
Abstract
Vernalization genes underlying dramatic differences in flowering time between spring wheat and winter wheat have been studied extensively, but little is known about genes that regulate subtler differences in flowering time among winter wheat cultivars, which account for approximately 75% of wheat grown worldwide. Here, we identify a gene encoding an O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) that differentiates heading date between winter wheat cultivars Duster and Billings. We clone this TaOGT1 gene from a quantitative trait locus (QTL) for heading date in a mapping population derived from these two bread wheat cultivars and analyzed in various environments. Transgenic complementation analysis shows that constitutive overexpression of TaOGT1b from Billings accelerates the heading of transgenic Duster plants. TaOGT1 is able to transfer an O-GlcNAc group to wheat protein TaGRP2. Our findings establish important roles for TaOGT1 in winter wheat in adaptation to global warming in the future climate scenarios.
Similar content being viewed by others


Introduction
Wheat (Triticum aestivum, 2n = 6× = 42, AABBDD) is the most widely grown crop worldwide and has become a staple crop of agriculture and human nutrition globally1. Flowering time or heading date is a critical trait of wheat that underlies its adaptation to diverse climatic environments and cropping seasons. Wheat is classified into two distinct types: winter wheat, which requires exposure of the seedlings to low temperatures to accelerate flowering (vernalization), and spring wheat, which lacks this vernalization requirement2,3. Winter wheat cultivars account for ~75% of the wheat grown worldwide and will become more important as demand for food increases in future as a result of the accelerating population growth.
The four wheat vernalization genes VRN1, VRN2, VRN3, and VRN4 have been cloned using a positional cloning approach. VRN1 is a promoter of flowering that is upregulated by vernalization4 and encodes a MADS-box transcription factor with high similarity to the Arabidopsis thaliana (Arabidopsis) meristem identity gene APETALA1 (AP1)5. The VRN2 gene is a dominant repressor of flowering that is downregulated by vernalization6 and encodes a putative zinc finger and a CCT domain (ZCCT) containing protein, which has no ortholog in Arabidopsis7. VRN3 also promotes flowering and is upregulated by vernalization8 and encodes a RAF kinase inhibitor similar to FLOWERING LOCUS T (FT) in Arabidopsis9,10. VRN4 is similar to VRN1 in sequence and function but exists in an ancient subspecies T. aestivum ssp. sphaerococcum from South Asia11. Each of these VRN genes was cloned on the basis of qualitative variation in the vernalization requirement between spring wheat and winter wheat. Winter wheat cultivars are also classified into three types according to the quantitative variation in the duration of low temperature required to reach a vernalization saturation point: weak winter types, semi-winter types, and strong winter types12. The positional cloning of a major quantitative trait locus (QTL) that regulates the duration of the low-temperature requirement between semi-winter and strong winter wheat cultivars revealed that the trait is controlled by recessive vrn1 alleles encoding two different vrn1 proteins13.
Wheat cultivars are also classified into two groups based on their photoperiod sensitivity: the photoperiod-insensitive type with early flowering and the photoperiod-sensitive type with late flowering. The photoperiod gene PPD-H1 in barley (Hordeum vulgare L.) was cloned using positional cloning approach14, and its sequence provides intuitive information on orthologous genes on three homoeologous chromosomes in common wheat (PPD-A1, PPD-B1, and PPD-D1). Molecular markers developed for these wheat PPD genes have accelerated their extensive use in breeding15,16,17,18.
All of these cloned and characterized vernalization and photoperiod genes confer dramatic phenotypic variation. The difference in heading date can be as large as a few months between spring and winter wheat4,6, a couple of months among photoperiod-sensitive and insensitive cultivars14, and up to 45 days among semi-winter and strong winter cultivars tested under constant temperatures13. The substantial effects caused by these genes have facilitated their cloning but limited their use in a given wheat growth area because spring wheat is not well-adapted to winter wheat areas and vice versa. Allelic variation in recessive vrn1, recessive vrn3, and PPD-sensitive alleles is exploited to regulate the developmental phases of winter wheat cultivars, and these tri-locus genetic combinations have allowed coarse tuning of the phenotypic variation among contemporary winter wheat cultivars in the Great Plains13,17. However, some winter wheat cultivars possess minor differences in heading date that cannot be explained by allelic variation in these known vernalization and photoperiod alleles/genes or that can be explained only by cloning additional genes.
Recent studies have shown that mean global surface air temperature has risen and will continue to increase in the future19,20. Winter wheat cultivars will become increasingly vulnerable to the resulting higher temperatures and climate warming, because a decrease in the duration of low temperatures may result in no or incomplete vernalization21,22,23,24,25,26,27,28. Delays in flowering time caused by changes in the climate might be minor, but minor differences can be critical for sustainable food production. To date, little is known about genes that mediate those minor differences in flowering time among winter wheat cultivars.
In this work, we use a map-based cloning approach to clone a gene encoding an O-GlcNAc transferase that can be used to finely regulate flowering time in winter wheat cultivars.
Results
A QTL for heading date was identified and cloned from a winter wheat population
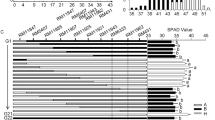
The two winter wheat cultivars Duster and Billings were identified to possess the same allele for each of three known developmental genes: vrn-A1b, PPD-D1b, and vrn-D3b that were genotyped according to their allelic variation13,17 (Supplementary Fig. 1) (Supplementary Method 1). However, Billings showed early development and Duster showed delayed development under greenhouse conditions without vernalization (Fig. 1a) and with vernalization (Supplementary Fig. 2a). Therefore, we sought genes responsible for the difference between the two winter wheat cultivars by phenotyping the Duster × Billings population of 260 doubled-haploid (DH) lines (Fig. 1b). This DH population was phenotyped under various environments and was genotyped using genotype-by-sequencing (GBS) markers29. We integrated the phenotypic data with those from the GBS markers and identified a QTL for heading date that mapped to the short arm of chromosome 6A. This QTL for heading date was thus referred to as QHd.osu-6A (Fig. 1c). The QHd.osu-6A locus explained 14.3% (LOD = 8.7), 11.7% (LOD = 7), and 10.2% (LOD = 6.1) of the total phenotypic variation in the population tested in the greenhouse (Fig. 1c). This locus also explained the subtler difference in heading date between the two alleles when the same population was tested in the field with natural vernalization (Supplementary Fig. 3). The result was confirmed in five critical DH lines without (Supplementary Fig. 2b) and with vernalization (Supplementary Fig. 2c). The co-localization of QHd.osu-6A for heading date from various environments revealed its consistent genetic effect in winter wheat.

a The two parental lines, Duster (left) and Billings (right), grown in a greenhouse with controlled temperatures and long days and without vernalization. b Representative DH plants segregating for heading date in the Duster × Billings DH population in the greenhouse and without vernalization. a, b Scale bar = 15 cm. c Mapping of QHd.osu-6A for heading date. Phenotypic data were obtained from the DH population grown in the greenhouse and vernalized (HD-VRN) or not vernalized in 2014 (HD-GH14) and 2015 (HD-GH15). The GBS marker data were deposited in the NCBI SRA (accession number SRP051982) in a previous study13. Two GBS markers, GBS7889 and GBS10048, linked with the QTL peak, are indicated in red. The vertical dashed line indicates that the two GBS markers are under the QTL peaks. The x-axis represents genetic distance (cM) within the genomic region covering QHd.osu-6A, and the y-axis represents the LOD value of QHd.osu-6A. The horizontal dashed line in the QTL curve of QHd.osu-6A represents a threshold log of the odds (LOD) value of 2.5. d Fine mapping of QHd.osu-6A using nine internal markers to genotype five DH lines and six F3:4 lines that contained crossovers at the targeted region. X in red indicates a crossover. Two markers flanking the candidate gene are indicated in red; three markers linked to the candidate gene are highlighted in gray. The final candidate is indicated in blue. The numbers under the markers are the physical locations (bp) of these markers according to the IWGSC RefSeq v1.0 genomic sequence. Heading date was phenotyped for days from planting to heading with six replicates for each of DH lines and 15–20 plants for in each of F3:4 lines. Source data underlying (c) are provided as a Source Data file.
Initially, we used five recombinant lines in the DH population to map the candidate genes for QHd.osu-6A within an ~2.1-Mb region between two flanking markers, GBS7889 and GBS10048 (Supplementary Fig. 3), according to the IWGSC RefSeq v1.0 genomic sequence of Chinese Spring (CS). Then, we developed 11 internal markers, including four regular PCR markers (Supplementary Fig. 4), three sequencing markers (Supplementary Fig. 5), and four Kompetitive allele-specific PCR (KASP) markers (Supplementary Fig. 6) to fine-map QHd.osu-6A. We also identified six new crossovers in the region between GBS7889 and GBS10048 (Fig. 1d) from 618 F2:4 plants derived from a Duster × Billings cross (Supplementary Method 2). We phenotyped eleven critical lines, including five DH lines and six recombinant lines, in the presence and absence of vernalization for fine mapping (Fig. 1d). The gene responsible for QHd.osu-6A was narrowed down to within a 382,146-bp genomic region flanked by two markers, 6A-KASP13 and 6A400M1. According to the IWGSC RefSeq v1.0 sequences, this region contained three candidate genes: TraesCS6A01G091100, TraesCS6A01G091200, and TraesCS6A01G091300 (Fig. 2a). Finally, we cloned the gene for QHd.osu-6A using the classic map-based cloning approach (Supplementary Method 2).

a The physical locations of QHd.osu-6A candidate genes. The QHd.osu-6A interval is delimited within 382,146 bp, flanked by the two markers 6A-KASP13 and 6A-400M1 that are indicated in green dots. The candidate gene, TraesCS6A01G091300, is indicated in a red dot, and the other two candidate genes in this region are indicated in yellow dots. b Allelic variation in TraesCS6A01G091300 among Chinese Spring (CS), Duster, and Billings. The sequence of TraesCS6A01G091300 in Billings is the same as in CS, but the sequence of Duster has SNPs across the gene, which are indicated with blue asterisks. A red asterisk indicates a SNP that changes amino acid. SSR denotes simple sequence repeats in the promoter region of TraesCS6A01G091300. The region of 168-bp insertion in intron 1 of the Billings allele is indicated by a red line. c, d Comparison of transcript levels of TraesCS6A01G091300 alleles in Duster and Billings in non-vernalized (GH) and vernalized plants (VRN) at two different stages. The RNA samples were collected from fully developed leaves at the fourth-leaf stage (0 wk) and after vernalization for 6 weeks (6 wk). Transcript levels of TraesCS6A01G091300 were analyzed using qRT-PCR and calculated using the 2–ΔΔCT method, where CT is the threshold cycle. The mean transcript level for each allele was analyzed using three biological replicates and two technical replicates, and a two-tailed unpaired Students’ t test showed no significant difference in expression between the two alleles (n = 6). The bars indicate standard error. Source data underlying (c, d) are provided as a Source Data file.
Next, we sequenced each of the two alleles, derived from Duster and Billings, respectively, for each of the three candidate genes. The sequences of TraesCS6A01G091100 (Supplementary Fig. 7) and TraesCS6A01G091200 (Supplementary Fig. 8), which encode pentatricopeptide repeat-containing proteins, were the same in Duster and Billings. Furthermore, the transcript levels of both genes did not differ significantly between the Duster and Billings alleles (Supplementary Fig. 9). Therefore, the two genes were excluded as candidates for QHd.osu-6A, and the third gene, TraesCS6A01G091300, was left as the sole candidate for QHd.osu-6A.
TraesCS6A01G091300 consists of eight exons and seven introns and shows numerous nucleotide differences within the sequenced region between Duster and Billings (which had the same sequence as CS). These included differences within the genic region, ~1.2 kb before the start codon and 121 bp after the stop codon (Fig. 2b, Supplementary Fig. 10). One single-nucleotide polymorphism (SNP) in the coding region resulted in the substitution of a valine residue at position 229 of the protein encoded by the Duster (late) allele for the isoleucine encoded by the Billings (early) allele (Supplementary Fig. 11).
TraesCS6A01G091300 is annotated in the IWGSC RefSeq v1.0 database to encode a transmembrane protein with 343 amino acids. When we used the predicted TraesCS6A01G091300 protein sequence to query nonredundant protein databases in GenBank, all of its hits in plants represented hypothetical or uncharacterized proteins except for an ortholog in Arabidopsis (NP_201101), which is annotated as a putative uridine diphosphate (UDP)-N-acetylglucosamine-N-acetylmuramyl-pyrophosphoryl-undecaprenol N-acetylglucosamine protein (an O-GlcNAc transferase, abbreviated OGT) (Supplementary Fig. 11). The Arabidopsis NP_201101 is functionally uncharacterized and shows no similarity or domain conservation with the known plant OGT protein, SECRET AGENT (AtSEC)30. The Arabidopsis SPINDLY (AtSPY) was reported to be an OGT protein31 but it was recently believed to be an O-fucosyltransferase32,33. TraesCS6A01G091300 is referred to as TaOGT1 in common wheat, with TaOGT1b representing the early allele in Billings and TaOGT1a representing the late allele in Duster.
A key regulatory DNA element differentiated transcript levels of the two TaOGT1 alleles
We formulated two mutually exclusive hypotheses to address how the two alleles of TaOGT1 generate differential phenotypes. One hypothesis was that the heading date was regulated by TaOGT1 at the transcriptional level as a result of SNPs in the promoter and the non-coding regions. The alternative hypothesis was that the heading date was regulated by TaOGT1 at the protein level, due to the point mutation in this protein. Therefore, we set out to test the two hypotheses and validated that the heading date was regulated by TaOGT1 at the transcriptional level.
We observed that the transcript level of TaOGT1b in Billings was significantly higher than that of TaOGT1a in Duster when each was grown at a variety of broad ambient temperatures either without (Fig. 2c) or with vernalization (Fig. 2d). The Billings TaOGT1b allele contained a 168-bp insertion in intron 1 compared to the Duster TaOGT1a allele (Fig. 2b, Supplementary Fig. 12). We developed two constructs to test whether the 168-bp fragment is involved in regulation of reporter gene expression in a transient GUS expression system. The construct TaOGT1b-Prom included 300 bp of the promoter sequence, exon 1, and 200 bp of intron 1 containing the 168-bp fragment from the Billings allele, and the TaOGT1a-Prom sequence included 300 bp of promoter sequence, exon 1, and 32 bp of intron 1 without the 168-bp fragment from the Duster allele (Fig. 3a). The two constructs contained the identified 300 bp promoter sequence excluding the SNPs in the promoter between the two TaOGT1 alleles and had the only difference for the presence and absence of the 168-bp fragment. The LUC gene was driven by the ubiquitin promoter, which was used as an internal transformation control to provide an estimate of the efficiency of transient expression. We co-transformed TaOGT1b-Prom and TaOGT1a-Prom, respectively, with the LUC construct into wheat protoplasts to assess their promoter activity. Protoplasts transformed with TaOGT1b-Prom showed much higher GUS expression than protoplasts transformed with TaOGT1a-Prom, which lacks the 168-bp fragment (Fig. 3b). This result provided experimental evidence that the 168-bp insertion/deletion (indel) was important for the transcriptional regulation of TaOGT1.

a A diagram depicting the constructs used to test regulatory elements in TaOGT1. The promoter (Prom), exon 1 (E1), and part of intron 1 (In1) from Duster TaOGT1a without the 168-bp fragment, and from Billings TaOGT1b with the 168-bp fragment, were used as a promoter of the GUS reporter gene. The endogenous control construct was created using the Ubiquitin promoter to drive the LUC gene. b Comparison of GUS/LUC activity ratio. The GUS and LUC activities were assayed in a transient expression in wheat protoplast system. The data are presented as mean ratio ± SEM, which was calculated from six independent protoplast transformation reactions (n = 6) for each construct. A two-tailed unpaired t test showed significant difference between the two constructs from the two TaOGT1 alleles. c The sequence of the 168-bp insertion in intron 1 of the Billings TaOGT1b allele. The CArG box is indicated in red. d–f EMSA interactions of the vrn1 and TaVRT2 proteins with the CArG-box DNA probe labeled with biotin. EMSA was performed for the vrn1b protein from 2174 (which has the same vrn1b as Billings and Duster) (d), vrn1a from Jagger (e), and TaVRT2 (f). Lane 1 indicates the biotin-labeled TaOGT1b DNA probe alone. Lane 2 represents vrn1 or TaVRT2 protein and DNA in the EMSA reactions. Lane 3 represents vrn1 or TaVRT2 protein and 100× protein competitors that were added to the EMSA reactions. These EMSA reactions were repeated three times. Source data underlying (b, d–f) are provided as a Source Data file.
We analyzed the sequence of the 168-bp DNA fragment of the TaOGT1b allele and identified a potential CArG-box (CATTTTTTGG) (Fig. 3c), which is a conserved DNA target site of MADS-domain proteins including VRN14,34,35,36 and TaVRT237,38,39. In an in vitro electrophoretic mobility shift assay (EMSA), both vrn-A1 (Fig. 3d, e) and TaVRT2 (Fig. 3f) proteins physically bound the 168-bp DNA fragment containing this CArG-box. We concluded that the Billings TaOGT1b allele has a direct target site for the MADS-box proteins in wheat, but the Duster TaOGT1a allele does not have this target site.
Constitutive overexpression of the Billing TaOGT1b allele in Duster accelerated heading of transgenic plants
We performed a transgenic complementation analysis to confirm that TaOGT1b from Billings accelerated flowering in transgenic Duster plants. We transformed the TaOGT1b allele into immature Duster embryos through particle bombardment of an expression construct in which the maize Ubiquitin promoter was fused with TaOGT1b in the expression vector pMDC32. This generated seven positive T0 plants, two of which (TaOGT1b-OE9 and TaOGT1b-OE12) were self-pollinated to produce T1 seeds for further studies. In each of the two T1 populations, TaOGT1b genetically segregated as a single-copy transgene. On average, the heading dates of transgenic plants were 21 days earlier in the unvernalized populations (Fig. 4a, b) and 6 days earlier in the vernalized population (Fig. 4c, d) than that of the non-transgenic sibling populations.

a TaOGT1b from Billings was overexpressed in Duster. A transgenic (right) and a non-transgenic plant (left) grown in the greenhouse without vernalization are shown. b Comparison of the mean heading date for transgenic and non-transgenic plants in the two T1 populations without vernalization. c A transgenic (right) and a non-transgenic plant (left) grown in the greenhouse with vernalization. a, c Scale bar = 15 cm. d Comparison of the mean heading date for transgenic and non-transgenic plants in the two T1 populations with vernalization. b, d Statistical comparison of the mean heading date was performed using a two-tailed unpaired Student’s t test to determine the significance level between transgenic and non-transgenic plants (n = 8). The error bars indicate standard error. e A UDP standard curve for the amount of UDP (x-axis) and the signal-to-background ratio (y-axis) based on the luminescence reading in the samples (n = 3). f Comparison of TaOGT1b enzyme activity in the total protein extract from the leaves of TaOGT1b-OE9 transgenic and non-transgenic plants. The x and y axes represent the amounts of protein (ng) and UDP (calculated from the UDP standard curve) in the samples (n = 3), respectively. Source data underlying (b, d–f) are provided as a Source Data file.
TaOGT1 had the O-GlcNAcylation enzyme activity
We characterized the OGT activity in the TaOGT1b-overexpressing transgenic plants. First, we tested whether TaOGT1 had similar activity to O-GlcNAc transferase, using a UDP detection kit. In this OGT detection reaction, a GlcNAc group from the UDP-GlcNAc donor is transferred to the side chain of serine and threonine on the OGT substrates, and the released UDP can be detected to indicate OGT activity (Fig. 4e). We attempted to express the full-length TaOGT1 protein in Escherichia coli for this experiment, but were unsuccessful, probably because it is a eukaryotic-membrane-like protein that is not expressed in E. coli40. Therefore, we extracted total protein from transgenic TaOGT1b-OE9 plants, which included overexpressed TaOGT1b and native TaOGT1a, and total protein from non-transgenic plants, which contained only native TaOGT1a. Protein extract from transgenic TaOGT1b-OE9 plants showed higher O-GlcNAc transfer activity than that extracted from non-transgenic plants (Fig. 4f). Using 20 ng of each total protein sample, we detected UDP concentrations of 0.167 μM in TaOGT1b-OE9 wheat plants but only 0.078 μM UDP in non-transgenic plants. Together, these results demonstrate that TaOGT1 has O-GlcNAcylation enzyme activity.
TaOGT1 O-GlcNAcylated TaGRP2
To identify the substrates of the TaOGT1 protein, TaOGT1b was used as a bait to screen a yeast-two hybrid (Y2H) library established using whole seedlings of winter wheat cultivar “2174”41. From the screening of ~2 × 107 cells, we identified two interacting proteins, both of which were annotated as nonspecific serine/threonine protein kinases: TraesCS1B02G364800, hereafter referred to TaK1 (Supplementary Fig. 13), and TraesCS4D02G196100, hereafter referred to TaK4 (Supplementary Fig. 14). We confirmed the direct interaction between TaOGT1b and TaK1 or TaK4 by co-transformation of yeast cells (Supplementary Fig. 15) and using a transient expression system in tobacco leaves (Supplementary Fig. 16), but neither was a direct substrate of O-GlcNAcylation by TaOGT1 in vitro (Supplementary Fig. 17).
The TaGRP2 protein (TraesCS4B02G020300) (Supplementary Fig. 18) is reported to be a glycine-rich RNA-binding protein (GR-RBP or GRP) that is O-GlcNAcylated during vernalization in wheat42. The direct interaction between TaOGT1b and TaGRP2 was observed in co-transformation of yeast cells (Supplementary Fig. 15) and in a transient expression system with bimolecular fluorescence complementation (BiFC) in tobacco leaves (Supplementary Fig. 16). We found that proteins extracted from TaOGT1b transgenic plants produced a higher O-GlcNAcylation signal of TaGRP2 than those from non-transgenic plants (Fig. 5a, Supplementary Fig. 19a), indicating that TaOGT1 could O-GlcNAcylate TaGRP2. The amount of GlcNAcylated TaGRP2 proteins by TaOGT1 in the GlcNAcylation reaction with TaK1 was significantly lower than that in the control reaction without TaK1 (Fig. 5b, Supplementary Fig. 19b), indicating that the GlcNAcylation of TaOGT1 on TaGRP2 was repressed by TaK1. However, TaK4 showed no effect on the GlcNAcylation of TaOGT1 on TaGRP2 (Fig. 5b). Surprisingly, the TaOGT1 protein was able to GlcNAcylate some E. coli proteins that were not purified from the TaGRP2, TaK1, or TaK4 protein samples (Fig. 5b, Supplementary Fig. 17), indicating that TaOGT1 has the capacity of GlcNAcylation on diverse proteins in different organisms.

a Comparison of OGT enzyme activity among different TaOGT1 proteins. TaOGT1 proteins from a transgenic plant (+), TaOGT1 proteins from a non-transgenic plant (−), and three different TaOGT1 protein fragments expressed in E. coli: aa 1–28, aa 1–192, and aa 172–343. Presence and absence of the particular protein in the reaction is indicated by “+” and “−”, respectively. The GlcNAcylated TaGRP2 protein was detected using the anti-O-GlcNAc antibody CTD110.6 and is highlighted by an arrow. b Effects of TaK1 and TaK4 on GlcNAcylation by TaOGT1. TaOGT1 was extracted from total protein from leaves of transgenic wheat plants, and TaK1 or TaK4 were added to the GlcNAcylation reaction. The upper image indicates GlcNAcylated TaGRP2 protein detected with the CTD110.6 antibody against O-GlcNAc, and the lower image indicates actin protein as an endogenous control, detected by an anti-actin antibody. c Comparison of the sequences of two GlcNAcylation sites. d Comparison of OGT enzyme activity of two protein fragments of aa 1–28 and aa 233–264. e Comparison of AtSEC and AtSPY proteins expressed in E. coli with TaOGT1 proteins from a transgenic plant (+) and a non-transgenic plant (−). TaGRP2 protein was used as a substrate in each GlcNAcylation reactions. The images of the same gels stained with Coomassie Brilliant Blue (CBB) are provided in Supplementary Fig. 19. These GlcNAcylation reactions were repeated three times. Source data underlying (a, b, d, e) are provided as a Source Data file.
TaOGT1 protein had two active sites
We found that the TaOGT1 protein contains two active sites for O-GlcNAcylation. We split TaOGT1 into three protein fragments, TaOGT1-P1 (aa 1–28), TaOGT1-P2 (aa 1–192), and TaOGT1-P3 (aa 172 to the final residue, aa 343), to facilitate their expression in E. coli. Notably, all three fragments could GlcNAcylate TaGRP2 (Fig. 5a). Sequence analysis suggested that a conserved domain similar to aa 1–28 in TaOGT1-P1 was present at aa 234–264 in TaOGT1-P3 (Fig. 5c, Supplementary Fig. 20). Therefore, we expressed the TaOGT1-P4 fragment (aa 234–264) and found that both TaOGT1-P1 and TaOGT1-P4 catalyzed the GlcNAcylation of TaGRP2 (Fig. 5d, Supplementary Fig. 19c), demonstrating that TaOGT1 contains two active sites. We observed no significant difference in the level of TaGRP2 O-GlcNAcylation by TaOGT1a and TaOGT1b, despite one amino acid difference between their protein sequences (Supplementary Fig. 21). Therefore, the difference in heading date between Duster and Billings was not caused by TaOGT1 at the protein level.
In Arabidopsis, AtSEC is reported to have the O-GlcNAcylation enzyme activity30, and AtSPY is reported to have the O-fucosyltransferase activity33,34 but not the O-GlcNAcylation enzyme activity31. We found that TaGRP2 was GlcNAcylated by AtSEC but not by AtSPY (Fig. 5e, Supplementary Fig. 19d). The results indicated that although TaOGT1 and AtSEC have no similarity or domain conservation in protein sequences, they showed a similar GlcNAcylation activity on the same substrate as TaGRP2.
TaOGT1 activity was associated with sugar content and the transcript levels of flowering time genes
To establish a signaling network, we tested which genes and metabolites were regulated in TaOGT1b-overexpressing transgenic plants, using non-transgenic plants as controls. We tested these plants in the greenhouse without vernalization and with vernalization for 1, 3, or 6 weeks. Both the transcript levels of TaOGT1 (overexpressing TaOGT1b from Billings plus native TaOGT1a in Duster) (Fig. 6a) and the enzymatic activity of TaOGT1 (Fig. 6b) increased in the TaOGT1b-overexpressing transgenic plants. Due to the elevated transfer of GlcNAc to nucleotides as a result of the increased TaOGT1 content, the content of glucose (Fig. 6c) and sucrose (Fig. 6d) decreased, but the content of fructose did not significantly change (Fig. 6e). Surprisingly, the transcript levels of vrn1 (Fig. 6f), vrn3 (Fig. 6g), PPD1 (Fig. 6h), and TaGRP2 (Fig. 6i) all increased in the TaOGT1b-overexpressing transgenic plants, compared with the non-transgenic plants. The transcript levels of either VRN2 (Fig. 6j) or TaVRT2 (Fig. 6k) did not change significantly in the TaOGT1b-overexpressing transgenic plants, compared with the non-transgenic plants. The effects of low temperature on the transcript levels of these genes tested in the same samples indicated that vrn1 (Fig. 6f) and vrn3 (Fig. 6g) were significantly upregulated, VRN2 (Fig. 6j) and TaVRT2 (Fig. 6k) were significantly downregulated, but TaGRP2 (Fig. 6i) was not significantly regulated by low temperature. The downregulation of TaOGT1 by low temperature was detectable in the 3 weeks-vernalized plants but not in the 6 weeks-vernalized plants (Fig. 6a). The significance levels of each gene and sugar regulated by TaOGT1 and low temperature are summarized in Supplementary Table 1. We analyzed the association of TaOGT1 enzymatic activity and TaOGT1 transcript level with content of each sugar and transcript level of each flowering-time gene in all transgenic and non-transgenic plants with and without vernalization (Supplementary Tables 2, 3). The most striking correlations included a positive correlation between TaOGT1 activity and transcript levels of vrn1 (R2 = 0.9498, p < 0.00001, Fig. 6l) and vrn3 (R2 = 0.8304, p < 0.0001, Fig. 6m). In addition, there was a negative correlation between sucrose content and PPD1 transcript levels (R2 = 0.5482, p = 0.0058, Fig. 6n). The results indicated that TaOGT1 enzymatic activity was associated with the transcript levels of the flowering-time genes and sugar content in the cytoplasmic matrix of wheat leaves.

a–k Comparison of gene expression and sugar content in TaOGT1b overexpressed transgenic plants (+) and non-transgenic plants (−). The plants were grown in the greenhouse (GH) without vernalization or were treated in a cold room (CR) with vernalization for 1 week (1 wk), 3 weeks (3 wk), or 6 weeks (6 wk). The same samples of fully developed leaves from three different plants with two technical replicates (n = 6) were used to determine TaOGT1 transcript levels (a); O-GlcNAc transferase activity (b); the contents (mg g−1) of glucose (c), sucrose (d), and fructose (e); and transcript levels of vrn1 (f), vrn3 (g), PPD1 (h), TaGRP2 (i), VRN2 (j), and TaVRT2 (k). Transcript levels were calculated using the 2–ΔΔCT method, where CT is the threshold cycle. The significant differences in mean expression for each gene were compared between the two alleles using a two-tailed unpaired Student’s t test (Supplementary Table 1); the bars indicate the standard error. l–n Correlation analyses of data for all transgenic and non-transgenic plants with and without vernalization (n = 12): l. UDP vs. vrn1; m. UDP vs. vrn3; n sucrose vs. PPD1. The significant correlations between two variables were shown in Supplementary Tables 2, 3. Source data underlying (a–k) are provided as a Source Data file.
Discussion
In this study, we cloned TaOGT1 and revealed the signaling networks that regulate heading date in winter wheat (Fig. 7). TaOGT1b promoted the heading of winter wheat cultivars under broad ambient temperatures because its intron contains a binding site for MADS-box transcriptional factors, including VRN1 and TaVRT2, which were tested in this study. VRN1 is a central regulator of heading date in different wheat species, subspecies, and cultivars4,13,36,43,44,45. TaVRT2 was reported to be a repressor of wheat development, whose transcript levels were decreased by vernalization37,38, but TaVRT2 was recently reported to be a promoter of wheat development, whose transcript levels were increased39. We tested the transcript levels of TaVRT2 and found that it was dramatically decreased by vernalization (Fig. 6k). TaOGT1 O-GlcNAcylates TaGRP2, which is a negative regulator of flowering via its direct binding to VRN1 pre-mRNA42,46. The O-GlcNAcylated modification on TaGRP2 is induced by vernalization46. Our results indicated that TaGRP2 was not significantly regulated by low temperature, but it was dramatically increased in the TaOGT1 overexpressed transgenic wheat plants, compared with non-transgenic plants (Fig. 6i). Due to the O-GlcNAcylation of TaOGT1 targets, the content of soluble glucose decreased, which promoted the expression of vrn1, vrn3, and PPD1. Therefore, TaOGT1 is a critical regulator of flowering in winter wheat cultivars.

Arrows indicate promotion and “−|” indicates repression. The genotypes of the winter wheat cultivars Billings and Duster contain recessive vrn1 and vrn3 alleles and a photoperiod-insensitive allele, PPD1. Italic names indicate genes, and non-italic names indicate proteins. Shaded gray circles represent the cytoplasmic matrix.
Previous studies showed that vrn1 genes are induced by low temperature in winter wheat4,8,43,44,45. The upregulation of vrn1 is due to indels in the promoter region that result in the loss of a recognition site by repressors4,6,36,47,48 or the gain of a binding site by the microRNA TamiR112349. The repressor binding sites are also located within vrn1 intron 150 and the 5′ untranslated region48. The upregulation of vrn3 by low temperature is due to a large insertion in the promoter region6. Overexpression of TaOGT1 potentially modifies the proteins involved in the interactions with the regulatory sites of these key flowering genes in wheat.
In Arabidopsis, AtSEC was identified based on their sequence similarity to OGTs in animals30. Although AtSEC is different in sequence from TaOGT1, AtSEC was able to O-GlcNAcylate TaGRP2. There are up to 34 unannotated or uncharacterized proteins in Arabidopsis that share similarity with wheat TaOGT1. The cloning and characterization of TaOGT1 suggest that orthologous and homologous proteins such as NP_201101 in Arabidopsis might be OGTs and may be involved in the O-GlcNAcylation of 262 proteins that are modified by O-GlcNAc in Arabidopsis51. There are 168 O-GlcNAc-modified proteins in wheat46, and as the characterized OGT in wheat, TaOGT1 can be used to reveal functions of the homoeologous and homologous OGTs in this species. Sugar may be involved in the transition from the vegetative to reproductive phase in wheat, based on the observation that the levels of monosaccharides such as glucose and fructose are significantly higher in cold-treated plants, as a result of starch degradation and sucrose metabolism52,53. However, evidence for a direct role of sugar in flowering is limited. Our findings provide experimental evidence that the transcript levels of vrn1, vrn3, PPD1, and TaGRP2 were upregulated by TaOGT1 overexpression in transgenic wheat. It is likely that the upregulation of these different flowering-time genes is due to the resulting reduced glucose content in the common cytoplasmic matrix in wheat leaves. In humans and animals, only 2–5% of all glucose is used to generate the UDP-GlcNAc sugar nucleotide, but O-GlcNAc can serve as a nutrient and stress sensor54,55. In Arabidopsis, trehalose-6-phosphate (T6P), which is present only in trace amounts, functions as a proxy for carbohydrate status to regulate flowering, and when the gene encoding T6P synthase 1 (TPS1) was knocked down, plants flowered extremely late56. Based on sequence identity, orthologous wheat genes of Arabidopsis TPS1 (AT1G78580) can be found (e.g., ACI16353 and KAF6991139), but none of them has been functionally characterized. The identification and characterization of TaOGT1 might represent a connection between wheat and Arabidopsis in carbohydrate signaling pathways via glucose catabolism. TPS1 catalyzes the formation of T6P from glucose-6-phosphate and UDP–glucose (UDPG)57, whereas OGT1 catalyzes the transfer of GlcNAc from UDP-GlcNAc to serine and threonine residues of proteins58. Both UDPG and UDP-GlcNAc are derived from glucose catabolism. In addition, the T6P pathway also regulates flowering via the florigen FT. The T6P pathway regulates flowering at two sites in the plant; in the leaves where FT expression is induced by TPS1, and at the shoot apical meristem, where flowering-time genes are regulated by TPS156.
TaOGT1 confers relatively minor effects on heading date compared with genes in vernalization and photoperiod pathways, but these minor effects nonetheless are critical for cultivars grown in the same growth area. Minor effects could also result from earliness per se (Eps) genes, such as Eps-3Am59 or Eps-Am160 in the diploid einkorn wheat Triticum monococcum, but no Eps gene has been cloned in common wheat. Fine selection for a gene with minor effects represents a challenge in conventional breeding. The presence of a constitutive 168-bp insertion in the TaOGT1b Billings allele is an intrinsic factor that is independent of the environment. However, it represents an instrumental molecular marker that fine-regulates flowering time and accelerates heading date in winter wheat cultivars by several days, to potentially help them adapt to future climate scenarios. The TaOGT1 gene would be a starting point to reveal functions of numerous orthologous and homologous proteins in flowering time and sugar metabolism in plant species.
Methods
Mapping and cloning of QHd.osu-6A
For gene mapping, we generated a doubled-haploid (DH) population of 260 lines from a cross between Duster (PI 644016) and Billings (PI 656843), two hard red winter wheat cultivars that are cultivated extensively across the southern Great Plains of the USA. Detailed information about the two cultivars and the DH population is provided in Supplementary Method 1. In previous studies, the Duster × Billings DH population was tested in field conditions at Stillwater Research Station, Oklahoma, for 2 years, and heading date was scored when a first head completely emerged from the boot in a plant. The data for heading date were analyzed in this study. The same DH population was grown in a greenhouse at day/night temperatures of 25/20 °C with long day conditions (16 h light/8 h darkness), and the population was either vernalized (Supplementary Method 2) or not vernalized. The phenotypes of plants in these two different treatments were integrated with GBS markers, which resulted in mapping QHd.osu-6A to the short arm of chromosome 6A, in a region that was not reported to affect flowering time; therefore, QHd.osu-6A was cloned using a positional cloning approach. The primers used for molecular markers are listed in Supplementary Table 4. Detailed procedures used to clone QHd.osu-6A are provided in Supplementary Method 2.
Generation of transgenic TaOGT1b wheat
Primers TaOGT1b-attB1-F1 and TaOGT1b-attB1-R1 (Supplementary Table 5) were designed to amplify the complete cDNA of TaOGT1b from Billings, and the cDNA was cloned into the pMDC32 vector containing the maize Ubiquitin promoter61, which has previously been successfully used to express genes in wheat6,62. The ubiquitin-TaOGT1b-pMDC32 construct was transformed into Duster by particle bombardment using a published protocol63. Seven individual transgenic Duster plants (T0) were generated, and two of these (TaOGT1b-OE9 and TaOGT1b-OE12) were propagated to give T1 populations for further experiments.
In an independent experiment, embryos from TaOGT1b-OE-9 T0 were dissected and grown in rooting medium at 22–23 °C in long days (16 h light/8 h darkness) for 2 weeks before the resulting seedlings were transferred to the soil for greenhouse analyses. At the seventh-leaf stage, 10 TaOGT1b-OE9 T1 plants were divided into two equal groups, of which one was vernalized and the other was not. After 1, 3, or 6 weeks of vernalization, the first complete leaf of each tiller was collected for analysis of gene expression, sugar content, and O-GlcNAcylation enzyme activity. The samples were collected at 17:00 h before additional lights were turned on at 18:00 h to make a long day condition, and the central part of the fresh leaves was analyzed as recommended64,65.
Transient promoter activity assays
The DNA fragments consisting of the promoter, exon 1, and intron 1 DNA from TaOGT1a and TaOGT1b were respectively fused to the uidA gene encoding GUS as a reporter, which were cloned into the pMDC32 vector. The primers used for cloning were GUS-HindIII-F1 and GUS-KpnI-R1 (Supplementary Table 5). The only difference between the DNA fragments was that the Billings TaOGT1b allele contained a 168-bp fragment in intron 1. The internal control for the TaOGT1::GUS assay consisted of the pMDC32 construct that contained the Ubiquitin promoter fused to the gene for luciferase (LUC) that was cloned using primers LUC attB1-F1 and attB1-R1 (Supplementary Table 5). This Ubiquitin::LUC construct was co-transformed with TaOGT1a-GUS and TaOGT1b-GUS into wheat protoplasts using PEG (40% PEG, 0.2 M mannitol and 100 mM CaCl2)66, and the GUS/LUC ratios were used to generate relative promoter activities67. The co-transformed protoplasts were incubated for 48 h at 25 °C in a reaction with 1 mM 4-methylumberlliferyl-β-d-glucuronide in lysis buffer (Sigma), and the reaction was terminated with 0.2 M Na2CO3 after 30 min. The GUS activity was assayed using a Synergy H1 reader (BIO-TEK Instruments. Winooski, VT), and LUC activity was assayed with the Luciferase 1000 Assay system (Promega, E4550). Ratios of GUS to LUC [GUS/LUC = (GUS-30 min − GUS-0 min) × 10/LUC] activities were used to define promoter activity67.
Quantification of gene expression
Quantitative real time PCR (qRT-PCR) was used to determine the transcript levels of TaOGT1, vrn1 (primers vrn1-F1 and vrn1-R2), VRN2 (primers VRN2-F and VRN2-R), vrn3 (primers vrn3-F and vrn3-R), PPD1 (primers PPD-F1 and PPD-R1), TaVRT2 (primers TaVRT2-F and TaVRT2-R), TaGRP2 (primers TaGRP2-F and TaGRP2-R), and Actin (primers Actin-F2 and Actin-R) from the same cDNA samples. The primers for the expression of these genes are listed in Supplementary Table 5. The transcripts of TaOGT1 in transgenic Duster plants included native TaOGT1a and transgenic TaOGT1b from Billings, whereas non-transgenic plants contained native TaOGT1a. Total RNA was extracted from leaves, qRT-PCRs were performed for gene expression, and detailed information is provided in Supplementary Method 3. Gene transcript levels were calculated by the 2–ΔΔCT method, where CT is the threshold cycle.
Electrophoretic mobility shift assay
Primers TaOGT1-168Ind-F1 and TaOGT1-168Ind-R1 (Supplementary Table 5) were used to amplify the gDNA fragment from Billings using Phusion High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA). The DNA fragment was used as a probe after labeling the 3′-OH end of the double-stranded DNA with Biotin using a Pierce™ Biotin 3′ end DNA labeling kit (Thermo Fisher Scientific, Waltham, MA). MBP-vrn-A1a protein from cultivar Jagger (aa 1–180) and MBP-vrn-A1b from cultivar 2174 (aa 1–180) were used for the electrophoretic mobility shift assay (EMSA)13. TaVRT2 was also cloned into pMAL-C2 vector using primers (primers TaVRT2-EcoRI-F1 and TaVRT2-BamHI-R1) (Supplementary Table 5), and expressed MBP-TaVRT2 proteins in E. coli (Supplementary Method 4) were used to perform electrophoretic mobility shift assay (EMSA) with the Light Shift Chemiluminescent EMSA Kit (Thermo Fisher Scientific, Waltham, MA).
TaOGT1 enzyme activity assay
The full-length cDNA of TaOGT1 was cloned into different expression vectors including pSUMO and pMAL-C2, but its protein could not be produced in E. coli, probably because it is a membrane-like protein that is not expressed well in this species40. Instead, total protein was extracted from leaves of transgenic TaOGT1b-OE9 wheat plants, which overexpressed TaOGT1b and expressed native TaOGT1a, and from wild-type plants, according to Rubio et al.68.
TaOGT1 activity was assayed using two approaches. First, O-GlcNAc transferase activity was measured using the UDP-GloTM Glycosyltransferase detection kit (Promega, Madison, WI). In an OGT detection reaction, GlcNAc from a UDP-GlcNAc donor is transferred to serine and threonine side chains on the substrates of the OGT, which releases UDP that can be detected to infer the activity of the OGT. The reagent converts the UDP to ATP and generates light that is detected using a luminometer Synergy H1 (BioTek Instruments, Winooski, VT). The luminescence was converted to UDP concentration using a UDP standard curve. Second, chemiluminescent signals were imaged using a Fluor Chem E imaging system with a CCD camera (Protein Simple, Santa Clara, CA). After a reaction in the GlcNAcylation reaction solution (12.5 mM MgCl2, 50 mM Tris–HCl, pH 7.5, and 1 mM DTT), proteins were separated by SDS–PAGE gel electrophoresis and transferred to a PVDF membrane (Bio-Rad, Hercules, CA). Immunoblotting using an antibody against O-GlcNAc (CTD110.6 mouse mAb HRP conjugate, Cell Signaling Technology, Danvers, MA) was used to detect O-GlcNAc modification of proteins46, and an antibody to actin (Abclonal, Woburn, MA) was used as a control.
Four truncated proteins of TaOGT1 were expressed in E. coli BL21 to determine different O-GlcNAcylation active sites: (1) aa 1–28 (using primers TaOGT1-EcoRI-F1 and TaOGT1-BamHI-R2); (2) aa 1–192 (using primers TaOGT1-NdeI-F1 and TaOGT1-BamHI-R3); (3) aa 172–343 (using primers TaOGT1-NdeI-F2 and TaOGT1-BamHI-R1); and (4) aa 233–264 (using primers TaOGT1-NdeI-F5 and TaOGT1-BamHI-R5). These TaOGT1 peptides were tested for their ability to GlcNAcylate TaGRP2, TaK1, and TaK4 proteins. The full-length cDNA of TaGRP2 (using primers TaGRP2-EcoRI-F1 and TaGRP2-BamHI-R1), TaK1 (using primers TaK1-NdeI-F1 and TaK1-BamHI-R1), and TaK4 (using primers TaK4-NdeI-F1 and TaK4-BamHI-R1) were cloned into pMAL-C2 vector and expressed in E. coli. The sequences of the primers for the cloning and expression of the constructs encoding these peptides are provided in Supplementary Table 5.
AtSEC and AtSPY from Arabidopsis (Col-0) were used to test the O-GlcNAcylation enzyme activity on TaGRP2. The full-length cDNA of AtSEC, which is the same as AT3G04240, was cloned into pSUMO vector using primers AtSEC-NdeI-F1 and AtSEC-BamHI-R1 (Supplementary Table 5). The full-length cDNA of AtSPY, which is the same as U62135, was cloned into pSUMO vector using primers AtSPY-EcoRI-F1 and AtSPY-BamHI-R1 (Supplementary Table 5). The proteins were expressed in E. coli (BL21), and the purified proteins were added in the O-GlcNAcylation reactions with TaGRP2.
Screening and identification of interacting proteins with TaOGT1
A prey yeast-two hybrid (Y2H) library was constructed using winter wheat cultivar “2174”41, which contains the same vrn-A1b, PPD-D1b, and vrn-D3b alleles as Billings. The TaOGT1b protein was used as a bait to screen a Y2H library (Supplementary Method 5). Positive clones encoding proteins TaK1 and TaK4 were selected for further studies. The protein interaction experiments were performed to confirm the protein interactions in co-transformation of yeast cells and in BiFC experiments (Supplementary Method 5).
Water-soluble carbohydrate (WSC) analysis
WSC in leaves were extracted using the protocol of Ruuska et al.69. Extracts from 5 mg dried and ground material were used to measure sucrose, glucose and fructose content using a Sucrose/Fructose/D-Glucose Assay Kit (Megazyme, Bray, Ireland) according to the manufacturer’s instructions. Contents of sucrose, glucose or fructose sugar were calculated according to a standard curve.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files. A reporting summary for this Article is available as a Supplementary Information file. The datasets and plant materials generated and analyzed during this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
IWGSC. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191 (2018).
Chouard, P. Vernalization and its relation to dormancy. Annu. Rev. Plant Physiol. 11, 191–238 (1960).
Pugsley, A. T. A genetic analysis of the spring-winter habit of growth in wheat. Aust. J. Agric. Res. 22, 21–31 (1971).
Yan, L. et al. Positional cloning of wheat vernalization gene. VRN1. Proc. Natl Acad. Sci. U.S.A. 100, 6263–6268 (2003).
Mandel, M. A., Gustafsonbrown, C., Savidge, B. & Yanofsky, M. F. Molecular characterization of the Arabidopsis floral homeotic gene. APETALA1. Nat. 360, 273–277 (1992).
Yan, L. et al. The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303, 1640–1644 (2004a).
Putterill, J., Robson, F., Lee, K., Simon, R. & Coupland, G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 80, 847–857 (1995).
Yan, L. et al. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl Acad. Sci. U.S.A. 103, 19581–19586 (2006).
Kardailsky, I. et al. Activation tagging of the floral inducer FT. Science 286, 1962–1965 (1999).
Kobayashi, Y., Kaya, H., Goto, K., Iwabuchi, M. & Araki, T. A pair of related genes with antagonistic roles in mediating flowering signals. Science 286, 1960–1962 (1999).
Kippes, N. et al. Identification of the VERNALIZATION 4 gene reveals the origin of spring growth habit in ancient wheats from South Asia. Proc. Natl Acad. Sci. U.S.A. 112, E5401–E5410 (2015).
Crofts, H. J. On defining a winter wheat. Euphytica 44, 225–234 (1989).
Li, G. et al. Vernalization requirement duration in winter wheat is controlled by TaVRN-A1 at the protein level. Plant J. 76, 742–753 (2013).
Turner, A., Beales, J., Faure, S., Dunford, R. P. & Laurie, D. A. The pseudo-response regulator Ppd-H1 provides adaptation to photoperiod in barley. Science 310, 1031–1034 (2005).
Beales, J., Turner, A., Griffiths, S., Snape, J. W. & Laurie, D. A. A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor. Appl. Genet. 115, 721–733 (2007).
Guo, Z., Song, Y., Zhou, R., Ren, Z. & Jia, J. Discovery, evaluation and distribution of haplotypes of the wheat Ppd-D1 gene. N. Phytol. 185, 841–851 (2010).
Chen, Y., Carver, B. F., Wang, S., Cao, S. & Yan, L. Genetic regulation of developmental phases in winter wheat. Mol. Breed. 26, 573–582 (2010).
Wang, S., Carver, B. F. & Yan, L. Genetic loci in the photoperiod pathway interactively modulate reproductive development of winter wheat. Theor. Appl. Genet. 118, 1339–1349 (2009).
Kerr, R. A. Climate change: Global warming is changing the world. Science 316, 188–190 (2007).
Franks, S. J. & Hoffmann, A. A. Genetics of climate change adaptation. Annu. Rev. Genet. 46, 185–208 (2012).
Miglietta, F., Tanasescu, M. & Marica, A. The expected effects of climate change on wheat development. Glob. Change Biol. 1, 407–415 (1995).
Humphreys, M. W. et al. A changing climate for grassland research. N. Phytol. 169, 9–26 (2006).
Kamran, A., Iqbal, M. & Spaner, D. Flowering time in wheat (Triticum aestivum L.): a key factor for global adaptability. Euphytica 197, 1075–1077 (2014).
Zanke, C. et al. Genetic architecture of main effect QTL for heading date in European winter wheat. Front. Plant Sci. 20, 207 (2014).
Lopes, M. S. et al. Optimizing winter wheat resilience to climate change in rain fed crop systems of Turkey and Iran. Front. Plant Sci. 9, 563 (2018).
Stewart, B. A., Thapa, S., Xue, Q. & Shrestha, R., Climate change. effect on winter wheat (Triticum aestivum L.) yields in the US Great Plains. J. Soil Water Conserv. 73, 601–609 (2018).
Kothari, K. et al. Potential climate change adaptation strategies for winter wheat production in the Texas High Plains. Agric. Water Manag. 225, 105764 (2019).
Yang, C., Frag, H., van Ieperen, W., Trindade, H. & Santos, J. A. Effects of climate change and adaptation options on winter wheat yield under rainfed Mediterranean conditions in southern Portugal. Climatic Change 154, 158–178 (2019).
Li, G. et al. Precisely mapping a major gene conferring resistance to Hessian fly in bread wheat using genotyping-by-sequencing. BMC Genom. 16, 108 (2015).
Hartweck, L. M., Scott, C. L. & Olszewski, N. E. Two O-linked N-acetylglucosamine transferase genes of Arabidopsis thaliana L. Heynh. have overlapping functions necessary for gamete and seed development. Genetics 161, 1279–1291 (2002).
Silverstone, A. L. et al. Functional analysis of SPINDLY in gibberellin signaling in Arabidopsis. Plant Physiol. 143, 987–1000 (2007).
Zentella, R. et al. The Arabidopsis O-fucosyltransferase SPINDLY activates nuclear growth repressor DELLA. Nat. Chem. Biol. 13, 479–485 (2017).
Wang, Y. et al. Nuclear localized O-Fucosyltransferase SPY facilitates PRR5 proteolysis to fine-tune the pace of Arabidopsis circadian clock. Mol. Plant. 13, 446–458 (2020).
Tilly, J. J., Allen, D. W. & Jack, T. The CArG boxes in the promoter of the Arabidopsis floral organ identity gene APETALA3 mediate diverse regulatory effects. Development 125, 1647–1657 (1998).
Yan, L. et al. Allelic variation at the VRN-1 promoter region in polyploid wheat. Theor. Appl. Genet. 109, 1677–1686 (2004b).
Pidal, B. et al. The CArG-box located upstream from the transcriptional start of wheat vernalization gene VRN1 is not necessary for the vernalization response. J. Hered. 100, 355–364 (2009).
Kane, N. A. et al. 2007. TaVRT2 represses transcription of the wheat vernalization gene TaVRN1. Plant J. 51, 670–680 (2007).
Kane, N. A. et al. TaVRT2, a member of the StMADS-11 clade of flowering repressors, is regulated by vernalization and photoperiod in wheat. Plant Physiol. 138, 2354–2363 (2005).
Xie, L. et al. TaVrt2, an SVP-like gene, cooperates with TaVrn1 to regulate vernalization-induced flowering in wheat. New Phytol. https://doi.org/10.1111/nph.16339 (2019).
Wagner, S., Bader, M. L., Drew, D. & De Gier, J. W. Rationalizing membrane protein overexpression. Trends Biotech. 24, 364–371 (2006).
Cao, S. & Yan, L. Construction of a high-quality Yeast two-hybrid (Y2H) library and its application in identification of interacting proteins with key vernalization regulator TaVRN-A1 in wheat. BMC Res. Note 6, 81–86 (2013).
Xiao, J. et al. O-GlcNAc-mediated interaction between VER2 and TaGRP2 elicits TaVRN1 mRNA accumulation during vernalization in winter wheat. Nat. Commun. 5, 4572 (2014).
Danyluk, J. et al. TaVRT-1, a putative transcription factor associated with vegetative to reproductive transition in cereals. Plant Physiol. 132, 1849–1860 (2003).
Murai, K., Miyamae, M., Kato, H., Takumi, S. & Ogihara, Y. WAP1, a wheat APETALA1 homolog, plays a central role in the phase transition from vegetative to reproductive growth. Plant Cell Physiol. 44, 1255–1265 (2003).
Trevaskis, B., Bagnall, D. J., Ellis, M. H., Peacock, W. J. & Dennis, E. S. MADS box genes control vernalization-induced flowering in cereals. Proc. Natl Acad. Sci. U.S.A. 100, 13099–13104 (2003).
Xu, S. et al. The protein modifications of O-GlcNAcylation and phosphorylation mediate vernalization response for flowering in winter wheat. Plant Physiol. 180, 1436–1449 (2019).
Golovnina, K. A., Kondratenko, E. Y., Blinov, A. G. & Goncharov, N. P. Molecular characterization of vernalization loci VRN1 in wild and cultivated wheats. BMC Plant Biol. 10, 168–182 (2010).
Chu, C. et al. A novel retrotransposon inserted in the dominant Vrn-B1 allele confers spring growth habit in tetraploid wheat (Triticum turgidum L.). G3 1, 637–645 (2011).
Yu, M., Carver, B. F. & Yan, L. TamiR1123 originated from a family of miniature inverted-repeat transposable elements (MITE) including one inserted in the Vrn-A1a promoter in wheat. Plant Sci. 215–216, 117–123 (2014).
Fu, D. et al. Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol. Genet. Genom. 273, 54–65 (2005).
Xu, S. et al. Proteomic analysis reveals O-GlcNAc modification on proteins with key regulatory functions in Arabidopsis. Proc. Natl Acad. Sci. U.S.A. 114, E1536–E1543 (2017).
Maruyama, K. et al. Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol. 164, 1759–1771 (2014).
Zhang, Z. et al. Natural variation in CTB4a enhances rice adaptation to cold habitats. Nat. Commun. 8, 14788 (2017).
Hart, G. W., Housley, M. P. & Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022 (2007).
Ponnu, J., Wahl, V. & Schmid, M. Trehalose-6-phosphate: connecting plant metabolism and development. Front. Plant Physiol. 2, 70 (2011).
Wahl, V. et al. Regulation of flowering by trehalose-6-phosphate signaling in Arabidopsis thaliana. Science 339, 704–707 (2013).
Paul, M. J., Primavesi, L. F., Jhurreea, D. & Zhang, Y. Trehalose metabolism and signaling. Annu. Rev. Plant Biol. 59, 417 (2008).
Hanover, J. A., Krause, M. W. & Love, D. C. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 1800, 80–95 (2010).
Gawroński, P. et al. A distorted circadian clock causes early flowering and temperature-dependent variation in spike development in the Eps-3A mutant of einkorn wheat. Genetics 196, 1253–1261 (2014).
Alvarez, M. A., Tranquilli, G., Lewis, S., Kippes, N. & Dubcovsky, J. Genetic and physical mapping of the earliness per se locus Eps-Am1 in Triticum monococcum identifies EARLY FLOWERING 3 (ELF3) as a candidate gene. Funct. Integr. Genom. 16, 365–382 (2016).
Christensen, A. H., Sharrock, R. A. & Quail, P. H. Maize polyubiquitin gene: structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol. Biol. 18, 675–689 (1992).
Liu, M. et al. The STENOFOLIA gene from Medicago alters leaf width, flowering time and chlorophyll content in transgenic wheat. Plant Biotech. J. 16, 186–196 (2018).
Sparks, C. A. & Jones, H. D. Genetic transformation of wheat via particle bombardment. in Cereal Genomics: Methods and Protocols (eds. Henry, J. R. & Furtado, A.) 201–218 (Humana Press, 2014).
Lunn, J. E. & Hatch, M. D. Primary partitioning and storage of photosynthate in sucrose and starch in leaves of C4 plants. Planta 197, 385–391 (1995).
Campbell, J. A., Hansen, R. W. & Wilson, J. R. Cost-effective colorimetric microtitre plate enzymatic assays for sucrose, glucose and fructose in sugarcane tissue extracts. J. Sci. Food Agric. 79, 232–236 (1999).
Shan, Q., Yan, P. W., Jun, L. & Cai, X. G. Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 9, 2395–2410 (2014).
Zhang, Y. et al. Natural alleles of GLA for grain length and awn development were differently domesticated in rice subspecies japonica and indica. Plant Biotech. J. 17, 1547–1559 (2019).
Rubio, V. et al. An alternative tandem affinity purification strategy applied to Arabidopsis protein complex isolation. Plant J. 21, 767–778 (2005).
Ruuska, S. A. et al. Large scale transcriptome analysis of the effects of nitrogen nutrition on accumulation of stem carbohydrate reserves in reproductive stage wheat. Plant Mol. Biol. 66, 15–32 (2007).
Acknowledgements
This project was supported by the Agriculture and Food Research Initiative Competitive Grants (2017-67007-25939) from the USDA National Institute of Food and Agriculture (NIFA), the NSF grant (OIA-1826820), and grants from the Oklahoma Center for Advanced Science and Technology (OCAST, AR17-020-03). M.F. received scholarships from the China Scholarship Council, Agricultural College of Nanjing Agricultural University, and the National Key Research and Development Program of China (2016YFD0100402) for her visit at the Oklahoma State University. H.J. was supported by the China 111 Project (B08025). This study was also supported by the Oklahoma Wheat Research Foundation and the Oklahoma Agricultural Experiment Station.
Author information
Authors and Affiliations
Contributions
M.F., F.M., H.J., G.L., C.P., R.N., and L.Y. performed the experiments and analyzed data. P.A. and B.C. phenotyped the population in the field and analyzed results. Z.M. advised M.F., and B.C. and L.Y. designed the experiments and directed the research. F.M. and L.Y. wrote the paper. All authors read and approved the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Jizeng Jia and other, anonymous, reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, M., Miao, F., Jia, H. et al. O-linked N-acetylglucosamine transferase is involved in fine regulation of flowering time in winter wheat. Nat Commun 12, 2303 (2021). https://doi.org/10.1038/s41467-021-22564-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-22564-8
This article is cited by
-
A molecular mechanism for embryonic resetting of winter memory and restoration of winter annual growth habit in wheat
Nature Plants (2024)
-
Genetic basis of geographical differentiation and breeding selection for wheat plant architecture traits
Genome Biology (2023)
-
Wheat genomic study for genetic improvement of traits in China
Science China Life Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
