
Abstract
Cortical and limbic brain areas are regarded as centres for learning. However, how thalamic sensory relays participate in plasticity upon associative learning, yet support stable long-term sensory coding remains unknown. Using a miniature microscope imaging approach, we monitor the activity of populations of auditory thalamus (medial geniculate body) neurons in freely moving mice upon fear conditioning. We find that single cells exhibit mixed selectivity and heterogeneous plasticity patterns to auditory and aversive stimuli upon learning, which is conserved in amygdala-projecting medial geniculate body neurons. Activity in auditory thalamus to amygdala-projecting neurons stabilizes single cell plasticity in the total medial geniculate body population and is necessary for fear memory consolidation. In contrast to individual cells, population level encoding of auditory stimuli remained stable across days. Our data identifies auditory thalamus as a site for complex neuronal plasticity in fear learning upstream of the amygdala that is in an ideal position to drive plasticity in cortical and limbic brain areas. These findings suggest that medial geniculate body’s role goes beyond a sole relay function by balancing experience-dependent, diverse single cell plasticity with consistent ensemble level representations of the sensory environment to support stable auditory perception with minimal affective bias.
Similar content being viewed by others


Introduction
Associative learning depends on the reliable integration of sensory stimuli from the environment and their association with specific aversive or appetitive outcomes to shape future behaviours. Many cortical and limbic brain areas have been identified as centres for associative learning. However, how thalamic sensory relays like the auditory thalamus (medial geniculate body, MGB), which provide direct sensory input to these areas, participate in plasticity upon associative learning, yet ensure stable long-term sensory coding remains unknown. Auditory fear conditioning, a well-studied classical conditioning paradigm, identified the amygdala as a core brain area for associative learning of stimulus-predicted (conditioned stimulus, CS, e.g., tone) aversive outcomes (unconditioned stimulus, US, e.g., mild foot shock)1,2,3,4. At its input site, amygdala response plasticity is thought to be driven by synaptic potentiation in basolateral amygdala (BLA) neurons5,6. However, early work demonstrated that the higher order MGB, a major auditory input hub to the amygdala7, is a site of CS-US integration and plasticity upon fear learning8,9,10,11,12,13,14,15. Enhanced responses to conditioned stimuli and increased synaptic drive from presynaptic MGB neurons to the BLA might act as an additional plasticity mechanism for associative fear learning. Nevertheless, the role of MGB in neuronal response plasticity upon fear learning has been controversially discussed1,16,17 and recent physiological studies of fear conditioning mostly omitted this site of sensory integration and response potentiation upstream of the amygdala and auditory cortex. It is currently unknown if individual MGB neurons exhibit complex response dynamics upon adaptive associative and defensive behaviours and how this potential heterogeneity is balanced with reliable representations of sensory inputs from the environment. Furthermore, we are currently lacking a concept of the ensemble level activity and dynamics in this widely projecting thalamic auditory relay site, which is crucial to delineate the distributed population code underlying associative learning and adaptive defensive behaviours18,19.
Here we used a combination of deep brain Ca2+ imaging, miniature microscopy and fear conditioning in freely moving mice to reveal the response dynamics and plasticity of large populations of auditory thalamus neurons20,21,22. We find that individual auditory thalamus neurons exhibit mixed selectivity of CS and US responses with highly diverse plasticity patterns during associative learning, while the ensemble representation of auditory stimuli remains stable along learning. These findings suggest that auditory thalamus plays a role beyond a classic relay function by balancing experience-dependent plasticity with stable ensemble level representations of the sensory environment to support stable auditory perception with minimal affective bias.
Results
Deep brain imaging of auditory thalamus during fear conditioning
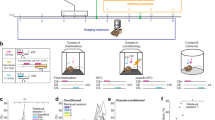
We established a gradient-index lens deep brain miniature microscope imaging approach of identified auditory thalamus neuronal populations in freely behaving mice (Fig. 1a, b)20,22. Using genetically-encoded Ca2+ sensors (Fig. 1c, AAV2/5.CaMKII.GCaMP6f)23, we tracked large populations of individual MGB neurons across a four-day auditory fear conditioning paradigm in freely moving mice. Similar to previous reports24, we found GABAergic fibres, typically originating in inferior colliculus and the thalamic reticular complex25,26, but virtually no GABAergic somata in MGB (Supplementary Fig. 1)27, indicating that we mainly imaged Ca2+ activity of thalamic relay neurons. We were able to follow 93 ± 4 GCaMP6f-expressing neurons per mouse (Fig. 1d, e, N = 24 mice) stably within and across sessions (see Methods). MGB neurons exhibited diverse, spontaneous activity patterns in freely moving animals (Fig. 1f) as well as cell-specific responses to pure tone auditory stimuli (Fig. 1g).

a Mouse with a head-mounted miniaturized microscope (left). Location of gradient refractive index (GRIN) lens in the medial geniculate body (MGB, right). b Example GCaMP6f expression in MGB. Similarly replicated expression patterns for all animals where GCaMP6f was injected in MGB (N = 24 mice). c High magnification of GCaMP6f-expressing MGB neurons from B. d Individual motion corrected fields of view (maximum intensity projection) of one example animal across a four-day fear conditioning paradigm (Hab, FC, Ext. 1, Ext. 2) as well as the maximum intensity projection across all days. Red circles indicate selected individual components. Replicated for all mice (N = 24) that underwent calcium imaging. e Average number of individual ICs/animal (93 ± 4 neurons, N = 24 mice). Boxplots show median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. f Changes in Ca2+ fluorescence of five individual neurons during the habituation session. Lines indicate CS tone presentations (red: 12 kHz, blue: 6 kHz). g Tone responses on habituation day 1 of all recorded MGB neurons in fear conditioning experiments (N = 855 neurons, N = 9 mice).
Next, we used a classic four-day fear conditioning and fear extinction paradigm28 (Fig. 2a, b), in which mice learn to associate a mild foot-shock unconditioned stimulus (US) with a predictive conditioned stimulus (CS+, 6 or 12 kHz pure tones, 200 ms pips, 27 pips per CS). After fear conditioning, mice exhibited enhanced freezing to the CS+ (61 ± 6%) when compared to a neutral CS− (42 ± 8%, N = 15, p < 0.01, Mann–Whitney test, Fig. 2b; Supplementary Fig. 2a), which extinguished upon repetitive CS+ presentation (Friedman test, p < 0.001, followed by Dunn’s multiple comparisons test, Extinction 1 early vs. Extinction 1 late p < 0.01, Extinction 1 early vs. Extinction 2 late p < 0.001, Extinction 2 early vs. Extinction 2 late p < 0.05). During fear conditioning, the total neuronal population (Fig. 2c) as well as individual MGB neurons (Fig. 2d, e) were strongly responsive to both the CS+ and the US. The proportion of US responsive neurons (75 ± 5%) was significantly higher than the proportion of CS+ (27 ± 3%) and CS− neurons (20 ± 2%, N = 9 mice, see “Methods” for classification of responsive neurons), while similar proportions of neurons were responsive to the CS+ and the CS− (Fig. 2f; Supplementary Fig. 2b, c, Friedman test, p < 0.001 followed by Dunn’s multiple comparisons test CS+ vs. CS− p > 0.05, CS+ vs. US p < 0.05, CS− vs. US p < 0.001). Furthermore, we found mixed selectivity in subpopulations of neurons that were responsive to combinations of tones and foot shocks, yet they were not enriched beyond chance level in the total population (Fig. 2g). These multisensory neurons were spatially intermingled in MGB and not locally clustered (Fig. 2h–j).

a Details of the 4-day fear conditioning paradigm. b Conditioned stimulus (CS) CS+ and CS− freezing (mean ± s.e.m.) during the habituation, fear conditioning as well as extinction days (Ext. 1, Ext. 2. e and l indicate early and late phases of extinction, i.e., the first four or last four CS+ of the session. Friedman test, p < 0.001, followed by Dunn’s multiple comparisons test, Ext.1e vs Ext.1 l p = 0.0069, Ext.1e vs Ext.2 l p = 0.0002, Ext.2e vs Ext.2 l p = 0.0281, N = 15 mice). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. c Population response of one example animal to the CS+ and US (unconditioned stimulus, mean ± s.e.m.). Blue dots indicate CS+ tone pips. Green bar indicates shock US. Example cell response to the CS+ (d) and US (e). Mean ± s.e.m. of five trials. Dots indicate CS+ tone pips. Inset represents average response to single pips. f Proportion of CS+, CS− and US responsive neurons. Friedman test, p < 0.001, followed by Dunn’s multiple comparisons test: CS+ vs. US, p = 0,029; CS− vs. US, p = 0.0005 (N = 9 mice). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. g Proportion of mixed selectivity CS± and US coding neurons. Red line indicates chance overlap level (N = 9 mice). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. h Example spatial map of unisensory and multisensory mixed selectivity CS and US coding neurons in MGB. i Relationship between within response group and across response group pairwise spatial distance between neurons (N = 855 cells, N = 9 mice). j Cumulative distribution function of pairwise distances between all, US-responsive, CS+ and CS− responsive neurons (N = 855 cells, N = 9 mice). k Mean Ca2+ activity (± s.e.m) of sound-correlated neurons during shock evoked sound events e.g., mouse escape sounds and low frequency harmonic vocalizations (LFH, orange), the first CS+ pip (blue) and the US (green) from N = 550 CS+/US or N = 4956 sound event trials from 110 cells out of N = 3 mice. *, **, *** indicate p values smaller than 0.05, 0.01 and 0.001, respectively.
To test if US responses are solely driven by movement of the animal or self-induced sounds, e.g., escape runs or low frequency harmonic vocalizations during the aversive foot shock, we correlated the activity of individual MGB neurons with movement speed or the occurrence of sounds in the context. First, a large proportion of MGB neurons exhibited an apparent correlation between movement speed and Ca2+ activity during the US. However, this is most likely due to the simultaneous occurrence and conflation of the 2 s aversive foot shock and the behavioural output (escape), given that the activity in the large majority of MGB neurons was not motion or speed correlated during the habituation period (Supplementary Fig. 3). In addition, US responses in MGB cannot be solely explained by the auditory environment, i.e., movement sounds or low frequency harmonic vocalization29,30 of the animal during the aversive foot shook, given that US and CS+ responses were typically substantially larger in sound correlated neurons than responses to self-evoked sounds of the animal (Fig. 2k; Supplementary Fig. 4). This indicates that MGB US responses are most likely driven by direct somatosensory input, pain signals or aversive state switches.
In summary, our data demonstrates that auditory thalamus neurons are strongly responsive to both pure auditory tones as well as aversive stimuli. This integration of CS and US inputs underlines that MGB neurons are ideal candidates for sensory plasticity upon associative learning9,17.
Neural response dynamics of MGB neurons upon fear conditioning
MGB neurons, particularly in the medial subdivision, have been shown to potentiate auditory CS responses upon fear learning17. However, their response diversity and dynamics on the population level upon associative learning remain unknown. To understand the learning-related dynamics of MGB neurons, we followed the activity of large populations of individual MGB neurons across the 4-day fear conditioning paradigm. Using a cluster analysis approach, we classified CS+ responsive neurons according to their response dynamics before and after fear conditioning and fear extinction (Fig. 3a, b). On the habituation day and on the two extinction days, we identified eight subgroups of CS+ responsive neurons. 7 ± 1% of cells show stable CS+ responses across days. The remainder could be separated in the following subgroups of plastic neurons: neurons that abolish their complete (21 ± 3%, CS down cells) or onset (9 ± 2%, on-down cells) CS+ response after fear conditioning, neurons with enhanced CS+ responses when the animal is in a high fear state (23 ± 5%, fear cells), neurons that are inhibited when the animal is in a high fear state (4 ± 1%, fear-down cells) as well as neurons that enhance or decrease their response when the animal extinguished the fear behaviour (14 ± 3%, extinction-up cells; 8 ± 2%, extinction-down cells). In addition, we identified cells that had stable, enhanced CS+ responses after fear learning (14 ± 4%, persistent cells, Fig. 3c). Similar subgroups were found for CS− responsive neurons. However, in contrast to the US-paired CS+, the group of CS− stable cells was most prominent across days (Supplementary Fig. 5a–c). In addition, we found similar proportions of CS+ (Supplementary Fig. 2b) and CS− responsive (Supplementary Fig. 2c) neurons during the habituation, fear conditioning and extinction days. However, the proportion of neurons that were plastic and changed their CS responses across days was significantly higher in the CS+ group (93 ± 1%) compared to the CS− group (60 ± 7%, 2-way ANOVA, p < 0.0001, followed by Tukey post hoc test, p < 0.05), while the proportion of stable neurons was higher in the CS− (40 ± 7%) compared to the CS+ group (7 ± 1%, 2-way ANOVA, p < 0.0001, followed by Tukey post hoc test, p < 0.05, Fig. 3d), indicating that neural response plasticity is learning-specific and more prominent for the paired conditioned stimulus than the control stimulus. To rule out that CS+ plasticity is not just a result of a general drift in tone responsiveness across days, we performed an unpaired conditioning paradigm where the foot shock is not temporally contingent with the tone CS+, such that the mice experience similar sensory stimuli to the fear conditioned animals but do not associate the CS+ with the US and fail to learn to freeze to the CS+ (Supplementary Fig. 6a, b). Unpaired conditioning did not affect the proportions of tone and foot shock responsive neurons in MGB (Supplementary Fig. 6c–h). However, compared to fear conditioned animals, the majority of neurons exhibited stable across-day CS+ tone responses in the unpaired condition (61 ± 9%), while the proportion of plastic neurons was significantly decreased (39 ± 9%, N = 5 mice, Fig. 3e). This data indicates that MGB single cell tone responses are by-and-large stable across days, while CS+ response dynamics of individual neurons after fear conditioning are due to associative learning-induced plasticity and not just a general drift of tone-responsiveness in MGB across days. Notably, for the minority of MGB neurons that exhibited CS+ plasticity, the relative proportion of fear cells is significantly decreased (Supplementary Fig. 6f) when compared to fear conditioned animals, indicating that the emergence of this population of neurons with potentiated CS+ responses is specific to auditory fear learning.

a Heat map of single cell CS+ responses on the habituation, extinction 1 and extinction 2 days. Cells were clustered into groups depending on their CS+ response pattern (N = 386 cells, N = 9 mice). b Average traces ± s.e.m of neuronal clusters in (a). c Proportion of individual plasticity groups within CS+ responsive cells/animal (Kruskal-Wallis test, p < 0.05, followed by Dunn’s multiple comparisons test; Stable vs cs down, p = 0.013, cs down vs fear down, p = 0.0003, cs down vs extinction down, p = 0. 035, fear vs fear down p = 0.0041; N = 9 mice). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. d Proportion of CS+ and CS− stable and plastic neurons (2-way ANOVA followed by Sidak multiple comparisons test, p < 0.001; stable CS− vs. stable CS+, p = 0.002; stable CS− vs. plastic CS+, p < 0.001; stable CS+ vs. plastic CS−, p < 0.001; stable CS+ vs. plastic CS+, p < 0.001; plastic CS− vs. plastic CS+, p = 0.002, N = 9 mice, data presented as mean values ± s.e.m.). Circles represent individual animals. e Proportion of CS+ stable and plastic neurons in a pseudoconditioning paradigm compared to fear conditioned animals (2-way ANOVA followed by Sidak multiple comparisons test, p < 0.001; stable unpaired vs plastic unpaired, p = 0.019; stable paired vs. plastic paired, p < 0.001; stable unpaired vs. stable paired, p < 0.001; plastic unpaired vs. plastic paired, p < 0.001, Unpaired group N = 5 mice, Paired group N = 9 mice, data presented as mean values ± s.e.m.). Circles represent individual animals. f Experimental paradigm. Auditory tuning was tested before and after fear conditioning. g Population response to individual 200 ms pips before (black) and after (orange) fear conditioning (mean ± s.e.m, N = 681 cells, N = 7 mice). h Proportion of tone-responsive cells before and after fear conditioning (Two-tailed, Wilcoxon signed-rank test, p = 0.0156, N = 7 mice, data presented as mean values ± s.e.m.). Lines represent individual animals. i Mean auditory responses ± s.e.m of one example neuron before (black) and after (orange) fear conditioning to different tone frequencies (numbers indicate kHz). CS+ : 8 kHz, CS−: 20 kHz. j Population statistics for BF tuning towards the CS+ (two-tailed Wilcoxon signed-rank test, p < 0.001, n = 284 neurons, 7 mice). Horizontal lines represent median. k Population statistics for BF tuning towards the CS− (two-tailed Wilcoxon signed-rank test, p > 0.05, N = 284 neurons, 7 mice). Horizontal lines represent median. l Heat maps of single cell US responses to the five US stimulations during the fear conditioning day (N = 634 cells, N = 9 mice). m Average response ± s.e.m of plasticity subtypes of US-responsive MGB neurons (N = 634 cells, N = 9 mice, see also Supplementary Fig. 3). n Proportion of individual plasticity groups within US responsive cells/animal (Kruskal-Wallis test, p < 0.01 followed by Dunn’s multiple comparisons test; Stable vs down, p = 0.043; down vs off-up, p = 0.0006; down vs. off-down, p < 0.001; down vs. inh, p = 0.0055; off-down vs inh type 1, p = 0.018; N = 9 mice). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. o Proportion of US (N = 5 mice, p > 0.05, Wilcoxon signed-rank test) and CS plastic cells (N = 5 mice, p > 0.05, two-tailed Wilcoxon signed-rank test) in the ventral (MGBv) and medial (MGBm) subdivisions of MGB. Insert: Schematic of GRIN lens location above the different MGB subdivisions. *, **, *** indicate P values smaller than 0.05, 0.01 and 0.001, respectively.
All-in-all, this data reveals a broad response diversity of MGB neurons upon fear learning that extends previous observations of fear conditioning potentiated neurons15,16,31. The plastic CS+ subgroups are similar to previously described functional groups in the amygdala21,22,28,32. While fear and extinction neurons are the most prominent groups, they appear alongside other distinct subgroups, indicating that diverse CS+ response plasticity occurs not only in the amygdala, but also upstream in auditory thalamus.
MGB neurons are diversely tuned to auditory frequencies9 and individual neurons were reported to change their frequency tuning upon associative learning11,12,31. To estimate changes in auditory frequency tuning in large populations of individual MGB neurons before and after fear conditioning, we presented 200 ms pips of at least eleven different pure tone frequencies (1–40 kHz) at 65–85 dB to freely moving mice while simultaneously imaging MGB Ca2+ activity (Fig. 3f). We found that the mean pip response of the MGB population across all frequencies was nearly doubled after fear conditioning (Fig. 3g). Furthermore, the proportion of tone responsive neurons was increased post conditioning (Fig. 3h, before: 22 ± 7%, after: 37 ± 6%, Wilcoxon signed-rank test, p < 0.05, N = 7 mice). Besides a general enhancement of pip responses across the whole population and all tones, we found that fear conditioning induces a specific enhancement of selective frequencies compared to the pre-conditioning state (Fig. 3i), which resulted in a shift of the best frequency towards the conditioned stimulus (Fig. 3j, pre FC: | Best frequency - CS+ | = 11 ± 0.45 kHz, post FC: | Best frequency - CS+ | = 8 ± 0.39 kHz, Wilcoxon signed-rank test, p < 0.001, N = 284 cells from N = 7 mice). This shift was specific for the CS+ and did not occur for the CS− (Fig. 3k, p > 0.05, Wilcoxon signed-rank test). This data indicates that auditory fear conditioning affects the auditory frequency tuning of MGB neurons in a stimulus specific manner. However, the absolute shift across the population is small (ca. 3 kHz), suggesting that MGB preserves a broad tuning range for reliable sensory representation, despite a stronger representation of the CS+.
In addition to plasticity of CS tone representations, we found that US responsive neurons can be subdivided into stable (8 ± 1%) and plastic cells (92 ± 1%) during fear conditioning. The majority of US responsive cells was plastic and exhibited dynamic intra-session representations of the US (Mann–Whitney test, p < 0.001). Using a similar cluster analysis approach, we identified neurons that demonstrated intra-session potentiation or depression and subtypes of inhibited as well as off-responsive neurons that potentially signal relief from the shock (Fig. 3l–n). However, despite its prominent diversity, the US response type was not predictive of CS plasticity in MGB neurons. US response and plasticity type did not overlap with CS response and plasticity type above chance levels (Supplementary Fig. 7), indicating that US inputs per se do not drive plasticity in MGB neurons. Nevertheless, adaptive US responses in MGB could act as an upstream teaching signal in addition to local circuit mechanisms33, which direct plasticity in downstream areas like the amygdala.
MGB is subdivided into a first order, auditory cortex-projecting nucleus (MGBv) as well as higher order nuclei (MGBd, MGBm), which send axons to cortical and limbic brain areas, e.g., the amygdala (Doron and LeDoux, 1999). To test if MGB CS and US plasticity types are different between first order and higher order nuclei, we subdivided the cells depending on their location in the GRIN lens field of view between MGBv and MGBm after anatomical verification of the lens front location for all mice where MGBv and MGBm were simultaneously imaged (N = 5 mice, Supplementary Fig. 8a, b). Similar to the total population of MGB neurons, large fractions of MGBv and MGBm neurons exhibited plasticity to the US or CS+ (Fig. 3o). Nevertheless, plastic cells were not significantly different between either subdivision (Fig. 3o) and the diversity of plasticity subtypes was similar in the first order (MGBv) vs. higher order (MGBm) area of auditory thalamus (Supplementary Fig. 8c–e).
Overall, we found that responses of individual MGB neurons to the CS and US are plastic upon fear conditioning. In addition to previously reported potentiated auditory neurons, we find highly diverse subtypes of CS or US plastic neurons that go beyond FC-driven response potentiation and are distributed similarly across both first order and higher order MGB subdivisions. US response subtypes were not predictive of CS plasticity, nor were the proportions of subgroups or their enrichment predictive of behavioural outcomes on an animal-by-animal basis (Supplementary Table 1), indicating that MGB neurons might play diverse roles in guiding memory formation during associative learning.
Amygdala-projecting MGB neurons are plastic upon associative fear learning
Higher order MGBm neurons project to different output targets including primary and secondary auditory cortex, striatum and the basolateral amygdala34,35,36,37. Enrichment of plastic neurons in the MGB → BLA pathway might be crucial for fear learning, given BLA’s key role in aversive memory formation38. Similar to previous reports39, we found BLA-projecting MGB neurons to be typically located in higher order MGB areas and particularly enriched in the medial subdivision of MGB (Fig. 4a, b, MGBm, 70 ± 7 vs. 23 ± 6% dorsal, MGBd or 7 ± 3% ventral, MGBv). By-and-large, 30% of MGBm and 10% of MGBd neurons were amygdala-projecting (Fig. 4c). In contrast, only a small fraction of MGBv neurons (2%) were retrogradely traced from the amygdala. Furthermore, 85 ± 2% of amygdala-projecting MGB neurons (Fig. 4d; Supplementary Fig. 9a, N = 6 mice) were positive for the higher-order MGB area marker calretinin40, suggesting that calretinin is a highly prevalent but not exclusive marker of BLA-projecting MGB neurons.

a Injection of AAV2-retro.hSyn1.mCherry.WPRE.hGHp(A) and latex beads in the basolateral amygdala (BLA). MGB was counterstained for calretinin (cyan) and NeuN (yellow) to quantify the BLA projectors (red). b Distribution of BLA-projecting neurons within MGB (N = 6 mice, Friedman test p < 0.001, followed by Dunn’s multiple comparisons test %MGBm vs. %MGBv, p = 0.0016). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. c Region-specific proportion of BLA-projecting neurons within MGB subdivisions (N = 4 mice, Friedman test, p < 0.01, followed by Dunn’s multiple comparisons test, %MGBm vs. %MGBv, p = 0.014). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. d Proportion of calretinin-positive BLA-projecting neurons (N = 4 mice). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. e Schematic of viral strategy and location of GRIN lens in MGB to image neuronal activity of MGB → BLA-projecting neurons. f MGB field of view with MGB → BLA-projecting neurons. Replicated in all animals that underwent calcium imaging (N = 6 mice). g Number of identified individual components per animal (69 ± 9, N = 6 mice). h Mean ± s.e.m population response of one example animal to the CS+ and CS−. Black dots indicate CS + tone pips. Bar indicates shock US. Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. i Proportion of CS + , CS− and US responsive neurons for the total MGB population and amygdala-projecting neurons (2-way ANOVA, main effect group, F(1,13) = 3.3, p > 0.05, N = 9 total MGB population mice and N = 6 MGB → BLA projection neurons mice, see also Fig. 2f). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. j Proportion of mixed selectivity CS± and US coding neurons for the total MGB population and amygdala projecting neurons 2-way ANOVA, F(1,13) = 3.9, p > 0.05, N = 9 mice for the total MGB population and N = 6 mice for the population of MGB → BLA projection neurons, see also Fig. 2. Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. g Dotted lines indicate chance overlap level. k Examples traces of groups of stable, onset down, fear and extinction neurons. l Proportion of individual plasticity groups within CS + responsive cells / animal (2-way ANOVA, F(1,13) = 1.2, p > 0.05, N = 9 mice for the total MGB population and N = 6 mice for the population of MGB → BLA projection neurons). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. m Proportion of individual plasticity groups within US responsive cells / animal (2-way ANOVA, F(1,13) = 0.5, p > 0.05, N = 9 mice for the total MGB population and N = 6 mice for the population of MGB → BLA projection neurons). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. ** indicate p values smaller than 0.01.
To test the physiological function and neuronal activity of amygdala-projecting MGB neurons in fear learning, we used a retrograde virus approach to specifically express GCaMP6f in MGB → BLA-projectors (see “Methods”, Fig. 4e; Supplementary Fig. 9b). On average, we could identify 69 ± 9 BLA-projecting GCaMP6f-positive MGB neurons per mouse (Fig. 4f, g, N = 6 mice). Similar to the total MGB population average response (Fig. 2c), MGB → BLA projectors were activated by the CS+ and the US (Fig. 4h). Across animals, 22 ± 5%, 10 ± 2% or 68 ± 7% of neurons were responsive to the CS+, CS− or US, respectively. These proportions are comparable to the total MGB population (Figs. 4i, 2-way ANOVA, p > 0.05). Furthermore, we did not find that combinations of CS+, CS− and US coding neurons were enriched above chance levels (Fig. 4j). Using a cluster analysis approach, we found the same subgroups of CS+ plasticity types in the subpopulation of MGB → BLA projecting neurons (Fig. 4k, l) across the conditioning paradigm, including stable cells, onset-down cells, CS-down cells, fear cells, fear-inhibited cells, extinction up cells, extinction down cells as well as persistent cells (Supplementary Fig. 9c–e). The proportions of the CS+ plasticity subgroups were similar to the total population in MGB (2-way ANOVA, p > 0.05). Analogous to the CS+ representation across days, we found comparable proportions of US plasticity types in MGB → BLA projectors when compared to the total population (Fig. 4m; Supplementary Fig. 9f, 2-way ANOVA, p > 0.05).
This data demonstrates that CS as well as US information is encoded by BLA-projecting MGB neurons, identifying MGB → BLA projectors not only as a source of CS tone inputs but also as a strong source of aversive US signals (see also9). However, CS and US plasticity is functionally diverse beyond response potentiation and, compared to the total MGB population, CS and US signalling is not enriched in this specific subpopulation of amygdala-projecting MGB neurons.
Activity in the MGB → BLA pathway is necessary for fear memory consolidation and stabilizes MGB plasticity
To test if activity in MGB → BLA projection neurons is necessary for fear learning8,41,42,43,44 and MGB plasticity, we specifically expressed the inhibitory opsin ArchT in MGB → BLA projection neurons (Fig. 5a, b, Supplementary Fig. 10a-e). Inhibition of MGB → BLA projectors during CS-US pairing on the conditioning day (Fig. 5c) had no effect on fear acquisition (Fig. 5d, mean freezing during the last two CS+ presentations, GFP: 65 ± 4%, N = 13 mice; MGB → BLA ArchT: 60 ± 6%, N = 9 mice; p > 0.05, Mann–Whitney test, Supplementary Fig. 10f). However, freezing levels were significantly reduced during the fear test 24 h later (Fig. 5e, mean freezing during the first four CS+, GFP: 48 ± 6%, N = 13 mice; MGB → BLA ArchT: 22 ± 5%, N = 9 mice; p < 0.01, Mann–Whitney test), indicating that activity in MGB → BLA projectors is necessary for the consolidation of fear memories. In addition, and similar to amygdala-projecting MGB neurons (Fig. 5a–e), we found that inhibition of neuronal activity in the total MGB population (Supplementary Fig. 10f–i) or in the first order ventral nucleus of MGB (Supplementary Fig. 10j) had no effect on fear acquisition during the conditioning session but supressed fear memory consolidation.

a Optogenetic approach to inhibit MGB → BLA projection neurons. b Example ArchT expression in MGB → BLA projection neurons. Replicated for all animals that underwent optogenetic inhibition of MGB → BLA neurons (N = 9 mice). c Experimental paradigm: MGB → BLA neurons are manipulated during the CS+ and US on the fear conditioning day. d Freezing of GFP and MGB → BLA ArchT-expressing animals at the end of the fear conditioning paradigm (mean freezing levels to the last two CS+, GFP: N = 13 mice, MGB → BLA ArchT: N = 9 mice, p > 0.05, two-tailed Mann–Whitney test). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. e Freezing of GFP and ArchT-expressing animals upon fear recall during early extinction 1 (Ext. 1, mean freezing during the first four CS+, GFP: N = 13 mice, MGB → BLA ArchT: N = 9 mice, two-tailed Mann–Whitney test, p = 0.0056). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. f All-optical approach to inhibit MGB → BLA projection neurons during simultaneous recording of total MGB population activity. g Example GCaMP6f expression in MGB (top) and ArchT expression in MGB → BLA projection neurons (bottom). Replicated for all animals that underwent the all-optical paradigm (N = 6 mice). h Mean MGB population activity ± s.e.m in response to CS+ and US stimuli upon optogenetic light presentation of tdTomato (tdTom, black, N = 5 mice) and ArchT animals (orange, N = 6 mice). i Proportion of MGB neurons with stable and plastic CS+ responses after fear conditioning in tdTom (N = 5) and ArchT (N = 6) mice (2-way ANOVA, F(1, 9) = 10.09, p < 0.05, Sidak’s multiple comparisons test, p = 0.0104 each). Boxplots represent median, 2nd, 3rd quartile, minimum and maximum. Cross indicates mean. *, ** indicate p values smaller than 0.05 and 0.01, respectively.
To test how activity in MGB → BLA neurons during fear learning affects across-day plasticity of the total MGB population, we used an all-optical miniaturized microscope imaging approach (Fig. 5f). We specifically expressed ArchT or tdTomato in MGB → BLA neurons (Fig. 5g) and suppressed their activity during the CS-US pairings on the fear conditioning day (Fig. 5c) while simultaneously imaging the activity of the total MGB population (CaMKII-driven GCaMP6f). Given that MGB → BLA neurons represent only a fraction of the total MGB neuronal population (ca. 11 ± 2% in total MGB, N = 4 mice, see also Fig. 4c), inhibiting MGB → BLA neurons during fear conditioning did not affect the mean CS+ and US response of the total imaged MGB population (Fig. 5h and Supplementary Fig. 11a–e). However, when comparing the CS+ plasticity across fear learning we find that inhibition of MGB → BLA neurons during fear conditioning enhances the proportion of plastic neurons after fear consolidation (Fig. 5i, stable neurons: tdTom = 27 ± 6%, ArchT = 7 ± 2%; plastic: tdTom = 73 ± 6%, ArchT = 93 ± 2%; 2-way ANOVA, F(1, 9) = 10.09, p < 0.05, Sidak’s multiple comparisons test, p < 0.05 for stable and plastic neurons; tdTom, N = 5, ArchT, N = 6 mice). Thus, suppression of activity in MGB → BLA circuits leads to a facilitation of plasticity in MGB–potentially as a compensation mechanism–suggesting that feedforward signalling from MGB to BLA during associative fear learning is crucial to stabilize plasticity in auditory thalamus.
Population coding and representation of the conditioned stimulus across days
Next, we tested if the CS+ and CS− can be decoded from Ca2+ activity based on MGB population activity. First, we trained a three-way quadratic decoder to distinguish between CS+, CS− and baseline activity within the same session (see “Methods”). To balance for different cell population sizes between animals, we randomly sub-selected 40 cells for each animal and averaged decoder accuracy across 50 independent runs. Furthermore, to account for different numbers of CS+ and CS− presentations, we only decoded the first four CS+ and CS− presentations. Within each individual session, the decoders achieved high classification accuracy (Fig. 6a, >80% compared to 33% chance level) indicating a distinct representation of the individual CSs by the MGB population. Surprisingly, decoding accuracy was higher in the population of MGB → BLA projectors compared to the total MGB population, except for the fear conditioning day. To test if CS tones can be accurately detected across days from MGB population activity, we next trained sets of two-way decoders to distinguish between baseline and CS+ or CS− responses for each experimental day and tested the trained decoders across days (Fig. 6b). Strikingly, we found that decoder accuracy is robust across days reaching decoding levels of ca. 70% or higher, for both the CS+ and the CS− (Fig. 6c). This is in contrast to amygdala population coding, where decoding levels for the CS+ drop to chance levels after fear conditioning21, indicating that MGB population representations of CS tones are stable despite associative learning. Furthermore, we found a drop in CS+ encoding in MGB → BLA projectors during the fear conditioning day, which recovered afterwards (Fig. 6c, see also Fig. 6a), indicating temporary changes in CS+ encoding during associative learning.

a Intraday three-way decoder of CS+, CS− and baseline population responses in CaMKII-positive (black) and identified amygdala-projecting MGB neurons (turquoise) reached a minimum mean accuracy of 81% across animals. Decoder accuracy dropped to chance levels for decoders trained on randomly label training sets (data presented as mean values ± s.e.m.). b Intra- and across day accuracy of decoders trained on CS+ or CS− vs. baseline responses, respectively. 1: Hab, 2: FC, 3: Ext. 1, 4: Ext. 2. c Quantification of intra and across day decoder accuracy for decoders trained on habituation day data. Mean decoder accuracy across days is >70% for CS+ and CS− population responses in CaMKII-positive and identified amygdala-projecting MGB neurons (MGB→BLA N = 6 mice, MGB N = 9 mice, data represent mean ± s.e.m.). Relative change in Euclidean population vector distance between the CS+ (d, e) or CS− (f, g) and the US within the fear conditioning session (d, f) or across the individual days of the behavioural paradigm (e, g). Statistics: d: Friedman test across the relative change in CS+ to US PVD of MGB → BLA-projectors (p < 0.01), Dunn-Sidak multiple comparisons test 1st and 3rd vs. 5th CS/US pairing, p < 0.05. G: Friedman test across the relative change in CS− to US PVD of the total BLA-population (p < 0.05), Dunn-Sidak multiple comparisons test FC vs. Ext.2: p < 0.05. All other data sets in d-g: p > 0.05. MGB population: N = 9 mice, MGB → BLA-projectors: N = 6 mice (data represent mean ± s.e.m.). h Relative change in PVD between the CS and US for Control and ArchT (2-way ANOVA followed by Sidak’s post hoc test, p < 0.005, 5th CS+ presentation Control vs. ArchT, p = 0.0438, Control N = 5 mice, ArchT N = 6 mice, data represent mean ± s.e.m.).
Finally, we compared the population vector distance (PVD) between the evoked population responses to the CS+ and CS− and the evoked population responses to the US (Fig. 6d–g). During fear conditioning, we found a decrease of the PVD between the CS+ and the US with consecutive CS-US pairings (Fig. 6d) for both the total MGB population as well as for MGB → BLA-projectors. However, the time courses of the PVD change were different and only the MGB → BLA population reached a significant change in PVD at the end of the session indicating different population dynamics for this subgroup of MGB neurons during associative learning (Friedman test, p < 0.01, 1st and 3rd versus 5th pairing: Dunn-Sidak multiple comparisons test, p < 0.05). Importantly, no changes were found between the evoked responses to the CS− and US during conditioning. In contrast to previous observations in the amygdala21, the PVD between the CS+ and the US changes were not preserved post conditioning on the extinction days and the population representation recovered to pre-conditioning levels similar to the observations using two-way or three-way decoders (Fig. 6a, c). The same was observed for the across-day population vector distance of the CS+ to itself (Supplementary Fig. 12). However, note that we found a significant difference between the CS− to US PVD of the total MGB population between the conditioning day and the second extinction day (FC versus Ext. 2: Friedman test, p < 0.05, Dunn-Sidak multiple comparisons test, p < 0.05). Nevertheless, we did not find a change in the CS− to US PVD between the habituation, fear conditioning and early extinction days indicating that the CS− representation is stable during high fear states in relation to the habituation day and shows no significant drift intraday during fear conditioning. Furthermore, the CS− drifts further away from the US in comparison to the habituation day which might be reflective of an enhanced safety signal after extinction.
In addition to the general lack of consolidation of population level changes across fear learning, the strength of PVD-changes between the CS+ and US were not predictive of learned freezing behaviour on an animal-by-animal basis (Supplementary Fig. 13).
Finally, using an all-optical approach to inhibit MGB → BLA projectors during fear learning (see Fig. 5), we find that the suppression of activity in MGB → BLA leads to a reduced shift in the population vector distance between the CS+ and the US on the fear conditioning day (Fig. 6h, 5th pairing, relative change control: −30 ± 13%, ArchT: −2 ± 3%, 2-way ANOVA, p < 0.05, Sidak multiple comparisons test, p < 0.05), which might be crucial for downstream population plasticity in BLA. However, suppression of MGB → BLA activity did not affect the post-conditioning CS-US PVD-change reset (Supplementary Fig. 11f–h).
Taken together, this data indicates that high dimensional representations of CS+ tones are stably encoded in MGB populations across associative fear learning, despite plastic changes in single cell response patterns. In contrast to the basolateral amygdala21, MGB population representations of sensory stimuli only transiently change during associative fear learning and reset overnight, which might be crucial for unbiased representations of stimuli from the environment.
Discussion
By imaging large populations of MGB neurons, we find that auditory thalamus is a site of diverse neuronal plasticity during associative fear learning on the level of single cells as well as the total MGB population. However, changes in MGB population level coding are only transient and do not consolidate overnight, which might be instructive for plastic changes in downstream structures during learning, yet allows for long-term stability of sensory coding across days.
On the level of individual MGB neurons, we observed associative learning-induced CS+ response potentiation that resembles classic studies which demonstrated that auditory thalamus exhibits enhanced responses to aversive conditioned tones14,15,16,45. However, recording simultaneously from large populations of individual neurons in MGB during fear conditioning, we find diverse plasticity patterns (at least 7) that are similar or go beyond previously reported plasticity types in cortical46 or limbic areas21,22,28,32 downstream of MGB as well as calretinin-positive subpopulations of lateral thalamus neurons44. Changes in CS responsiveness of individual MGB neurons upon fear learning and extinction were bidirectional, i.e., potentiated or depressed, and depended on the behavioural state of the animal. For example, we find different functional types that are enhanced or depressed particularly in high fear or extinction states. This extends the notion of unidirectional response potentiation in MGB upon associative learning and demonstrates that auditory thalamus neurons exhibit heterogeneous, adaptive signalling of threat-predicting auditory stimuli of the environment. Plasticity in MGB is specific to auditory fear conditioning and not just a general drift of single cell responses across days. However, by-and-large, the proportion of individual plasticity subtypes was not correlated with the strength of post-conditioning freezing or discrimination levels47 of the animals (Supplementary Table 1, Supplementary Fig. 13b), suggesting that individual functional neuronal types in MGB are per se not predictive of an animals’ behavioural output. Nevertheless, the heterogeneity of the neuronal response plasticity might be necessary to enhance the computational capacity and memory specificity of MGB48,49,50,51. Furthermore, precise behavioural outputs might be orchestrated by heterogenous and complex activity in MGB in coordination with a distributed downstream network. For example, MGB neurons project onto amygdala principal neurons and several amygdala interneuron subclasses33,44,52,53,54,55, suggesting that the diversity of MGB plasticity is tuned to the precise MGB → BLA connectivity. This in turn would be crucial for the specific activation of distinct amygdala circuits and amygdala memory engrams to recruit input specific behavioural outputs44,56,57,58.
Besides auditory stimuli, MGB neurons signal the aversive foot shock (US) during fear conditioning9,15. Indeed, we found that the proportion of foot shock encoding neurons exceeds the number of tone CS+ encoding neurons in auditory thalamus of freely moving animals. The large proportion of US encoding neurons in MGB and the strength of the US signal on the population level could not be explained by movement of the animal or self-vocalization-induced activation of MGB59,60. Aversive US responses are considered to be more prominent in higher order auditory thalamus (MGBm)9. Our imaging sites covered both first order (MGBv) and higher-order (MGBm) MGB, and US encoding neurons were equally present in both sites. This demonstrates that multisensory encoding is not an exclusive feature of higher-order areas of MGB in freely moving animals, but can also occur in the first order ventral subdivision that projects to auditory cortex, suggesting that auditory thalamus conveys aversive US information to a broad range of cortical and limbic downstream areas during associative fear learning. Strikingly, US responses were heterogeneous across the population of MGB neurons during fear conditioning. Besides stable US responders, we identified several plasticity types of US responsive neurons, including short term facilitating, depressing or off-responsive neurons. This functional diversity of US neurons indicates that first and higher order auditory thalamus can signal distinct types of instructive information, for example adaptive teaching signals for associative fear learning as well as relief or safety signals upon termination of the US. Future studies need to address if and how these non-uniform adaptive MGB US signals are relayed to specific circuits elements in downstream areas like the amygdala or auditory cortex55,56,57,61,62,63,64,65,66,67.
CS and US coding neurons are spatially intermingled in auditory thalamus and a large fraction of MGB neurons exhibit mixed selectivity for both, the CS tone and US foot shock. The convergence of diverse CS and US responses in individual MGB neurons renders auditory thalamus an ideal site for neuronal plasticity in associative learning9,17, which is supported by the finding of large numbers of different subgroups of plastic neurons upon fear conditioning. Nevertheless, similar to observations downstream in the basolateral amygdala21,22, the convergence of CS and US responses in MGB neurons was not predictive of the response plasticity of a given neuron. Instead, MGB neurons exhibit manifold functional classes and outcomes of CS/US conversion upon learning (e.g., CS/US responsive cells can become potentiated fear cells or CS down cells), suggesting that heterogeneous fear conditioning-induced auditory response plasticity in MGB is most likely governed by multiple cellular or circuit mechanisms of neuronal plasticity. Indeed, we find subsets of neurons in all groups of CS plastic neurons that were not US responsive during FC (see Supplementary Fig. 7). Converging subthreshold CS and US inputs in dendrites, which cannot be detected by extracellular measurement of somatic neuronal activity during fear conditioning, yet might induce local dendritic plasticity mechanisms68,69,70,71, could be a potential source of CS response adaptation. Alternatively, subsets of MGB neurons might not require converging CS and US input15 to drive functional plasticity upon associative learning, arguing for additional plasticity mechanisms that go beyond classical Hebbian plasticity and coincidence detection on a millisecond timescale72, and might additionally involve slower, neuromodulatory mechanisms73,74,75 similar to amygdala circuits21,76 or consolidation during sleep14.
Furthermore, brain-wide distributed interacting circuit mechanisms could play a role in the formation of single cell plasticity upon associative fear learning, not only in MGB but across multiple fear-related brain areas16,28,77,78,79,80,81,82. The detailed computations within this distributed network19 and the role of auditory thalamus are poorly understood. Plasticity and adaptive changes in MGB depend on uni- or multi-synapse feedback circuits from distinct brain areas including the amygdala16,83 and cortex84. Nevertheless, our data supports the notion that learning-induced modifications of neuronal activity in MGB could drive plastic neuronal responses in downstream areas17. This suggests that at least a part of the heterogeneous response plasticity in amygdala or cortex during associative fear learning could be inherited in a feedforward fashion from adaptive changes in the thalamic relay independent of and in addition to local synaptic and circuit mechanisms33,61,63.
Both, first order auditory cortex-projecting MGBv as well as higher order areas of MGB (including calretinin-positive lateral thalamus→BLA neurons44) are necessary for memory consolidation and we found similar proportions of CS and US plastic neurons in both subregions of MGB. Auditory cortex is involved in the formation of fear memories to complex sound stimuli46 and plasticity in auditory cortex-projecting MGBv neurons might play an instructive role. Higher order areas of MGB are more broadly-projecting areas of auditory thalamus and target among others auditory cortex, striatum and amygdala34,35,37,44,85,86. Given the amygdala’s prominent role in associative fear learning and our finding that activity in the MGB → BLA pathways is crucial for fear memory consolidation (note that we did not find a recently observed effect of MGB → BLA calretinin-positive neurons on fear memory acquisition on the conditioning day44, which might due to differences in the strength of the conditioning protocols), we hypothesized that CS and US plastic neurons are specifically enriched in MGB → BLA projection neurons when compared to the total population including less plastic MGBv neurons11,12,31. Using a retrograde viral approach, we specifically imaged BLA-projecting MGB neurons to distinguish these cells from the general population. Surprisingly, we found that the proportion of plastic neurons was not enhanced in amygdala-projecting neurons and was similar to the proportion of plastic neurons in the total MGB population. This lack of enrichment of neurons with dedicated functions in associative learning suggests that the MGB → BLA pathway is most likely not a labelled line, but that MGB potentially propagates experience-dependent changes of neuronal activity in associative fear learning to a wider brain network, including auditory cortex and striatum34. This is reminiscent of recent findings showing that heterogenous behaviour-related neural activity of projection neurons of a given brain area is broadcast simultaneously and in parallel to different downstream targets irrespective of the output pathway22,87. Interestingly, this broadcasting might be achieved by the same subsets of individual MGB neurons given that higher order MGB → BLA neurons also project to auditory cortex (Supplementary Fig. 14)44,86. Finally, perturbing activity in the MGB → BLA feedforward pathway during fear conditioning led to enhanced, aberrant across-day single cell plasticity patterns in the total MGB population after fear conditioning. MGB “over-plasticity” could be a compensatory mechanism due to the lack of amygdala activity and polysynaptic amygdala feedback to MGB, e.g., via the reticular nucleus of the thalamus83, which might be crucial to fine-tune single cell plasticity in MGB, further supporting the notion that distributed activity across brain areas and networks is necessary to stabilize neuronal plasticity and facilitate precise memory formation as well as behavioural output16,18,19,88. Future work including simultaneous multi-site recordings89, targeted activity-dependent neural manipulations90 and computational neuroscience tools will be required to delineate how this heterogeneous, widely distributed population code is established, subsequently interpreted by different downstream regions18,19 and related to complex behavioural output.
Locally, on the level of the auditory thalamus, CS responses could be decoded reliably from the population level responses of the total MGB ensemble. Within a given day of the fear conditioning paradigm, we could train decoders that reliably distinguish between CS+, CS− or baseline activity with high accuracy. Strikingly, we could also train decoders that accurately classified baseline vs. CS+ or CS− presentations across all experimental sessions and along associative fear learning. This suggests that MGB ensembles exhibit stable population level tone representation across days, despite the plasticity of CS responses of individual cells during fear conditioning, which can be stable over weeks45. This data is supported by the observation that the MGB population vector difference between the CS+ and US decreases during fear conditioning, yet recovers to baseline levels on the next day after the conditioning session. Thus, MGB exhibits stable tone representations on the population level across associative learning, which will be crucial for reliable representations of sensory stimuli from the environment, for example, in light of changing stimulus statistics49 in complex environments and plastic single cell responses (see above) or behaviour-driven changes in response amplitudes91. This is in stark contrast to fear-biased population level changes in sensory representation in the amygdala that are further stabilized and consolidated after learning, and prevent the decoding of tone responses across fear conditioning21. While the population code of the amygdala stabilizes “fear hi-jacking” of the sensory representation, MGB exhibits transient changes in population level encoding, which provides a clean slate for future perception that is unaffected by a valence bias. Nevertheless, the transient population level changes during fear conditioning in MGB, which are dependent on MGB → BLA projection neuron activity, might be crucial to guide long term population level changes in the amygdala or other downstream areas upon associative learning21,34,92.
Taken together, our data indicates that auditory thalamus is ideally positioned to exhibit a complex role in guiding neuronal plasticity and valence assignment during associative learning that goes beyond the classical role of auditory processing and response potentiation during conditioning and potentially extrapolates to a broad set of behavioural functions93. Delineating the neural circuit mechanisms that underlie these highly dynamic representations of uni- and multisensory stimuli in MGB and their experience-dependent plasticity will open new avenues to understand the role of early, pre-cortical sensory relays like auditory thalamus in the formation of sensory percepts and memories that mediate complex behaviours.
Methods
Animals
In total, 8 to 11-week-old C57Bl/6JRj mice were used throughout the study. Behavioural experiments were performed with male mice. All experiments were done in accordance with institutional guidelines (University of Basel, Tierschutz) and were approved by the Cantonal Veterinary Office of Basel-Stadt, Switzerland. Animals were housed on a 12-h light / dark cycle at an ambient mean temperature and humidity of 22 C and 55%, respectively. Food and water were provided ad libitum.
Surgeries, virus injection and GRIN lens as well as optical fibre implantation
Virus was injected with the help of a stereotaxic apparatus (Kopf Instruments) in the medial geniculate body (for imaging experiments 500 nl, AAV2/5.CaMKII.GCaMP6f.WPRE.SV40, Penn Vector Core, for optogenetic experiments 500 nl AAV2/5.CAG.flex.ArchT.GFP, UNC Vector Core, AAV2/5.CaMKII.EGFP.WPRE.hGHp(A), VVF Zürich, AAV2/5.CaMKII.eArchT3.0.2 A.EGFP.WPRE, VVF Zurich or AAV2/5.hSyn.dlox.EGFP.dlox.WPRE, VVF Zurich, for all-optical experiments 500 nl, AAV2/5.CAG.flex.ArchT.tdTomato, UNC Vector Core or AAV2/5.CAG.dlox.tdTomato.dlox.WPRE, VVF Zurich, for tracing experiments 500 nl, AAV2/5.hSyn.flex.axonGCaMP6s94, Addgene; coordinates: AP: −3.28, ML: −1.9, DV: −3.1 mm,) or basolateral amygdala (300 nl, AAV2/9.CaMKII.GCaMP6f.WPRE.SV40, Penn Vector Core; rAAV2-retro.EF1a.GCaMP6f.WPRE, Georg Keller, FMI Vector Core, Basel, Switzerland, for tracing experiments 300 nl, rAAV2-retro.hSyn1.mCherry.WPRE.hGHp(A), VVF Zurich; or 50 nl CTB 555, Invitrogen, for optogenetic experiments 300 nl rAAV2-retro.hSyn1.mCherry.icre.WPRE.hGHp(A), VVF Zürich, for all-optical experiments 300 nl AAV2-retro.CaMKII.iCre.WPRE.hGHp(A), VVF Zurich; coordinates AP: −1.7, ML: −3.6, DV: −3.6 mm) of 8-11 week old mice with a glass pipette and a pressure ejection system (Picospritzer) under isoflurane anaesthesia (1 − 2%) and buprenorphine (0.1 mg/kg) and ropivacaine (65 mg/kg) analgesia. The rAAV2-retro helper was a gift from Alla Karpova & David Schaffer (Addgene plasmid #81070). Virus for retrograde labelling of MGB neurons was supplemented with blue non-retrograde polymer microspheres (1:2400, Duke Scientific Corp.) to label BLA injection sites. For miniature microscope experiments, one week after virus injection, a gradient refractive index (GRIN) lens (0.5 or 0.6 mm diameter, Inscopix) was implanted during a second surgery (anaesthesia and analgesia see above). A 0.8 mm diameter craniotomy was drilled above the MGB and a small track was cut with a 0.7 mm sterile needle. The GRIN lens was then slowly advanced into the brain (coordinates: AP: −3.28, ML: −1.9, DV: −3.0 mm), fixed to the skull with light curable glue (Loctite 4305, Henkel) and the skull was sealed with Scotchbond (3 M), Vetbond (3 M) and dental acrylic (Paladur, Kulzer). A titanium head bar (custom made) was attached to fix the animal during the miniature microscope base plate mounting procedure. For optogenetic experiments, virus (see above) was injected bliaterally in the basolateral amygdala and medial geniculate body as described above. One week later, optical fibres (0.4 mm, 0.5 NA, Thorlabs) were implanted bilaterally above the medial geniculate body (Coordinates: AP: −3.28, ML: −1.9, DV: −2.9 mm). Optical fibres, the wound and skull were fixed and sealed in a similar manner to GRIN lens implantations. Animals were provided with analgesia (buprenorphine, ropivacaine) and their well-being was monitored throughout the entire experimental period.
Behavioural paradigms and analysis
Behavioural experiments were performed during the animal’s light period. A four-day auditory fear conditioning paradigm was performed in a habituation / test context (days 1, 3, 4) and a fear conditioning context (day 2). Mice were presented with 5 intermingled CS+ and CS− during habituation (6 kHz and 12 kHz, intermingled) in a round plexiglass context. CSs were composed of 27 tone pips (200 ms, 75 dB) presented at a rate of 1.1 Hz (Tucker-Davis Technologies, TDT 78 or RZ6). Fear conditioning was performed in a ca. 25 cm square plexiglass box and a shock grid floor (Coulbourn, Noldus). The CS+ (6 kHz and 12 kHz, counterbalanced) was terminated by a 2 s 0.65 mA foot shock 1.1 s after the last tone pip. During the extinction sessions (day 3 and day 4, habituation context), 4 CS− and 12 CS+ were presented. For optogenetic experiments, animals were habituated to the optical fibre attachment procedure for 3 days before the start of the fear conditioning paradigm. On the fear conditioning day, optical fibres were attached to the optical fibre implant via a ceramic mating sleeve (Thorlabs). MGB ArchT-expressing neurons were continuously inhibited during the five CS-US pairings (starting 2 s before CS onset until 2 s after US offset) with a 565 nm LEDs (M565D2, Thorlabs). Optical stimulation was controlled with a custom-built stimulation setup consisting of an Arduino board (Arduino Uno REV3, Arduino) and LED drivers (LEDD1B, Thorlabs). The light intensity measured at the optical fibre tip was 19 mW and optical fibre implants had a typical attenuation of 30%. Optogenetic experiments were performed and analyzed in a blinded fashion. Behavioural experiments were performed and analysed using Cineplex 3.4.1 (Plexon Inc) or Ethovision 14 (Noldus). Behavioural tracking based on the centre of mass of the mouse was performed using inbuilt functions of Cineplex and Ethovision. Freezing was initially detected automatically based on periods of absence of movement (threshold: 1 s) and then manually controlled and adjusted for non-freezing episodes (e.g., grooming) post hoc.
Plasticity of auditory tuning of MGB neurons was tested with a three-day paradigm. On day one, the animals were exposed to 165–195 presentations of 200 ms pure tone pips ranging from 1 to 40 kHz at 65, 75 and 85 dB. Pure tones were presented as a series of three pips at a frequency of 0.5 Hz in a round plexiglass context. The different combinations of tone frequency and sound pressure levels were presented as randomized trials (five repetitions per combination) with a minimum intertrial-interval of 11 s. On the consecutive day, the animals underwent a fear conditioning paradigm as described above (counterbalanced CS+ frequencies across animals: 8, 16 or 20 kHz). On the post-learning test day (day three), mice were exposed to the same presentation of pure tone pips as on day 1. MGB neural activity was imaged throughout the four-day fear conditioning and three-day auditory tuning paradigm using a miniature microscope.
Miniature microscope imaging
The miniature microscope (nVista2.0 or nVoke, Inscopix) was fixed to the base plate on the mouse’s head before the experiment using head-fixation at the head bar on a flying saucer style running wheel. Mice were initially habituated to this procedure. MGB Ca2+ fluorescence was imaged continuously during the behavioural session with the following settings (nVista Software Version: 2.0.4 or nVoke Software Version: 2.1.10): Framerate: 20 Hz, blue LED-Power: 50–70%, Gain: 1.0–2.5, nVoke stimulation LED power: 10 mW/mm2. Image size: 1024 × 1024 or 1080 × 1080 pxl. LED power and gain were adjusted according to GCaMP expression levels and the same settings were used across days for individual mice. For all-optical imaging and optogenetic stimulation experiments (nVoke, Inscopix), the stimulation LED was switched on 2 s before the start of the CS and terminated 2 s after the end of the US.
Image analysis
Raw image data was analysed as previously described21,22,95. Briefly, movies from all behavioural sessions were spatially down sampled (2x), bandpass filtered (Fourier transform) and normalized by the filtered image (ImageJ). The movies from all days were then concatenated into a single file and motion corrected using Turboreg (min. three rounds)96. Only movies that motion-corrected successfully across days with final spatial dislocations of < 2 μm were used for Ca2+ trace extraction. Principal and independent component analysis-based detection of individual regions of interest (ROIs) was performed on down sampled (5 Hz) ΔF/F movies. ROIs were truncated at 50% peak intensity and limited to a size of 30 pixels (ca. 60 μm). ROIs were initially oversampled (300 ICs) and then overlaid with the maximum intensity projection of the 4-day movie. ROIs that did not match individual neurons were discarded. We typically retained 97 ± 5 ICAs per animal for CaMKII-GCaMP6f, N = 19 mice, 69 ± 9 ICAs per animal for rAAV2-retro.EF1a.GCaMP6f, N = 6 mice, 90 ± 2 ICAs per animal for tdTomato nVoke experiments, N = 5 mice and 94 ± 2 ICAs per animal for ArchT nVoke experiments, N = 6 mice. These ICs were then applied to the 20 Hz motion corrected raw fluorescence movie to extract single cell Ca2+ traces for further processing. The detected ICs from the concatenation method overlap > 99% with a similar individual day detection and post-hoc alignment method and was thus deemed more suitable and efficient (Supplementary Fig. 15).
Ca2+ data analysis
All analysis was based on linearly detrended and z-scored Ca2+ traces of individual neurons. Ca2+ traces were baselined to the time periods preceding CS or US onset. To identify CS− and US-responsive neurons and their plasticity types across days, 30 s CS and 2.8 s US responses were analysed using a combined statistical and supervised cluster analysis approach as previously described22.
Auditory tuning curves were calculated based on the mean Ca2+ response during the 250 ms time window after pip onset. Cells were classified as tone-responsive to individual frequency pips if their mean response exceeded 0.5 zS for at least two of the frequencies tested. The best frequency (BF) of a neuron was defined as the frequency that prompted the maximal Ca2+ response averaged across trials. The difference in BF to the CS+ for comparison across fear conditioning is calculated on an animal-by-animal basis in absolute values as ΔBF = | BF - CS+ | .
CS+, CS− and baseline responses were decoded from MGB Ca2+ activity by fitting three-way (CS+ vs. CS− vs. baseline) or two-way (CS+ vs. baseline or CS− vs. baseline) quadratic discriminant analysis classifiers. We classified CS+ and CS− responses based on the first four presentations to balance for uneven numbers of CS+/− presentations across habituation, fear conditioning and extinction days. Baseline responses were sampled from the 30 s periods preceding the CS+ and CS−. Classifiers were trained on the mean response of five consecutive pip responses within one CS (or the baseline period), such that each training set contained 20 input variables per condition (i.e., 40 for two-way decoders and 60 for three-way decoders). The mean response was calculated based on a 300 ms time window after pip onset and classifiers were trained using a tenfold cross-validation procedure. Decoder accuracy was calculated as the mean of the diagonal of the confusion matrix. Classifiers were trained for each individual animal and are presented as mean decoding accuracy across animals. To balance for unequal cell numbers between the different animals, we randomly selected 40 neurons from each animal and calculated the mean accuracy from 50 independent runs.
The population vector distance (PVD) between CS and US responses was calculated based on binned (0.275 s bins to accommodate the 1.1 Hz pip frequency) 30 s CS and 4 s US responses. PVD was calculated as the Euclidean distance between each CS bin and the mean binned US response and then averaged for each 30 s CS. Intraday PVD changes were normalized to the PVD of the first CS and across day PVD changes were calculated as the mean intraday PVD change for all CSs and normalized to the mean PVD of the habituation day.
Sound recordings and analysis
Acoustic signals of the fear conditioning context were recorded with a PCB Precision Condenser Microphone (Model 377C01) microphone and a RZ6 Auditory Processor (Tucker-Davis Technologies) 50 cm above the fear conditioning context at 195 kHz simultaneous to miniature microscope imaging during the fear conditioning session. Sound waves were high-pass filtered at 1 kHz and spectrograms were computed using short-time Fourier transforms (spectrogram function, Signal Processing Toolbox, MATLAB, Mathworks). To detect sound-level correlated neuronal activity, the cross-correlation coefficient between the squared acoustic signal binned in 50 ms and the corresponding Ca2+ signal of individual neurons was computed with a maximum lag of 500 ms. Cells were classified as sound-correlated if they exceeded a maximal cross-correlation coefficient of 0.2. Acoustic event onsets were detected based on the peak of the differentiated squared and binned sound wave. Acoustic events were not distinguished between animal movement-related sounds and vocalizations of the animal.
Histology
After completion of the behavioural experiment, mice were transcardially perfused with ca. 5 ml phosphate buffered saline (PBS, ThermoFisher) followed by 40 ml 4% paraformaldehyde (PFA) in PBS (pH = 7.4). Brains were removed and stored overnight in 4% PFA. Of 150 μm coronal slices were prepared using a vibratome (Campden Instruments) and immunostained for calretinin using the following solutions and protocol: carrier solution: 1% normal horse serum (NHS, Vector Laboratories) with 0.5% Triton (ThermoFisher) in phosphate buffered saline (PBS, ThermoFisher), blocking solution: 10% NHS with 0.5% Triton in PBS. After several rounds of PBS washes, slices were blocked for two hours at room temperature and incubated in primary antibody in carrier solution (goat anti-calretinin, 1:1000, Swant; rabbit anti-NeuN, 1:3000, Abcam; rabbit anti-GABA, 1:500, SigmaAldrich) overnight at 4 °C. Slices were washed again in PBS and incubated for 2 h at room temperature in secondary antibody in carrier solution (donkey anti-goat 647, 1:1000, ThermoFisher; donkey anti-rabbit 405, 1:1000, Abcam; donkey anti-rabbit 555, 1:1000, ThermoFisher). After four final washes, slices were mounted on slides and cover slipped using 22 × 50 mm, 0.16–0.19 mm thick cover glass (FisherScientific). Images were acquired with a LSM710 confocal microscope (Zeiss) and stitched with Zen 2.1 (black, Zeiss). Confocal images were post processed with ImageJ (Version: 2.0). Cells were manually counted using the cell counter plugin (https://imagej.nih.gov/ij/plugins/cell-counter.html) for ImageJ.
Statistical analysis
Statistical analysis was performed using Matlab (Mathworks) and Prism 8 (Graphpad). Unless otherwise indicated, normal distribution of the data was not assumed and non-parametric tests were performed. Values are presented as mean ± SEM unless stated otherwise. Box and whisker plots indicate median, interquartile range as well as the minimum to maximum value of the distribution. Statistical tests are mentioned in the text or figure legends. *, **, *** indicate p values smaller than 0.05, 0.01 and 0.001, respectively.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The source data underlying the main figures and Supplementary information are available as Source Data file.
Code availability
Custom code used for analysis in this manuscript is available upon request.
References
Fanselow, M. S. & LeDoux, J. E. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron 23, 229–232 (1999).
Maren, S. & Fanselow, M. S. The amygdala and fear conditioning: has the nut been cracked? Neuron 16, 237–240 (1996).
Rogan, M. T., Stäubli, U. V. & LeDoux, J. E. Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390, 604–607 (1997).
Schafe, G. E. et al. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J. Neurosci. 20, 8177–8187 (2000).
Humeau, Y., Shaban, H., Bissière, S. & Lüthi, A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 426, 841–845 (2003).
McKernan, M. G. & Shinnick-Gallagher, P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611 (1997).
Rogan, M. T. & LeDoux, J. E. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron 15, 127–136 (1995).
Apergis-Schoute, A. M., Debiec, J., Doyère, V., LeDoux, J. E. & Schafe, G. E. Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: a role for presynaptic plasticity in the fear system. J. Neurosci. 25, 5730–5739 (2005).
Bordi, F. & LeDoux, J. E. Response properties of single units in areas of rat auditory thalamus that project to the amygdala. I. Acoustic discharge patterns and frequency receptive fields. Exp. Brain Res. 98, 261–274 (1994).
Bordi, F. & LeDoux, J. E. Response properties of single units in areas of rat auditory thalamus that project to the amygdala. II. Cells receiving convergent auditory and somatosensory inputs and cells antidromically activated by amygdala stimulation. Exp. Brain Res. 98, 275–286 (1994).
Edeline, J. M. & Weinberger, N. M. Subcortical adaptive filtering in the auditory system: associative receptive field plasticity in the dorsal medial geniculate body. Behav. Neurosci. 105, 154–175 (1991).
Edeline, J. M. & Weinberger, N. M. Thalamic short-term plasticity in the auditory system: associative returning of receptive fields in the ventral medial geniculate body. Behav. Neurosci. 105, 618–639 (1991).
Han, J.-H. et al. Increasing CREB in the auditory thalamus enhances memory and generalization of auditory conditioned fear. Learn. Mem. 15, 443–453 (2008).
Hennevin, E., Maho, C., Hars, B. & Dutrieux, G. Learning-induced plasticity in the medial geniculate nucleus is expressed during paradoxical sleep. Behav. Neurosci. 107, 1018–1030 (1993).
Ryugo, D. K. & Weinberger, N. M. Differential plasticity of morphologically distinct neuron populations in the medial geniculate body of the cat during classical conditioning. Behav. Biol. 22, 275–301 (1978).
Maren, S., Yap, S. A. & Goosens, K. A. The amygdala is essential for the development of neuronal plasticity in the medial geniculate nucleus during auditory fear conditioning in rats. J. Neurosci. 21, RC135:1-6 (2001).
Weinberger, N. M. The medial geniculate, not the amygdala, as the root of auditory fear conditioning. Hear. Res. 274, 61–74 (2011).
Headley, D. B., Kanta, V., Kyriazi, P. & Paré, D. Embracing complexity in defensive networks. Neuron 103, 189–201 (2019).
Mobbs, D., Headley, D. B., Ding, W. & Dayan, P. Space, time, and fear: survival computations along defensive circuits. Trends Cogn. Sci. 24, 228–241 (2020).
Ghosh, K. K. et al. Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878 (2011).
Grewe, B. F. et al. Neural ensemble dynamics underlying a long-term associative memory. Nature 543, 670–675 (2017).
Gründemann, J. et al. Amygdala ensembles encode behavioral states. Science 364, 1–9 (2019).
Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Hackett, T. A., Clause, A. R., Takahata, T., Hackett, N. J. & Polley, D. B. Differential maturation of vesicular glutamate and GABA transporter expression in the mouse auditory forebrain during the first weeks of hearing. Brain Struct. Funct. 221, 2619–2673 (2016).
Rouiller, E. M., Colomb, E., Capt, M. & de Ribaupierre, F. Projections of the reticular complex of the thalamus onto physiologically characterized regions of the medial geniculate body. Neurosci. Lett. 53, 227–232 (1985).
Winer, J. A., Marie, Saint, Larue, R. L., Oliver, D. T. & GABAergic, D. L. feedforward projections from the inferior colliculus to the medial geniculate body. Proc. Natl Acad. Sci. 93, 8005–8010 (1996).
Jager, P. et al. Inhibitory interneurons distribute widely across the mouse thalamus and form ontogenetic spatial clusters. bioRxiv (2019) https://doi.org/10.1101/651745.
Herry, C. et al. Switching on and off fear by distinct neuronal circuits. Nature 454, 600–606 (2008).
Grimsley, J. M. S., Hazlett, E. G. & Wenstrup, J. J. Coding the Meaning of Sounds: Contextual Modulation of Auditory Responses in the Basolateral Amygdala. J. Neurosci. 33, 17538–17548 (2013).
Williams, W. O., Riskin, D. K. & Mott, K. M. Ultrasonic sound as an indicator of acute pain in laboratory mice. J. Am. Assoc. Lab. Anim. Sci. 47, 8–10 (2008).
Edeline, J.-M. & Weinberger, N. M. Associative retuning in the thalamic source of input to the amygdala and auditory cortex: Receptive field plasticity in the medial division of the medial geniculate body. Behav. Neurosci. 106, 81–105 (1992).
Amano, T., Duvarci, S., Popa, D. & Pare, D. The fear circuit revisited: contributions of the basal amygdala nuclei to conditioned fear. J. Neurosci. 31, 15481–15489 (2011).
Krabbe, S. et al. Adaptive disinhibitory gating by VIP interneurons permits associative learning. Nat. Neurosci. 22, 1834–1843 (2019).
Chen, L., Wang, X., Ge, S. & Xiong, Q. Medial geniculate body and primary auditory cortex differentially contribute to striatal sound representations. Nat. Commun. 10, 418 (2019).
Kimura, A., Donishi, T., Sakoda, T., Hazama, M. & Tamai, Y. Auditory thalamic nuclei projections to the temporal cortex in the rat. Neuroscience 117, 1003–1016 (2003).
Kwon, J.-T. et al. Optogenetic activation of presynaptic inputs in lateral amygdala forms associative fear memory. Learn. Mem. 21, 627–633 (2014).
Smith, P. H., Uhlrich, D. J. & Manning, K. A. Evaluation of medial division of the medial geniculate (MGM) and posterior intralaminar nucleus (PIN) inputs to the rat auditory cortex, amygdala, and striatum. J. Comp. Neurol. 527, 1478–1494 (2019).
LeDoux, J. E. Emotion circuits in the brain. Annu. Rev. Neurosci. 23, 155–184 (2000).
Doron, N. N. & LeDoux, J. E. Organization of projections to the lateral amygdala from auditory and visual areas of the thalamus in the rat. J. Comp. Neurol. 412, 282–409 (1999).
Lu, E., Llano, D. A. & Sherman, S. M. Different distributions of calbindin and calretinin immunostaining across the medial and dorsal divisions of the mouse medial geniculate body. Hear. Res. 257, 16–23 (2009).
Antunes, R. & Moita, M. A. Discriminative Auditory Fear Learning Requires Both Tuned and Nontuned Auditory Pathways to the Amygdala. J. Neurosci. 30, 9782–9787 (2010).
Han, X. et al. A High-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front. Syst. Neurosci. 5, 1–8 (2011).
Pereira, A. G., Farias, M. & Moita, M. A. A new thalamo-cortical-amygdala circuit is involved in processing a natural auditory alarm cue. bioRxiv (2019) https://doi.org/10.1101/716910.
Barsy, B. et al. Associative and plastic thalamic signaling to the lateral amygdala controls fear behavior. Nat. Neurosci. 23, 625–637 (2020).
Edeline, J.-M., Neuenschwander-El Massioui, N. & Dutrieux, G. Discriminative long-term retention of rapidly induced multiunit changes in the hippocampus, medial geniculate and auditory cortex. Behav. Brain Res. 39, 145–155 (1990).
Dalmay, T. et al. A critical role for neocortical processing of threat memory. Neuron 104, 1180–1194.e7 (2019).
Ghosh, S. & Chattarji, S. Neuronal encoding of the switch from specific to generalized fear. Nat. Neurosci. 18, 112–120 (2015).
Duarte, R. & Morrison, A. Leveraging heterogeneity for neural computation with fading memory in layer 2/3 cortical microcircuits. PLOS Comput. Biol. 15, e1006781 (2019).
Blackwell, J. M. et al. Stable encoding of sounds over a broad range of statistical parameters in the auditory cortex. Eur. J. Neurosci. 43, 751–764 (2016).
Tripathy, S. J., Padmanabhan, K., Gerkin, R. C. & Urban, N. N. Intermediate intrinsic diversity enhances neural population coding. Proc. Natl Acad. Sci. 110, 8248–8253 (2013).
Sweis, B. M., Mau, W., Rabinowitz, S. & Cai, D. J. Dynamic and heterogeneous neural ensembles contribute to a memory engram. Curr. Opin. Neurobiol. 67, 199–206 (2021).
Polepalli, J. S., Gooch, H. & Sah, P. Diversity of interneurons in the lateral and basal amygdala. Npj Sci. Learn 5, 1–9 (2020).
Wolff, S. B. E. et al. Amygdala interneuron subtypes control fear learning through disinhibition. Nature 509, 453–458 (2014).
Asede, D. et al. Sensory inputs to intercalated cells provide fear-learning modulated inhibition to the basolateral amygdala. Neuron 86, 541–554 (2015).
Lucas, E. K., Jegarl, A. M., Morishita, H. & Clem, R. L. Multimodal and site-specific plasticity of amygdala parvalbumin interneurons after fear learning. Neuron 91, 629–643 (2016).
Kim, J., Pignatelli, M., Xu, S., Itohara, S. & Tonegawa, S. Antagonistic negative and positive neurons of the basolateral amygdala. Nat. Neurosci. 19, 1636–1646 (2016).
Namburi, P. et al. A circuit mechanism for differentiating positive and negative associations. Nature 520, 675–678 (2015).
Gründemann, J. Distributed coding in auditory thalamus and basolateral amygdala upon associative fear learning. Curr. Opin. Neurobiol. 67, 183–189 (2021).
Schneider, D. M., Sundararajan, J. & Mooney, R. A cortical filter that learns to suppress the acoustic consequences of movement. Nature 561, 391–395 (2018).
Šuta, D., Popelá, J. & Syka, J. Coding of communication calls in the subcortical and cortical structures of the auditory system. Physiol. Res. 57 (Suppl 3), S149–S159 (2008).
Abs, E. et al. Learning-related plasticity in dendrite-targeting layer 1 interneurons. Neuron 100, 684–699.e6 (2018).
Aizenberg, M., Mwilambwe-Tshilobo, L., Briguglio, J. J., Natan, R. G. & Geffen, M. N. Bidirectional regulation of innate and learned behaviors that rely on frequency discrimination by cortical inhibitory neurons. PLOS Biol. 13, e1002308 (2015).
Gillet, S. N., Kato, H. K., Justen, M. A., Lai, M. & Isaacson, J. S. Fear learning regulates cortical sensory representations by suppressing habituation. Front. Neural Circuits 11, 112 (2018).
Krabbe, S., Gründemann, J. & Lüthi, A. Amygdala inhibitory circuits regulate associative fear conditioning. Biol. Psychiatry 83, 800–809 (2018).
Lee, S.-C., Amir, A., Haufler, D. & Pare, D. Differential recruitment of competing valence-related amygdala networks during anxiety. Neuron 96, 81–88.e5 (2017).
Letzkus, J. J. et al. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480, 331–335 (2011).
Senn, V. et al. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81, 428–437 (2014).
Weber, J. P. et al. Location-dependent synaptic plasticity rules by dendritic spine cooperativity. Nat. Commun. 7, 1–14 (2016).
Harvey, C. D. & Svoboda, K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450, 1195–1200 (2007).
Humeau, Y. & Lüthi, A. Dendritic calcium spikes induce bi-directional synaptic plasticity in the lateral amygdala. Neuropharmacology 52, 234–243 (2007).
Branco, T. & Häusser, M. The single dendritic branch as a fundamental functional unit in the nervous system. Curr. Opin. Neurobiol. 20, 494–502 (2010).
Markram, H., Lübke, J., Frotscher, M. & Sakman, B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215 (1997).
Brzosko, Z., Mierau, S. B. & Paulsen, O. Neuromodulation of spike-timing-dependent plasticity: past, present, and future. Neuron 103, 563–581 (2019).
Izhikevich, E. M. Solving the distal reward problem through linkage of STDP and dopamine signaling. Cereb. Cortex 17, 2443–2452 (2007).
Likhtik, E. & Johansen, J. P. Neuromodulation in circuits of aversive emotional learning. Nat. Neurosci. 22, 1586–1597 (2019).
Johansen, J. P. et al. Hebbian and neuromodulatory mechanisms interact to trigger associative memory formation. Proc. Natl Acad. Sci. 111, E5584–E5592 (2014).
Gehrlach, D. A. et al. Aversive state processing in the posterior insular cortex. Nat. Neurosci. 22, 1424–1437 (2019).
Herry, C. & Johansen, J. P. Encoding of fear learning and memory in distributed neuronal circuits. Nat. Neurosci. 17, 1644–1654 (2014).
Klavir, O., Genud-Gabai, R. & Paz, R. Functional connectivity between amygdala and cingulate cortex for adaptive aversive learning. Neuron 80, 1290–1300 (2013).
Likhtik, E., Stujenske, J. M., Topiwala, M. A., Harris, A. Z. & Gordon, J. A. Prefrontal entrainment of amygdala activity signals safety in learned fear and innate anxiety. Nat. Neurosci. 17, 106–113 (2014).
Ozawa, T. et al. A feedback neural circuit for calibrating aversive memory strength. Nat. Neurosci. 20, 90–97 (2017).
Penzo, M. A. et al. The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459 (2015).
Aizenberg, M. et al. Projection from the amygdala to the thalamic reticular nucleus amplifies cortical sound responses. Cell Rep. 28, 605–615.e4 (2019).
He, J. Corticofugal modulation of the auditory thalamus. Exp. Brain Res. 153, 579–590 (2003).
Jones, E. G. Viewpoint: the core and matrix of thalamic organization. Neuroscience 85, 331–345 (1998).
Pardi, M. B. et al. A thalamocortical top-down circuit for associative memory. Science 370, 844–848 (2020).
Weglage, M., Waernberg, E., Lazaridis, I., Tzortzi, O. & Meletis, K. Complete representation of action space and value in all striatal pathways. bioRxiv (2020) https://doi.org/10.1101/2020.03.29.983825.
Yuste, R. From the neuron doctrine to neural networks. Nat. Rev. Neurosci. 16, 487–497 (2015).
Jun, J. J. et al. Fully integrated silicon probes for high-density recording of neural activity. Nature 551, 232–236 (2017).
Emiliani, V., Cohen, A. E., Deisseroth, K. & Hausser, M. All-optical interrogation of neural circuits. J. Neurosci. 35, 13917–13926 (2015).
Williamson, R. S., Hancock, K. E., Shinn-Cunningham, B. G. & Polley, D. B. Locomotion and task demands differentially modulate thalamic audiovisual processing during active search. Curr. Biol. 25, 1885–1891 (2015).
Zhang, X. & Li, B. Population coding of valence in the basolateral amygdala. Nat. Commun. 9, 5195 (2018).
Gilad, A., Maor, I. & Mizrahi, A. Learning-related population dynamics in the auditory thalamus. bioRxiv (2020) https://doi.org/10.1101/2020.03.31.017632.
Broussard, G. J. et al. In vivo measurement of afferent activity with axon-specific calcium imaging. Nat. Neurosci. 21, 1272–1280 (2018).
Mukamel, E. A., Nimmerjahn, A. & Schnitzer, M. J. Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760 (2009).
Thévenaz, P., Ruttimann, U. E. & Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. Publ. IEEE Signal Process. Soc. 7, 27–41 (1998).
Acknowledgements
We thank Georg Keller, Tingjia Lu and Daniela Gerosa Erni for virus production and V. Jayaraman, R. Kerr, D. Kim, L. Looger, K. Svoboda and the HHMI Janelia GENIE Project for making GCaMP6 available, Ed Boyden and MIT for making ArchT available as well as Alla Karpova and David Schaffer who gifted the rAAV2-retro helper (Addgene plasmid # 81070 V). pAAV-hSynapsin1-FLEX-axon-GCaMP6s was a gift from Lin Tian (Addgene viral prep # 112010-AAV5). We thank Raymond Strittmatter and Robert Häring for mechanical workshop and electrical engineering support. We thank Yael Bitterman, Sabine Krabbe, Andreas Lüthi and the Gründemann lab for comments on the manuscript. Funding: Research was supported by the Swiss National Science Foundation (Ambizione Fellowship to J.G., SNF Professorship to J.G.), the European Research Council (Starting Grant, J.G.), the Forschungsfonds Nachwuchsforschende of the University of Basel (M.H.), the Forschungspreis der Schweizerischen Hirnliga (J.G.) and the Department of Biomedicine at the University of Basel. Artworks in parts of some figures were created with BioRender.com. Mouse brain anatomy figures and panels were modified and reprinted from The Mouse Brain in Stereotaxic Coordinates, Paxinos, Franklin, panels 45 and 47, 2001. Mouse cartoons with miniature microscopes from Gründemann et al.22. Reprinted with permission from AAAS.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.A.T., M.H. and J.G.; Methodology and imaging experiments, J.A.T., M.H. and J.G.; Experimental support, J.A.F., D.A.G., M.T., C.M.B., S.M.I. and T.L.; Analysis, J.A.T., M.H., C.M.B. and J.G.; Original draft, J.A.T. and J.G.; Revisions, all authors; Supervision and funding, J.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taylor, J.A., Hasegawa, M., Benoit, C.M. et al. Single cell plasticity and population coding stability in auditory thalamus upon associative learning. Nat Commun 12, 2438 (2021). https://doi.org/10.1038/s41467-021-22421-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-22421-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
