Stone, F. G. A. & Wilkinson, G.) vol. 12, pp. 1097–1135 (Pergamon Press, ed. 1,1995).
Zhu, Y., Wang, Q., Cornwall, R. G. & Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev. 114, 8199–8256 (2014).
Wong, O. A. & Shi, Y. Organocatalytic oxidation. Asymmetric epoxidation of olefins catalyzed by chiral ketones and iminium salts. Chem. Rev. 108, 3958–3987 (2008).
Katsuki, T. & Sharpless, K. B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 102, 5974–5976 (1980).
Martin, V. S. et al. Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity? J. Am. Chem. Soc. 103, 6237–6240 (1981).
Wang, X., Kitamura, M. & Maruoka, K. New, chiral phase transfer catalysts for effecting asymmetric conjugate additions of α-alkyl-α-cyanoacetates to acetylenic esters. J. Am. Chem. Soc. 129, 1038–1039 (2007).
Li, H., Song, J., Liu, X. & Deng, L. Catalytic enantioselective C−C bond forming conjugate additions with vinyl sulfones. J. Am. Chem. Soc. 127, 8948–8949 (2005).
Badiola, E. et al. Enantioselective construction of tetrasubstituted stereogenic carbons through Brønsted base catalyzed Michael reactions: α’-hydroxy enones as key enoate equivalent. J. Am. Chem. Soc. 136, 17869–17881 (2014).
Sawamura, M., Sudoh, M. & Ito, Y. An enantioselective two-component catalyst system: Rh-Pd-catalyzed allylic alkylation of activated nitriles. J. Am. Chem. Soc. 118, 3309–3310 (1996).
Zhang, L., Fu, N. & Luo, S. Pushing the limits of aminocatalysis: enantioselective transformations of α-branched β-ketocarbonyls and vinyl ketones by chiral primary amines. Acc. Chem. Res. 48, 986–997 (2015).
Liu, Y., Han, S.-J., Liu, W.-B. & Stoltz, B. M. Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc. Chem. Res. 48, 740–751 (2015).
Li, C., Ragab, S. S., Liu, G. & Tang, W. Enantioselective formation of quaternary carbon stereocenters in natural product synthesis: a recent update. Nat. Prod. Rep. 37, 276–292 (2020).
Long, R., Huang, J., Gong, J. & Yang, Z. Direct construction of vicinal all-carbon quaternary stereocenters in natural product synthesis. Nat. Prod. Rep. 32, 1584–1601 (2015).
Douglas, C. J. & Overman, L. E. Catalytic asymmetric synthesis of all-carbon quaternary stereocenters. Proc. Natl Acad. Sci. USA. 101, 5363–5367 (2004).
Trost, B. M. & Jiang, C. Catalytic enantioselective construction of all-carbon quaternary stereocenters. Synthesis 2006, 369–396 (2006).
Zhang, L., Fu, N. & Luo, S. Pushing the limits of aminocatalysis: enantioselective transformations of α-branched β-ketocarbonyls and vinyl ketones by chiral primary amines. Acc. Chem. Res. 48, 986–997 (2015).
Sonawane, R. P. et al. Enantioselective construction of quaternary stereogenic centers from tertiary boronic esters: methodology and applications. Angew. Chem. Int. Ed. 50, 3760–3763 (2011).
Acknowledgements
We thank V. B. Birman for the valuable discussion of the project. Financial support was provided by NSFC (21871260 and 21502192), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), Fujian Natural Science Foundation (2018J05035), and China Postdoctoral Science Foundation (2018M630734).
Author information
Authors and Affiliations
Contributions
X.F. and W.X. conceived the project. S.N. and H.Z. conducted most of the experiments. P.R.B. and G.Z. initiated the experimental section of the project. X.F. supervised the project and wrote the manuscript with the consultation of all co-authors. J.L. tested all single crystal and HRMS data. S.Y. checked all data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niu, S., Zhang, H., Xu, W. et al. Access to enantioenriched compounds bearing challenging tetrasubstituted stereocenters via kinetic resolution of auxiliary adjacent alcohols. Nat Commun 12, 3735 (2021). https://doi.org/10.1038/s41467-021-23990-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-23990-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
