
Abstract
Heterocycles 2-pyridone and uracil are privileged pharmacophores. Diversity-oriented synthesis of their derivatives is in urgent need in medicinal chemistry. Herein, we report a palladium/norbornene cooperative catalysis enabled dual-functionalization of iodinated 2-pyridones and uracils. The success of this research depends on the use of two unique norbornene derivatives as the mediator. Readily available alkyl halides/tosylates and aryl bromides are utilized as ortho-alkylating and -arylating reagents, respectively. Widely accessible ipso-terminating reagents, including H/DCO2Na, boronic acid/ester, terminal alkene and alkyne are compatible with this protocol. Thus, a large number of valuable 2-pyridone derivatives, including deuterium/CD3-labeled 2-pyridones, bicyclic 2-pyridones, 2-pyridone-fenofibrate conjugate, axially chiral 2-pyridone (97% ee), as well as uracil and thymine derivatives, can be quickly prepared in a predictable manner (79 examples reported), which will be very useful in new drug discovery.
Similar content being viewed by others
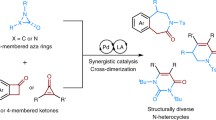
Introduction
2-Pyridone is an important class of electron-deficient heterocycle, widely found in bioactive natural products (Fig. 1A)1,2,3,4, eg. (−)-maximiscin, (+)-hosieine A, and (+)-lyconadin A. Recently, 2-pyridones have even been utilized as pivotal ligands in transition metal catalysis5,6. More importantly, 2-pyridone unit is recognized as a privileged pharmacophore prevalent in pharmaceutical agents (Fig. 1A)7,8,9,10,11,12, for instance, huperzine A (anti-Alzheimer)9, milrinone (anti-heart failure)10, SD-560 (anti-fibrosis)11, and camptothecin (antitumor)12. The latest example is tazemetostat (TazverikTM), the first EZH2 inhibitor approved by FDA in early 2020 for epithelioid sarcoma treatment13. In addition, several promising 2-pyridone-based EZH2 inhibitors are in clinical trials14,15. As revealed by crystallographic studies16,17, the 2-pyridone moiety acts as the common warhead of EZH2 inhibitors, thus it is crucial for their EZH2 enzyme inhibition activities (Fig. 1B). Following the success of tazemetostat, there is an urgent need from pharmaceutical industry for diversity-oriented synthesis (DOS)18,19 of 2-pyridone derivatives library, therefore to find new generation of EZH2 inhibitors via high-throughput biochemical screening (HTS)14,15.

A Structurally related natural products, ligands, and pharmaceuticals. B EZH2 inhibitors and their mode of action.
To date, significant progresses have been made for the preparation of 2-pyridone derivatives (Fig. 2A)20,21,22,23. Besides the classical strategies regarding intrinsic electrophilic substitutions24,25, pyridines hydrolysis26, and de novo construction of the 2-pyridone ring from acyclic precursors21,27,28, transition metal catalysis played an increasingly important role in developing step-economic methods20,21,22,23. Specifically, the catalytic site-selective C–H functionalization of 2-pyridones has become an emerging strategy23,29,30,31,32. However, these methods are commonly single-tasked, thus the obtained 2-pyridones are of limited diversity20,21,22,23. Additionally, some of them usually require specially functionalized substrates20,21,22,24,25,26,27,28,32 or harsh reaction conditions21,27, significantly limiting their scopes. Therefore, development of general and efficient approaches for rapid synthesis of 2-pyridone derivatives library from readily available starting materials is a highly desirable yet challenging subject14,15.

A Typical current strategies for 2-pyridones synthesis. B Diversity-oriented-synthesis of 2-pyridones via the Catellani strategy.
Palladium/norbornene (Pd/NBE) cooperative catalysis (namely, the Catellani reaction)33 is recognized as a powerful strategy for expeditious synthesis of highly substituted arenes34,35,36,37,38,39,40,41. Owing to diversified and orthogonal dual-functionalization (ortho/ipso) of aryl halides (mainly aryl iodides), the Catellani reaction has become a versatile tool for quickly building aromatics library39,40,41. However, current scope of this chemistry is mainly limited to aromatic substrates, and its application to partially aromatic scaffolds is rarely reported42,43,44,45, because the vinylic C–H bonds are generally more challenging to functionalize than aryl C–H bond46,47,48. In 2018, the Yamamoto group reported a unique two-component vinylogous Catellani annulation for the assembly of tricyclic benzo-fused products, involving partially aromatic 4-iodo-2-quinolones and 4-iodo-coumarin as substrates43. Later on, the Dong group successively realized two elegant alkenyl Catellani reactions, utilizing alkenyl (pseudo)halides44 and alkenes with a directing group45 as substrates, respectively. Inspired by their research, we envisaged to apply this challenging partially aromatic Catellani strategy for diversity-oriented synthesis of 2-pyridones. As shown in Fig. 2B, 4-iodo-2-pyridone (1), electrophilic alkyl/aryl halide (2), and terminating reagent (3) would engage in sequential ortho-C–H activation (intermediate I to II), functionalization (II–IV), and ipso-termination (intermediate IV to product 4). Such a sequence would enable controllable formation of two vicinal chemical bonds in a single operation (good step-economy). Hence, this Catellani-type dual-functionalization strategy would generate structurally very diversified 2-pyridones, thereby meeting the increasing needs from medicinal chemists14,15. Although this hypothesis is mechanistically feasible, the following challenges are foreseeable. First, although the iodinated 2-pyridones are easily accessible49, owing to their potential chelating ability5,6, they are challenging substrates and have never been used in Catellani reactions before34,35,36,37,38,39,40,41. Second, several potential side reactions, eg. cyclopropanation (via intermediates I and IV, Fig. 2B)43,44,47,48, direct coupling of 1 and 3 etc, will be competitive pathways. Third, a versatile NBE mediator needs to be identified, to promote the vinylic ortho-C–H activation and prevent the side reactions as well.
In this wok, we develop a palladium/norbornene cooperative catalysis-enabled diversity-oriented functionalization of heterocycles 2-pyridone and uracil. Readily available alkyl halides/tosylate and aryl bromides are utilized as ortho-alkylating and -arylating reagents, respectively. Widely accessible H/DCO2Na, boronic acid/ester, alkene and alkyne are employed as ipso-terminating reagents. A large number of useful derivatives of these heterocycles, including deuterium/CD3-labeled 2-pyridones, bicyclic 2-pyridones, 2-pyridone-fenofibrate conjugate, axially chiral 2-pyridone (97% ee), as well as uracil and thymine derivatives, can be quickly prepared in a predictable manner.
Results and discussion
Ortho-alkylation of 2-pyridones
Reaction design and optimization
Our efforts commenced with a model reaction, using 1-benzyl-4-iodo-5-methyl-2-pyridone (1a), ethyl 4-bromobutanoate (2a), and styrene (3a) as the reactants, to optimize the reaction conditions (see Supplementary Tables 1–5). Partial screening results were summarized in Table 1. The optimal catalyst, base, solvent, and temperature combination was Pd(OAc)2 (5 mol%), K2CO3 (2.5 equiv), dioxane, and 105 °C. Interestingly, phophine ligand was demonstrated dispensable for this reaction (see Supplementary Table 3)33,50,51,52,53,54. Nevertheless, a significant mediator effect was observed. With the promotion of simple N1 (1.0 equiv), the reaction furnished the desired product 4a in 27% yield (entry 1). Readily available norbornene derivatives55,56,57,58,59 N2−N6 showed similar reactivity as N1, and delivered 4a in 20−40% yields (entries 2–6). Gratifyingly, the Yu’s mediator (N7)56 significantly improved the reacton efficiency to deliver 4a in 60% yield (entry 7), while the Dong’s mediator (N8)44 further increased the yield to 80% (entry 8). Thus, N8 was identified as the optimal mediator (for additional mediator screening, see Supplementary Table 4). Further optimization indicated that the amount of N8 could be reduced to 50 mol% without deleterious effects (entry 9). Since 1a had multiple side reaction pathways, e.g., direct Heck reaction, deiodination, annulation with [NBE] etc, and 3a (1.5 equiv) was always in excess after the reaction, we therefore changed the molar ratio of reactants (entries 10−11). Good results were obtained with the ratio of 1a: 2a: 3a = 1.2:1.5:1.0, and yield of 4a was increased to 92% (entry 11). Finally, the optimal conditions involved increasing the reaction time to 36 h, which furnished 4a in 98% yield (95% isolated yield; entry 12).
Substrate scope
With the optimal reaction conditions confirmed, we first examined the scope of 2-pyridone, with bromide 2a and styrene 3a as the reaction partners. As shown in Table 2A, a series of 4-iodo-2-pyridones with substitution at C3 or C5 position, including methyl, fluoro, chloro, methyl ether, and ester group, reacted smoothly to provide the alkylated products (4a−4j) in 60–95% yields. The N-substitution of 2-pyridones could be benzyl (4a), methyl (4b), 2,4,6-trimethylbenzyl (Mesityl, 4c), methoxymethyl (MOM, 4d), and p-methoxybenzyl (PMB, 4g). Surprisingly, even the one with free N–H delivered the corresponding product 4e in 69% yield with simultaneous nucleophilic N-alkylation. For ortho-unsubstituted 4-iodo-2-pyridone, the dialkylated product 4l was obtained in 39% yield. Notably, after minor modification of the standard conditions, biologically relevant 6-iodo uracil also became a suitable substrate to afford the desired product (4m) in moderate yield. The practicality and robustness of this protocol are evident from the 3.0 mmol scale experiment, which led to a gram-scale preparation of product 4a (1.22 g, 98% yield), alongside the recovery of mediator N8 in 87% yield.
Then, the scope of alkylating reagent (2) was examined (Table 2B). Common methylation reagents including trimethylphosphate (2b)60, iodomethane (2c)61,62, methyl tosylate (2d)60, and its deuterated sibling (2e)60 were good reactants to deliver the (deuterated)methylation products (4n−o) in good to excellent yields (77−92%). Other simple alkylating reagents, eg. ethyl bromide (2g), butyl bromide (2h), and benzyl chloride (2i) gave the desired products (4p−r) in excellent yields (91−93%). Bromides containing an array of functional groups including methoxyl (4t), cyano (4v), chloro (4w), bromo (4x), amide (4y), and protected amino (4z) gave moderate to excellent yields (45−96%). Notably, free hydroxyl group (4u) was also compatible with this protocol.
Next, the scope of terminating reagent 3 was also explored. As shown in Table 3A, a wide range of olefins with electron-poor, -rich, or -neutral property were suitable substrates, including acrylates (4a′ and 4e′), acrylamide (4b′), vinyl phosphonic ester (4c′), phenyl vinyl sulfone (4d′), vinylsilane (4f′), olefinic alcohol (4g′), and styrenes (4h′−j′), providing the desired 2-pyridones in excellent yields (80−92%). Notabley, the reaction of trimethyl(vinyl)silane proceeded to afford the desilylation product (4f′) in 82% yield. The reactions with a complex styrene derived from antihyperlipidemic drug medicine fenofibrate proceeded uneventfully to afford 4j′ in 90% yield. Besides olefins, other types of terminating reagents were also applicable under the standard reaction conditions (Table 3B), including H/DCO2Na (4k′ and 4l′)62,63, MeB(OH)2 (4m′)61, PhBpin (4n′), and terminal alkyne (4o′)64, and the corresponding 2-pyridone products were obtained in 52−80% yields.
Ortho-arylation of 2-pyridones
Encouraged by the success of ortho-alkylation of 2-pyridones, we proceeded to explore the ortho-arylation. Gratifyingly, after minor modification of previous standard conditions, including the use of mediator N9 59 and a solvent change to DME (see Supplementary Tables 6-7), we were able to achieve this goal. Notably, the loading of Pd(OAc)2 could be impressively lowered to 1 mol%. As shown in Table 4A, an array of ortho-substituted 4-iodo-2-pyridones reacted smoothly with methyl 2-bromobenzoate (2A) and 3a to provide the arylated products (4A−H) in moderate to good yields (34−94%). Interestingly, the well-known ortho-effect of Catellani-type arylation34 was not observed for ortho-unsubstituted 4-iodo-2-pyridone, since the bisarylated product 4I was obtained in 69% yield. Then, the scope of arylation reagent was explored (Table 4B). Generally, aryl bromides with one ortho-electron-withdrawing group were good reagents, including ester (2A−B), amide (2D−E), nitro (2K) groups, even the reactive carboxylic acid (2C) and acetyl (2F) groups. Polysubstituted aryl bromides (2G−K) were also competent arylating reagents to deliver the desired 2-pyridones in 35–89% yields. As to the scope of terminating reagent, it was similar to the alkylation protocol (see Supplementary Fig. 5). Inspired by recent studies on Pd/chiral norbornene asymmetric catalysis65,66,67, we tested the synthesis of axially chiral 2-pyridone using enantiopure (+)-N9 (99% ee)66,67 as the mediator and sterically demanding 2,6-disubstituted aryl bromide 2L as the arylating reagent. Pleasingly, the desired product 4A* was obtained in 73% yield and excellent enantiomeric excess (97% ee), after minor modification of the reaction conditions (Table 4C and see Supplementary Table 8 for details).
Two-component annulation, N-deprotection, and follow-up transformations of the obtained 2-pyridones
Next, we focused on illustrating the synthetic utility of these protocols. First, an annulation process was explored based on a speculated two-component Catellani process (Fig. 3A). It was found that 1a reacted with the bifunctional reagent 5 and 6 bearing a bromide and an olefin moiety, to afford the cyclized products 7 and 8 in excellent yields. Thus, it provided an efficient method for the assembly of bicyclic 2-pyridone derivatives68,69,70. Then, N-deprotection of the obtained various 2-pyridones were performed to set the stage for further manipulations (Fig. 3B, C). For example, N-Bn deprotection of 4l′ proceeded smoothly under the catalytic hydrogenation conditions to deliver 9 in 77% yield71. The N-PMB group of 4g was readily removed in heated CF3CO2H to afford 1072, which could be quickly transformed into a OTf-substituted pyridine derivative (11) after treatment with Tf2O and pyridine73. In addition, BBr3 mediated N-MOM deprotection of 4d took place at a low temperature74, and the obtained intermediate 12 readily led to pyridine-fused lactone 13 through a facile two-step procedure75. Interestingly, BBr3 mediated N-MOM deprotection of the ortho C–H arylation product 14 provided two products: the normal one 15 in 27% yield and the methyl benzoate hydrolyzed product 16 in 42% yield, which were further transformed into complex pyridine derivatives 17 and 18 respectively in just one step with good yields.

A Two-component annulation. B N-deprotection and follow-up transformations of the obtained 2-pyridones 4l′, 4g, and 4d. C N-deprotection and follow-up transformations of the obtained 2-pyridone 14.
Diversity-oriented functionalization of uracils
Additionally, the successful functionalization of iodinated uracil (4m, Table 2A) and biology-oriented synthesis (BIOS)76 philosophy prompted us to further uncover the inherent value of our method by synthesizing biorelevant uracil and thymine derivatives (Fig. 4). Delightfully, with readily available iodinated uracils 1m and 1n as substrates, 2d and 2A as the electrophilic reagents, the 6-deuterated thymine derivative (80% deuteration) (19), and other five novel thymine analogs and derivatives (20−24) were facilely prepared in moderate yields, using ipso-hydrogenation (19), -Suzuki (20), -Heck (21−22), or -Sonogashira termination (23−24) respectively. Furthermore, the reaction of 1m with benzyl choride and methylboronic acid afforded 25 in 50% yield, resulting in a one-step formal synthesis of anti-HIV-1 agent 26 (previous method required 6 steps with only 6% overall yield)77. These results revealed the modularity of our method in diversity-oriented synthesis of useful uracil and thymine derivatives, which will be very attractive for developing new antiviral agents (Fig. 4B)78,79,80.

A Preparation of thymine derivatives and analogs. B Selected antiviral agents derived from uracil with C-5 and C-6 substitutions.
In summary, we have developed a palladium/norbornene cooperative catalysis enabled diversity-oriented functionalization of heterocycles 2-pyridone and uracil. The success of this research depends on the use of two unique norbornene derivatives as the mediator. Readily available alkyl halides/tosylate and aryl bromides are utilized as ortho-alkylating and -arylating reagents, respectively. Widely accessible ipso-terminating reagents, including H/DCO2Na, boronic acid/ester, terminal alkene and alkyne are compatible with this protocol. Thus, a large number of useful derivatives of these heterocycles, including deuterium/CD3-labeled 2-pyridones, bicyclic 2-pyridones, 2-pyridone-fenofibrate conjugate, axially chiral 2-pyridone (97% ee), as well as uracil and thymine derivatives, can be quickly prepared in a predictable manner, which will be very attractive for developing new generation of EZH2 inhibitors and antiviral agents. This work constitutes not only a nice extention of the Catellani reaction, but also a valuable addition to the toolbox of medicinal chemists
Methods
General procedure for Ortho-alkylating of 2-pyridone
To an oven-dried Schlenk tube equipped with a magnetic stir bar were added Pd(OAc)2 (5 mol%), norbornene derivatives N8 (0.05 mmol, 50 mol%), alkenyl iodide 1 (0.12 mmol, 1.2 equiv) and potassium carbonate (0.25 mmol, 2.5 equiv), and anhydrous 1,4-dioxane (1 mL) in the glove box. Then alkylating reagent 2 (0.15 mmol, 1.5 equiv) and terminating reagent 3 (0.1 mmol, 1.0 equiv) were added, and the mixture was heated to 105 °C. After completion of the reaction (monitored by TLC (thin layer chromatography), the mixture was cooled to r.t., filtered through a thin pad of celite, eluting with EtOAc (10 mL), and the combined filtrate was concentrated in vacuo. The residue was directly purified by column chromatography on silica gel or purified by PTLC (preparative thin layer chromatography) to give the desired product.
General procedure for Ortho-arylation of 2-pyridone
To an oven-dried Schlenk tube equipped with a magnetic stir bar were added Pd(OAc)2 (1 mol%), norbornene derivatives N9 (0.05 mmol, 50 mol%), alkenyl iodide 1 (0.1 mmol, 1.0 equiv) and potassium carbonate (0.25 mmol, 2.5 equiv), and anhydrous DME (1 mL) in the glove box. Then alkylating reagent 2 (0.15 mmol, 1.5 equiv) and terminating reagent 3 (0.15 mmol, 1.5 equiv) were added, and the mixture was heated to 105 °C. After completion of the reaction (monitored by TLC (thin layer chromatography), the mixture was cooled to r.t., filtered through a thin pad of celite, eluting with EtOAc (10 mL), and the combined filtrate was concentrated in vacuo. The residue was directly purified by column chromatography on silica gel or purified by PTLC (preparative thin layer chromatography) to give the desired product.
Data availability
The authors declare that all relevant data supporting the findings of this study are available within the paper and its supplementary information files.
References
Jessen, H. J. & Gademann, K. 4-Hydroxy-2-pyridone alkaloids: structures and synthetic approaches. Nat. Prod. Rep. 27, 1168–1185 (2010).
Du, L. et al. Crowdsourcing natural products discovery to access uncharted dimensions of fungal metabolite diversity. Angew. Chem. Int. Ed. 53, 804–809 (2014).
Pouny, I. et al. Cytisine-like akaloids from Ormosia Hosiei Hemsl. & E. H. Wilson. Phytochemistry 107, 97–101 (2014).
Kobayashi, J., Hirasawa, Y., Yoshida, N. & Morita, H. Lyconadin A, a novel alkaloid from Lycopodium Complanatum. J. Org. Chem. 66, 5901–5904 (2001).
Wang, P. et al. Ligand-accelerated non-directed C–H functionalization of arenes. Nature 551, 489–493 (2017).
Zhu, R. Y., Li, Z. Q., Park, H. S. & Yu, J.-Q. Ligand-enabled γ-C(sp3)–H activation of ketones. J. Am. Chem. Soc. 140, 3564–3568 (2018).
Li, Q., Mitscher, L. A. & Shen, L. L. The 2-pyridone antibacterial agents: Bacterial Topoisomerase inhibitors. Med. Res. Rev. 20, 231–293 (2000).
Medina-Franco, J. L., Martínez-Mayorga, K., Juárez-Gordiano, C. & Castillo, R. Pyridin-2(1H)-ones: a promising class of HIV-1 non-nucleoside reverse transcriptase inhibitors. ChemMedChem 2, 1141–1147 (2007).
Xu, Z. Q. et al. Treatment with Huperzine aimproves cognition in vascular dementia patients. Cell Biochem. Biophys. 62, 55–58 (2012).
Yano, M. et al. Effect of milrinone on left ventricular relaxation and Ca2+ uptake function of cardiac sarcoplasmic reticulum. Am. J. Physiol. Heart Circ. Physiol. 279, 1898–1905 (2000).
Liu, J. F. & Dong, Y. Patent: WO 2009/035598 A1.
Wall, M. E. & Wani, M. C. Camptothecin and taxol: from discovery to clinic. J. Ethnopharmacol. 51, 239–254 (1996).
U.S. Food and Drug Administration. Novel Drug Approvals for 2020. https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020 (2020).
Stazi, G., Zwergel, C., Mai, A. & Valente, S. EZH2 inhibitors: a patent review (2014–2016). Expert Opin. Ther. Pat. 7, 797–813 (2017).
Fioravanti, R., Stazi, G., Zwergel, C., Valente, S. & Mai, A. Six years (2012–2018) of researches on catalytic EZH2 inhibitors: the boom of the 2-Pyridone compounds. Chem. Rec. 12, 1818–1832 (2018).
Brooun, A. et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun. 7, 11384–11396 (2016).
Vaswani, R. G. et al. Identification of (R)‑N‑((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)‑1H‑indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B‑Cell lymphomas. J. Med. Chem. 59, 9928–9941 (2016).
Burke, M. D. & Schreiber, S. L. A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed. 43, 46–58 (2004).
Galloway, W. R. J. D., Isidro-Llobet, A. & Spring, D. R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 1, 80–93 (2010).
Varela, J. A. & Saa, C. Construction of pyridine rings by metal–mediated [2 + 2 + 2] cycloaddition. Chem. Rev. 103, 3787–3802 (2003).
Torres, M., Gil, S. & Parra, M. New synthetic methods to 2-pyridone rings. Curr. Org. Chem. 9, 1757–1779 (2005).
Dudnik, A. V., Gulevich, A. S., Chernyak, N. & Gevorgyan, V. Transition metal-mediated synthesis of monocyclic aromatic heterocycles. Chem. Rev. 113, 3084–3213 (2013).
Hirano, K. & Miura, M. A lesson for site-selective C–H functionalization on 2-Pyridones: radical, organometallic, directing group and steric controls. Chem. Sci. 9, 22–32 (2018).
Cline, R. E., Fink, R. M. & Fink, K. Synthesis of 5-Substituted pyrimidines via formaldehyde addition. J. Am. Chem. Soc. 81, 2521–2527 (1959).
Paquette, L. A. & Farley, W. C. Chlorination of conjugated dienamides: new application of the principle of least motion. J. Org. Chem. 32, 2725–2731 (1967).
Schrcder, P. et al. Neuritogenic militarinone-inspired 4-hydroxypyridones target the stress pathway kinase MAP4K4. Angew. Chem. Int. Ed. 54, 12398–12403 (2015).
Jessen, H. J., Schumacher, A., Shaw, T., Pfaltz, A. & Gademann, K. A unified approach for the stereoselective total synthesis of pyridone alkaloids and their neuritogenic activity. Angew. Chem. Int. Ed. 50, 4222–4226 (2011).
Fujii, M., Nishimura, T., Koshiba, T., Yokoshima, S. & Fukuyama, T. 2-pyridone synthesis using 2-(phenylsulfinyl)acetamide. Org. Lett. 15, 232–234 (2013).
Nakao, Y., Idei, H., Kanyiva, K. S. & Hiyama, T. Direct alkenylation and alkylation of pyridone derivatives by Ni/AlMe3 catalysis. J. Am. Chem. Soc. 131, 15996–15997 (2009).
Chen, Y., Wang, F., Jia, A. & Li, X. Palladium-catalyzed selective oxidative olefination and arylation of 2-pyridones. Chem. Sci. 3, 3231–3236 (2012).
Nakatani, A., Hirano, K., Satoh, T. & Miura, M. Nickel-catalyzed direct alkylation of heterocycles with α-bromo carbonyl compounds: C3-selective functionalization of 2-pyridones. Chem. Eur. J. 19, 7691–7695 (2013).
Odani, R., Hirano, K., Satoh, T. & Miura, M. Copper-mediated C6-selective dehydrogenative heteroarylation of 2-pyridones with 1,3-azoles. Angew. Chem. Int. Ed. 53, 10784–10788 (2014).
Catellani, M., Frignani, F. & Rangoni, A. A complex catalytic cycle leading to a regioselective synthesis of o,o’-disubstituted vinylarenes. Angew. Chem. Int. Ed. 36, 119–122 (1997).
Catellani, M., Motti, E. & Della Ca’, N. Catalytic sequential reactions involving palladacycle-directed aryl coupling steps. Acc. Chem. Res. 41, 1512–1522 (2008).
Ye, J. & Lautens, M. Palladium-catalysed norbornene-mediated C–H functionalization of arenes. Nat. Chem. 7, 863–870 (2015).
Zhu, H., Ye, C. & Chen, Z. Recent advances in the norbornene mediated palladium-catalyzed domino-type catellani reaction. Chin. J. Org. Chem. 35, 2291–2300 (2015).
Della, Ca’,N., Fontana, M., Motti, E. & Catellani, M. Pd/Norbornene: a winning combination for selective aromatic functionalization via C–H bond activation. Acc. Chem. Res. 49, 1389–1400 (2016).
Nairoukh, Z., Cormier, M. & Marek, I. Merging C–H and C–C bond cleavage in organic synthesis. Nat. Rev. Chem. 1, 35–51 (2017).
Liu, Z.-S., Gao, Q., Cheng, H.-G. & Zhou, Q. Alkylating reagents employed in Catellani-type reactions. Chem. Eur. J. 24, 15461–15476 (2018).
Cheng, H.-G., Chen, S., Chen, R. & Zhou, Q. Palladium(II)-initiated catellani-type reactions. Angew. Chem. Int. Ed. 58, 5832–5844 (2019).
Wang, J. & Dong, G. Palladium/norbornene cooperative catalysis. Chem. Rev. 119, 7478–7528 (2019).
Blaszykowski, C., Aktoudianakis, E., Bressy, C. D. & Lautens, M. Preparation of annulated Nitrogen-containing heterocycles via a one-pot palladium-catalyzed alkylation/direct arylation sequence. Org. Lett. 8, 2043–2045 (2006).
Yamamoto, Y., Murayama, T., Jiang, J., Yasui, T. & Shibuya, M. The vinylogous Catellani reaction: a combined computational and experimental study. Chem. Sci. 9, 1191–1199 (2018).
Wang, J., Dong, Z., Yang, C. & Dong, G. Modular and regioselective synthesis of all-carbon tetrasubstituted olefins enabled by an alkenyl Catellani reaction. Nat. Chem. 11, 1106–1112 (2019).
Wu, Z., Fatuzzo, N. & Dong, G. Distal alkenyl C–H functionalization via the palladium/norbornene cooperative catalysis. J. Am. Chem. Soc. 142, 2715–2720 (2020).
Wang, K., Hu, F., Zhang, Y. & Wang, J. Directing group-assisted transition-metal-catalyzed vinylic C–H bond functionalization. Sci. China Chem. 58, 1252–1265 (2015).
Catellani, M. & Chiusoli, G. P. Competitive processes in palladium-catalyzed C–C bond formation. J. Organomet. Chem. 233, C21–C24 (1982).
Khanna, A., Premachandra, I. D. U. A., Sung, P. D. & Van Vranken, D. L. Palladium-catalyzed Catellani aminocyclopropanation reactions with vinyl halides. Org. Lett. 15, 3158–3161 (2013).
Montgomery, J. I. et al. Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections. J. Med. Chem. 55, 1662–1670 (2012).
Catellani, M., Motti, E. & Minari, M. Symmetrical and unsymmetrical 2,6-dialkyl-1,1’-Biaryls by combined catalysis of aromatic alkylation via palladacycles and Suzuki-type coupling. Chem. Commun. 2, 157–158 (2000).
Catellani, M., Motti, E. & Baratta, S. A novel palladium-catalyzed synthesis of phenanthrenes from ortho-substituted aryl iodides and diphenyl- or alkylphenylacetylenes. Org. Lett. 3, 3611–3614 (2001).
Motti, E., Faccini, F., Ferrari, I., Catellani, M. & Ferraccioli, R. Sequential unsymmetrical aryl coupling of o-substituted aryl iodides with o-bromophenols and reaction with olefins: palladium-catalyzed synthesis of 6H-dibenzopyran derivatives. Org. Lett. 8, 3967–3970 (2006).
Della Ca’, N., Sassi, G. & Catellani, M. A Direct palladium-catalyzed route to selectively substituted carbazoles through sequential C–C and C–N bond formation: synthesis of carbazomycin A. Adv. Synth. Catal. 350, 2179–2182 (2008).
Motti, E. et al. A sequential Pd/norbornene-catalyzed process generates o-biaryl carbaldehydes or ketones via a redox reaction or 6H-dibenzopyrans by C–O ring closure. Org. Lett. 14, 5792–5795 (2012).
Chen, S. et al. The discovery of a Palladium(II)-initiated borono-Catellani reaction. Angew. Chem. Int. Ed. 57, 7161–7165 (2018).
Shen, P.-X., Wang, X.-C., Wang, P., Zhu, R.-Y. & Yu, J.-Q. Ligand-enabled meta-C–H alkylation and arylation using a modified norbornene. J. Am. Chem. Soc. 137, 11574–11577 (2015).
Qian, G. et al. Modular one-step three-component synthesis of tetrahydroisoquinolines using a Catellani strategy. Angew. Chem. Int. Ed. 57, 10980–10984 (2018).
Wang, J., Li, R., Dong, Z., Liu, P. & Dong, G. Complementary site-selectivity in arene functionalization enabled by overcoming the ortho constraint in Palladium/norbornene Catalysis. Nat. Chem. 10, 866–872 (2018).
Li, R. & Dong, G. Direct annulation between aryl iodides and epoxides through Palladium/norbornene cooperative catalysis. Angew. Chem. Int. Ed. 57, 1697–1701 (2018).
Gao, Q. et al. Modular dual-tasked C–H methylation via the Catellani strategy. J. Am. Chem. Soc. 141, 15986–15993 (2019).
Wilson, J. E. Palladium-catalyzed Catellani-type couplings using methylating reagents for the synthesis of highly substituted ortho-methyl-arenes and heteroarenes. Tetrahedron Lett. 57, 5053–5056 (2016).
Dong, Z., Lu, G., Wang, J., Liu, P. & Dong, G. Modular ipso/ortho difunctionalization of aryl bromides via Palladium/norbornene cooperative catalysis. J. Am. Chem. Soc. 140, 8551–8562 (2018).
Guo, L. et al. Cascade alkylation and deuteration with aryl iodides via Pd/Norbornene catalysis: an efficient method for the synthesis of congested deuterium-labeled arenes. Chem. Commun. 55, 8567–8570 (2019).
Lei, C., Jin, X. & Zhou, J. Palladium-catalyzed alkynylation and concomitant ortho alkylation of aryl iodides. ACS Catal. 6, 1635–1639 (2016).
Shi, H., Herron, A. N., Shao, Y., Shao, Q. & Yu, J.-Q. Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 558, 581–585 (2018).
Li, R., Liu, F. & Dong, G. Palladium-catalyzed asymmetric annulation between aryl iodides and racemic epoxides using a chiral norbornene co-catalyst. Org. Chem. Front. 5, 3108–3112 (2018).
Liu, Z.-S. et al. Construction of axial chirality via Palladium/chiral norbornene cooperative catalysis. Nat. Catal. 3, 727–733 (2020).
Lautens, M. & Piguel, S. A new route to fused aromatic compounds by using a Palladium-catalyzed alkylation – alkenylation sequence. Angew. Chem. Int. Ed. 39, 1045–1046 (2000).
Liu, Z.-S. et al. Palladium/norbornene cooperative catalysis to access tetrahydronaphthalenes and indanes with a quaternary center. ACS Catal. 8, 4783–4788 (2018).
Liu, Z.-S. et al. 5-norbornene-2-carboxylic acid: another catalytic mediator for catellani-type reactions. Tetrahedron 75, 1774–1780 (2019).
Chavan, S. P., Pathak, A. B. & Kalkote, U. R. A Practical formal synthesis of camptothecin. Tetrahedron Lett. 48, 6561–6563 (2007).
Lipshutz, B. H. & Amorelli, B. Total Synthesis of piericidin A1. Application of a modified negishi carboalumination-nickel-catalyzed cross-coupling. J. Am. Chem. Soc. 131, 1396–1397 (2009).
Xu, X.-H., Wang, X., Liu, G.-K., Tokunaga, E. & Shibata, N. Regioselective synthesis of heteroaryl triflones by LDA (lithium diisopropylamide)-mediated anionic thia-fries rearrangement. Org. Lett. 14, 2544–2547 (2012).
Chen, J. et al. Synthesis of isoquinolinone-based tricycles as novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. Bioorg. Med. Chem. Lett. 24, 2669–2673 (2014).
Yamada, S. & Abe, M. Selective deprotection and Amidation of 2-pyridyl esters via N-methylation. Tetrahedron 66, 8667–8671 (2010).
Wetzel, S., Bon, R. S., Kumar, K. & Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. 50, 10800–10826 (2011).
Chen, Y., Guo, Y., Yang, H., Wang, X. & Liu, J. Synthesis of i-(alkoxymethyl)-5-benzyl-6-methyluracil as potential non-nucleoside HIV-1 RT inhibitors. Synth. Commun. 36, 2913–2920 (2006).
Clercq, E. D. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2, 704–720 (2004).
El-Brollosy, N. R. et al. Synthesis of novel N-1 (allyloxymethyl) analogues of 6-benzyl-1-(ethoxymethyl)-5-isopropyluracil (MKC-442, Emivirine) with improved activity against HIV-1 and its mutants. J. Med. Chem. 45, 5721–5726 (2002).
Čerňová, M., Čerňa, I., Pohl, R. & Hocek, M. Regioselective direct C–H arylations of protected uracils: synthesis of 5- and 6-aryluracil bases. J. Org. Chem. 76, 5309–5319 (2011).
Acknowledgements
We are grateful to the National Key Research and Development Program of China (No. 2016YFB0101203) and start-up funding from Wuhan University for financial support. We thank Profs. Wen-Bo Liu, Suming Chen, and YongJia Tong (Wuhan University) for sharing their instruments, Dr. Ze-Shui Liu, Yu Hua, and Lan Zhou for affording some of the substrates, Jinxiang Ye for repeating some of the experiments, Tao Yang for assistance with the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Q.Z. conceived the idea, guided the project, and wrote the manuscript. Y.S., C.W., Q.G., C.L., L.L, X.Z., and S.L. performed the experiments and analyzed the data. Y.S., C.W., and H.-G.C. participated in the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Q.Z., Y.S., and C.W. have filed two provisional patient applications (202010807097.2, 202010807122.7). All other authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Etienne Derat and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shang, Y., Wu, C., Gao, Q. et al. Diversity-oriented functionalization of 2-pyridones and uracils. Nat Commun 12, 2988 (2021). https://doi.org/10.1038/s41467-021-23058-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-23058-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
