
Abstract
T follicular helper (TFH) cells are specialized effector CD4+ T cells critical to humoral immunity. Whether post-transcriptional regulation has a function in TFH cells is unknown. Here, we show conditional deletion of METTL3 (a methyltransferase catalyzing mRNA N6-methyladenosine (m6A) modification) in CD4+ T cells impairs TFH differentiation and germinal center responses in a cell-intrinsic manner in mice. METTL3 is necessary for expression of important TFH signature genes, including Tcf7, Bcl6, Icos and Cxcr5 and these effects depend on intact methyltransferase activity. m6A-miCLIP-seq shows the 3′ UTR of Tcf7 mRNA is subjected to METTL3-dependent m6A modification. Loss of METTL3 or mutation of the Tcf7 3′ UTR m6A site results in accelerated decay of Tcf7 transcripts. Importantly, ectopic expression of TCF-1 (encoded by Tcf7) rectifies TFH defects owing to METTL3 deficiency. Our findings indicate that METTL3 stabilizes Tcf7 transcripts via m6A modification to ensure activation of a TFH transcriptional program, indicating a pivotal function of post-transcriptional regulation in promoting TFH cell differentiation.
Similar content being viewed by others


Introduction
The production of high-affinity antibodies and generation of memory cells are critical for establishing protective immunity1,2. During acute viral infection, T follicular helper (TFH) cells provide help to cognate antigen-presenting B cells to facilitate the formation of germinal centers (GCs) and the development of long-lived plasma cells and memory B cells3. After priming by dendritic cells (DCs) in the T-cell zone, CD4+ T cells upregulate the expression of chemokine receptor CXCR5, the costimulatory receptor ICOS and the transcriptional repressor B-cell lymphoma 6 (Bcl-6), which together coordinate early fate commitment of TFH cells and their migration to the follicles. After interacting with cognate antigen-presenting B cells, these activated CD4+ T cells mature into TFH cells and further differentiate into GC TFH cells, providing help to GC B cells for humoral immunity4,5.
TFH differentiation is a complicated biological process6, which is tightly controlled by multiple transcription factors. As a master regulator of TFH differentiation, Bcl-6 represses the TH1, TH2, and TH17 lineage-specific transcription factors to promote the differentiation of activated CD4+ T cells into TFH cells7,8,9. On the contrary, Blimp1 (encoded by Prdm1) negatively modulates Bcl-6 transcription by binding to the Bcl6 promoter and acting as a repressor, thus preventing TFH differentiation and promoting the formation of non-TFH effector helper T cells9,10. Accumulative studies have demonstrated that TCF-1 (encoded by Tcf7) plays key roles in TFH differentiation by directly acting upstream of the Bcl-6–Blimp1 axis11,12,13. Although the regulation of these transcription factors has been intensely investigated at the transcriptional level, it remains unknown if post transcriptional mechanisms are involved in the balanced expression of these key factors during TFH differentiation.
N6-methyladenosine (m6A) is the most prevalent modification on eukaryote mRNAs, catalyzed by m6A methyltransferases14. Methyltransferase like 3 protein (METTL3, encoded by Mettl3) is the core catalytic subunit of m6A methyltransferases, and METTL14 is an allosteric activator of METTL315,16. The recruitment of m6A-binding proteins is actively involved in almost every stage of mRNA metabolism, from processing in the nucleus to translation and decay in the cytoplasm, adding another layer of regulatory mechanisms in gene expression17. Early studies showed that m6A-tagged transcripts had shorter half-life18, and binding of YTHDF2 to m6A promoted m6A-modified mRNA decay18,19. Recently, it was discovered that a group of novel m6A-binding proteins, IGF2BPs, stabilized methylated transcripts by guarding them against degradation20,21. The distinct roles of YTHDF2 and IGF2BPs suggest that the impact of m6A modification on mRNA stability is highly dependent on cell context.
Several studies have uncovered critical roles of m6A modification in immune regulation. It has been documented that m6A methylation is essential for normal hematopoiesis and leukemia development22,23,24. m6A methylation regulates T-cell homeostasis, as well as suppressive functions of Treg cells by targeting Socs genes25,26. In the context of viral infection, m6A represses type I interferon production in an innate antiviral state27,28. Despite these profound effects of m6A modification on immunoregulation, its role in TFH differentiation has not been determined.
In this study, by conditional targeting the Mettl3 gene in T cells, we demonstrate that METTL3-mediated m6A modification is critical for TFH cell differentiation. Mechanistically, METTL3 deficiency impairs the stability of m6A-modified Tcf7 mRNA, resulting in compromised activation of TFH transcriptional program.
Results
METTL3 controls TFH differentiation and GC reactions
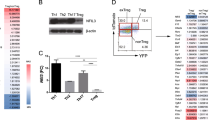
To investigate the role of METTL3 in TFH differentiation, Mettl3-floxed mice were crossed with Cd4-Cre mice to generate conditional deletion of METTL3 in T cells (Mettl3fl/flCd4-Cre mice), and the deletion efficiency of METTL3 in splenic CD4+ T cells was validated by quantitative RT-PCR (Supplementary Fig. 1a). We then infected Mettl3fl/flCd4-Cre mice and their wild-type control littermates (Ctrl) with LCMV-Armstrong strain. The frequency and numbers of CD44+CXCR5+ TFH cells were significantly diminished in Mettl3fl/flCd4-Cre mice compared with those in their control littermates on day 8 post viral infection (Fig. 1a, b). In contrast, METTL3-deficient CD4+ T cells were dramatically skewed toward the CD44+CXCR5− TH1 proportion, albeit the numbers of TH1 cells were also decreased with the ablation of METTL3 (Fig. 1a, b). It is worth mentioning that ablation of METTL3 resulted in more severe defects in TFH cells (21.4-fold change) than TH1 cells (1.9-fold change) upon acute viral infection (Fig. 1b). Given T-bet is expressed at a relatively high level on TH1 lineage and directs its commitment29, we also analyzed T-bet expression on both TH1 and TFH cells. We found TH1 cells expressed a much higher level of T-bet than TFH cells, and both TFH and TH1 cells downregulated T-bet expression in the absence of METTL3 (Supplementary Fig. 1b). Moreover, Mettl3fl/flCd4-Cre mice exhibited remarkably lower percentages and absolute cell numbers of GC TFH cells (identified as PD-1hiCXCR5+, ICOShiCXCR5+, or Bcl-6hiCXCR5+) than those of their control littermates (Fig. 1c, d). Accordingly, the expression levels of CXCR5, PD-1, ICOS, and Bcl-6 were dramatically lower on METTL3-deficient TFH cells than on wild-type cells (Supplementary Fig. 1c, d). These results indicated that METTL3 deficiency severely impairs TFH differentiation.

a, b Flow cytometry analysis of CD44+CXCR5+ TFH cells and CD44+CXCR5– TH1 cells, gated on splenic CD4+ T cells from Ctrl and Mettl3fl/flCd4-Cre mice on day 8 post infection (8 dpi). Summary of the frequency and cell numbers of indicated cell subsets are shown in b (n = 5 per group). c, d Flow cytometry analysis of PD-1hiCXCR5+ GC TFH cells (top panel), ICOShiCXCR5+ GC TFH cells (middle panel), and Bcl-6hiCXCR5+ GC TFH cells (bottom panel), gated on splenic CD44hiCD62LloCD4+ T cells from Ctrl and Mettl3fl/flCd4-Cre mice on 8 dpi. Summary of the frequency and cell numbers of indicated cell subsets are shown in d (n = 5 per group). e, f Flow cytometry analysis of splenic GL-7+Fas+ GC B cells (top panel) and IgDloCD138+ plasma cells (bottom panel) on 8 dpi. Summary of the frequency and cell numbers of GC B cells and plasma cells are shown in f (n = 3 per group). g Immunofluorescent staining of spleens from Ctrl and Mettl3fl/flCd4-Cre on 8 dpi. Green: PNA; Red: IgD; scale bar: 10 μm. h Analysis of LCMV-specific IgG concentration in serum on 8 dpi (top) and on 56 dpi (bottom) by ELISA (day 8: n = 12 for Ctrl group, n = 10 for Mettl3fl/flCd4-Cre group; day 56: n = 5 per group). Data are representative of at least three independent experiments. Error bars indicate standard error of the mean. P value was calculated by unpaired two-tailed Student’s t test.
Given the main function of TFH cells is to provide cognate B-cell help, which is a fundamental aspect of humoral immunity and generation of immunological memory1. We next examined whether METTL3 deficiency in CD4+ T cells affects GC formation during viral infection. A robust reduction in the proportions and numbers of GL-7+Fas+ GC B cells (Fig. 1e, f) and PNA+Fas+ GC B cells (Supplementary Fig. 1e, f) were observed in Mettl3fl/flCd4-Cre mice, compared with those in their wild-type counterparts. Furthermore, the frequency and cell numbers of IgDloCD138+ plasma cells were also much lower in Mettl3fl/flCd4-Cre mice than those in wild-type mice (Fig. 1e, f). The immunohistochemical analysis further confirmed substantially reduced PNA+ GCs within B-cell follicles in spleens from Mettl3fl/flCd4-Cre mice (Fig. 1g). To assess the consequences of defective TFH and GC responses in Mettl3fl/flCd4-Cre mice, we measured the LCMV-specific serum concentration of immunoglobulin G (IgG). The LCMV-specific IgG concentration was significantly lower in Mettl3fl/flCd4-Cre mice than that in their wild-type control littermates on day 8 and day 56 post viral infection (Fig. 1h). Collectively, these results suggested that METTL3 is required for TFH differentiation and GC responses.
To further validate these findings, we immunized Mettl3fl/flCd4-Cre mice and their wild-type control littermates by intraperitoneal administration of keyhole limpet hemocyanin (KLH) emulsified in Complete Freund’s Adjuvant (CFA). On day 8 post immunization, Mettl3fl/flCd4-Cre mice exhibited impaired development of TFH cells and GC TFH cells, but not TH1 cells (Supplementary Fig. 2a, b). Meanwhile, the percentages and cell numbers of GC B cells and plasma cells (Supplementary Fig. 2c, d) were also impaired in Mettl3fl/flCd4-Cre mice compared with their wild-type counterparts. These data jointly indicated METTL3 promotes TFH differentiation upon different antigen stimulation.
Meanwhile, the differentiation of other T helper lineages was also examined by using KLH immunization model. We found that both GATA3+ and IL-4-producing cells, as well as Foxp3+ cells were not altered in Mettl3fl/flCd4-Cre mice (Supplementary Fig. 2e–h), indicating TH2 and Treg cell differentiation are not affected in the absence of METTL3 upon KLH immunization. Interestingly, both RORγt+ and IL-17a-producing cells were significantly decreased in METTL3-deficient mice (Supplementary Fig. 2e–h), revealing METTL3 is essential for TH17 cell differentiation in vivo upon protein immunization.
METTL3 intrinsically regulates TFH differentiation
We next focused on whether METTL3 regulates TFH differentiation in a cell-intrinsic manner using both bone marrow chimeric and adoptive transfer mice models. We first generated bone marrow chimeric mice by reconstituting lethally irradiated wild-type recipient mice (CD45.1+) with a mixture donor of bone marrow cells from Mettl3fl/flCd4-Cre mice (CD45.2+) and wild-type (CD45.1+) competitor mice (Supplementary Fig. 3a). On day 8 post LCMV-Armstrong infection, Mettl3fl/flCd4-Cre mice-derived CD4+ T cells had much lower cell numbers of CD44+CXCR5+ TFH cells than those of competitor bone marrow-derived CD4+ T cells (Supplementary Fig. 3b, c). Correspondingly, CD4+ T cells originated in Mettl3fl/flCd4-Cre mice also exhibited a reduction of PD-1hiCXCR5+ GC TFH cells (Supplementary Fig. 3b, c). The expression levels of CXCR5, PD-1, ICOS, and Bcl-6 were significantly lower on METTL3-deficient TFH cells than those on competitor cells (Supplementary Fig. 3d).
To further rule out the effect of external factors on TFH differentiation, we adoptively transferred CD45.1+ SMARTA CD4+ T cells into CD45.2+ Mettl3fl/flCd4-Cre or their wild-type control littermates (Fig. 2a). On day 8 post viral infection, we observed the percentages and numbers of CD44+CXCR5+ TFH cells derived from CD45.1+ SMARTA CD4+ T cells were similar in the Mettl3fl/flCd4-Cre mice and their wild-type counterparts (Fig. 2b, c), indicating the microenvironment in the Mettl3fl/flCd4-Cre mice did not impair TFH differentiation. In contrast, METTL3-deficient CD4+ T cells displayed compromised TFH differentiation compared with that of congenic wild-type CD4+ T cells (Fig. 2b, c). Correspondingly, METTL3-deficient CD4+ T cells also exhibited defects in their ability to differentiate into CD44+CXCR5− TH1 cells (Fig. 2b, c). Besides, transferred CD45.1+ SMARTA cells also profoundly promoted the GC B and plasma cells differentiation (Fig. 2d, e), GC formation (Fig. 2f), and excessive production of LCMV-specific IgG antibody (Fig. 2g) in Mettl3fl/flCd4-Cre recipients. Together, our data demonstrated METTL3 is intrinsically required for TFH cell differentiation and functions as a key role in GCs formation.

a Scheme of adoptive transfer model. 5 × 106 CD45.1+ SMARTA cells were adoptively transferred into CD45.2+ Mettl3fl/flCd4-Cre mice or Ctrl host mice, followed by LCMV-Armstrong infection within 24 h. b, c Flow cytometry analysis of CD45.1+ SMARTA or CD45.2+ host T cell-derived CD44+CXCR5+ TFH and CD44+CXCR5– TH1 cells, gated on splenic CD4+ T cells from host mice on day 8 post viral infection. Frequency and numbers of CD44+CXCR5+ TFH cells and CD44+CXCR5– TH1 cells are shown in c (n = 5 per group). d, e Flow cytometry analysis of splenic PNA+Fas+ GC B cells (top panel) and IgDloCD138+ plasma cells (bottom panel) from host mice on 8 dpi. Summary of the frequency and cell numbers of GC B cells and plasma cells are shown in e (n = 6 per group). f Immunofluorescent staining of spleens from Ctrl or Mettl3fl/flCd4-Cre host mice on 8 dpi. Green: PNA; Red: IgD; scale bar: 10 μm. g Analysis of LCMV-specific IgG concentration in serum on 8 dpi by ELISA (n = 5 for Ctrl group, n = 5 for Mettl3fl/flCd4-Cre group, n = 6 for Ctrl host group, n = 6 for Mettl3fl/flCd4-Cre host group). Data are representative of at least three independent experiments. Error bars indicate standard error of the mean. P value was calculated by unpaired two-tailed Student’s t test (e) or two-way ANOVA coupled with multiple comparisons (c, g).
METTL3 is essential for the early initiation of TFH cells
During acute viral infection, effector CD4+ T cells’ commitment to the TFH lineage or TH1 lineage emerges before the initiation of GCs12. To investigate whether METTL3 is required for early TFH specification in vivo, naive Ctrl or Mettl3fl/flCd4-Cre SMARTA CD4+ T cells were labeled with cell-trace violet (CTV) and adoptively transferred into congenic recipient mice, followed by LCMV-Armstrong infection (Supplementary Fig. 4a). On day 3 post viral infection, SMARTA CD4+ T cells of both genotypes showed similar upregulation of T-cell activation markers CD69 and CD44, and downregulation of CD62L (Supplementary Fig. 4b, c). In vitro cell culture analysis also revealed that Mettl3fl/flCd4-Cre SMARTA CD4+ T cells displayed no obvious defects in T-cell activation (Supplementary Fig. 4d). Ctrl SMARTA CD4+ T cells exhibited vigorously proliferation post viral infection, whereas METTL3-deficient cells displayed a delayed proliferation (Fig. 3a). Lineage commitment to TFH lineage has occurred on day 3 post viral infection, as detected by Bcl-6+CXCR5+ cells30. Consistently, wild-type SMARTA cells developed robust numbers of Bcl-6+CXCR5+ TFH cells, whereas METTL3-deficient cells exhibited much lower percentages and cell numbers of Bcl-6+CXCR5+ TFH cells (Fig. 3b). Tracking the cell division showed Mettl3fl/flCd4-Cre SMARTA cells were predominantly in third and fourth divisions, while Ctrl SMARTA cells had advanced to fifth and sixth divisions (Fig. 3c, d), reflecting the impaired proliferation of activated CD4+ T cells in the absence of METTL3. Consistently, Mettl3fl/flCd4-Cre SMARTA cells exhibited a reduction of proportions and cell numbers of Bcl-6+CXCR5+ TFH cells in indicated cell divisions compared with that of Ctrl SMARTA cells (Fig. 3e, f). Interestingly, as a critical regulator for early TFH differentiation31, TCF-1 expression in CXCR5+ cells was also dramatically reduced due to ablation of METTL3 (Fig. 3g, h). Moreover, we also observed compromised CD25−CXCR5+ TFH cells in Mettl3fl/flCd4-Cre SMARTA cells by using the combination of CXCR5 and CD25 to identify TFH cells in activated CD4+ T cells (Supplementary Fig. 4e, f), and apoptosis of TFH cells was elevated in the absence of METTL3 (Supplementary Fig. 4g, h). We further assessed the expression levels of genes that are associated with TFH cells, including Tcf7, Cxcr5, Bcl6, Pdcd1, and Icos. The expression levels of these genes were substantially decreased in Mettl3fl/flCd4-Cre SMARTA TFH cells compared with those of Ctrl cells (Fig. 3i). Correspondingly, the expressions of Prdm1 and Id2, both well-known for promoting TH1 differentiation9,32, were much lower in Ctrl TFH cells than that of Mettl3fl/flCd4-Cre SMARTA cells (Fig. 3i). These results thus demonstrated that METTL3 is indispensable for early TFH commitment during acute viral infection.

a Flow cytometry analysis of cells from the wild-type recipient mice (CD45.1+) given adoptive transfer of naive CTV-labeled Ctrl or Mettl3fl/flCd4-Cre SMARTA cells, followed by LCMV-Armstrong infection and analysis 3 days later as CTV dilution by the transferred cells. b Flow cytometry analysis of Bcl-6+CXCR5+ TFH cells gated on SMARTA CD4+ T cells from recipient mice as in a. Summary of the frequency and cell numbers of TFH cells are shown on the right (n = 3 per group). c, d Flow cytometry analysis of different cell divisions gated on SMARTA CD4+ T cells from recipient mice as in a. The frequency of third to sixth divisions is summarized in d (n = 3 per group). e, f Contour plots display Bcl-6+CXCR5+ TFH cells in indicated divisions, among SMARTA cells from recipient mice as in a. Summary of the frequency and cell numbers of TFH cells in indicated divisions are shown in f (n = 3 per group). g, h Flow cytometry analysis of the expression of TCF-1 in CXCR5+ cells in indicated divisions gated on SMARTA CD4+ T cells from recipient mice as in a. Quantification of geometric mean fluorescence intensity (gMFI) of TCF-1 in indicated divisions is shown in h (n = 3 per group). i Quantitative RT-PCR analysis of mRNA abundance of TFH cell-related genes in CD25–CXCR5+ TFH cells from recipient mice as in a, relative expression was normalized to Ctrl cells (n = 6 per group). Data are representative of two independent experiments. Error bars indicate standard error of the mean. P value was calculated by unpaired two-tailed Student’s t test (b, i) or two-way ANOVA coupled with multiple comparisons (f, h).
METTL3 orchestrates the transcriptional profiles of TFH cells
We next investigated how METTL3 deficiency affects the transcriptional profiles of TFH cells. Considering CD4+ T cells generally differentiate into TH1 cells or TFH cells upon acute viral infection33 and METTL3 deficiency affects CXCR5 expression, we hence used CD44 and SLAM, which is expressed at low level on TFH cells9, to identify TH1 and TFH cells in activated CD4+ T cells. CD44+SLAMhi TH1 cells and CD44+SLAMlo TFH cells were sorted from Mettl3fl/flCd4-Cre mice and their Ctrl littermates on day 8 post viral infection and subjected to RNA-seq analysis. Compared with those in wild-type cells, 515 upregulated genes and 252 downregulated genes in METTL3-deficient TFH cells (Fig. 4a) and 763 upregulated genes and 332 downregulated genes (Supplementary Fig. 5a) in METTL3-deficient TH1 cells were identified, respectively (≥2-fold expression change, adjusted P value <0.01). In particular, the upregulated genes in TFH cells were significantly enriched in the T-cell differentiation and defense response to virus; the downregulated genes in TFH cells were apparently enriched in T-cell proliferation and differentiation (Fig. 4b). Then we selected a TFH cell and a GC TFH cell signature gene set11 for gene set enrichment analysis (GSEA). Both the TFH lineage gene set and the GC TFH-associated gene set were enriched in wild-type TFH cells, but not in METTL3-deficient TFH cells (Fig. 4c). When applying GSEA to exhibit the signature genes of TH1 (ref. 12), TH2 (ref. 34), TH17 (ref. 35), and Treg34 cells, we found that all of which were enriched in METTL3-deficient TFH cells (Supplementary Fig. 5b). Similarly, we also observed TFH, TH2, TH17, and Treg-related gene sets were positively enriched in METTL3-deficient TH1 cells (Supplementary Fig. 5c). These analyses indicated that loss of METTL3 leads to disordered gene profiles of both TFH and TH1 transcription program.

a Volcano map depicting genes upregulated (red) or downregulated (blue) 2-fold or more in TFH cells on 8 dpi. Heatmap of differentially expressed genes is shown on the right. b Gene Ontology (GO) terms of the differentially expressed genes in Mettl3fl/flCd4-Cre TFH cells compared with Ctrl TFH cells (Upregulated: top panel; Downregulated: bottom panel). c Gene set enrichment analysis (GSEA) of TFH and GC TFH gene set in Mettl3fl/flCd4-Cre TFH cells relative to expression in Ctrl TFH cells as in a. d GSEA analysis of “TCF-1-activated genes in TFH cells” and “TCF-1-suppressed genes in TFH cells” (GSE65693) in Mettl3fl/flCd4-Cre TFH cells relative to expression in Ctrl TFH cells as in a. Heatmaps representation of top 30 ranking genes in the leading edge are shown, respectively. e Quantitative RT-PCR analysis of selected interested genes in a, relative expression was normalized to that in Ctrl TFH cells (n = 7 for Ctrl group, n = 8 for Mettl3fl/flCd4-Cre group except Bcl6 gene (n = 7 per group)). Data are from one experiment with triplicates (a–d) or pooled from two experiments (e). Error bars indicate standard error of the mean. P value was calculated by unpaired two-tailed Student’s t test (e).
We next validated expression changes of key TFH genes and found that Cxcr5, Bcl6, Pdcd1, and Icos were significantly lower in METTL3-null TFH cells than in wild-type cells (Fig. 4e). In addition, the expression of Tcf7 was lower in METTL3-deficient TFH cells than in wild-type TFH cells (Fig. 4e). Among the genes with low expression, Tcf7 was of interest because of its role in Tfh cell differentiation, so we also performed GSEA analysis of a gene set containing TCF-1-activated genes in TFH cells13. Interestingly, remarkable enrichment was exhibited in wild-type TFH cells, whereas the TCF-1-suppressed gene set13 was notably enriched in METTL3-deficient cells, which suggested that METTL3 and TCF-1 share a common subset of target genes in TFH cells (Fig. 4d). Meanwhile, the expression of Lef1 mRNA was also decreased in METTL3-deficient TFH cells compared with wild-type cells (Fig. 4e). On the other hand, the expression of Prdm1, known as the antagonist of Bcl6, and Gzmb was higher in METTL3-null TFH cells than that in wild-type TFH cells (Fig. 4e). The expression levels of Bcl2l11 and Bax, both known as pro-apoptotic regulators36,37, were significantly elevated in METTL3-null TFH cells compared with those in the wild-type TFH cells (Fig. 4e), which corresponded to accelerated apoptosis in Mettl3fl/flCd4-Cre TFH cells (Supplementary Fig. 4g, h). In addition, the transcripts encoding other essential transcription factors, receptors and ligands for TFH differentiation, including Batf, Slamf6, and Il6ra, were downregulated in Mettl3fl/flCd4-Cre TFH cells compared with those in wild-type cells (Fig. 4e). Besides, we observed elevated expression of Socs1 mRNA, a known target of METTL325,26, in Mettl3fl/flCd4-Cre TFH cells (Fig. 4e). Collectively, these results suggested that METTL3 regulates TFH differentiation program by activating TFH program but suppressing TH1 lineage-associated genes.
METTL3 promotes TFH differentiation in an m6A catalytic activity-dependent manner
m6A methylation is catalyzed by a multicomponent methyltransferase complex, while METTL3 functions as the predominant catalytic subunit15,16. Next, we analyzed whether METTL3-mediated TFH differentiation is dependent on its m6A methyltransferase activity. To achieve this goal, we generated wild-type METTL3 (Mettl3-WT) and catalytic domain mutant METTL3 (D395A and W398A; Mettl3-Mut) constructs15 (Fig. 5a). These METTL3 proteins were then expressed in Mettl3fl/flCd4-Cre SMARTA CD4+ T cells with a retrovirus transduction system (Fig. 5b). On day 5 post infection, forced expression of Mettl3-WT and Mettl3-Mut both resulted in elevated METTL3 expression in Mettl3fl/flCd4-Cre SMARTA cells compared with the empty-vector (EV) retrovirus (Fig. 5c). EV retrovirus-infected Mettl3fl/flCd4-Cre SMARTA CD4+ T cells exhibited reduced expansion than EV retrovirus-infected Ctrl cells, which could be restored by overexpression of Mettl3-WT but not Mettl3-Mut (Fig. 5d). Moreover, EV retrovirus-infected Mettl3fl/flCd4-Cre SMARTA CD4+ T cells remained defective in the generation of CD44+CXCR5+ TFH cells, while Mettl3-WT but not Mettl3-Mut retrovirus promoted differentiation of Mettl3fl/flCd4-Cre SMARTA CD4+ T cells into a TFH fate (Fig. 5e). In addition, forced expression of Mettl3-WT could restore both the cell numbers of Mettl3fl/flCd4-Cre TFH cells and TH1 cells (Fig. 5f). Accordingly, ectopic expression of Mettl3-WT could largely rectify defective PD-1hiCXCR5+ GC TFH cells in METTL3-deficient cells (Fig. 5g, h). We also observed that the expression of TFH cell-related genes, including Cxcr5, Tcf7, Bcl6, Pdcd1, and Icos, were restored in Mettl3fl/flCd4-Cre cells with inducing Mettl3-WT expression (Fig. 5i). The data collectively indicated that the m6A-catalytic activity of METTL3 is necessary for TFH differentiation.

a Graphic representation of the wild-type (Mettl3-WT) and catalytic domain dead (Mettl3-Mut; DPPW to APPA) METTL3 constructs. b Scheme of retrovirus transduction experiment. SMARTA cells were transduced with indicated structures by using a retrovirus transduction system. Then, the transduced cells were adoptively transferred into congenic CD45.2+ wild-type mice followed by LCMV-Armstrong infection, and analyzed on day 5 post viral infection. c Flow cytometry analysis of METTL3 gMFI in GFP+CD4+ SMARTA cells from recipient mice. Quantitation of METTL3 gMFI is shown on the right (n = 3 per group). d Summary of the frequency and cell numbers of retrovirus-transduced SMARTA CD4+ T cells in host mice (n = 3 per group). e, f Flow cytometry analysis of CD44+CXCR5+ TFH populations and CD44+CXCR5– TH1 subsets gated on SMARTA GFP+CD4+ T cells from host mice adoptively transferred with empty vector (EV), Mettl3-WT, or Mettl3-Mut retrovirus-introduced SMARTA cells. Summary of the frequency and cell numbers of TFH cells and TH1 cells are shown in f (n = 3 per group). g, h Flow cytometry analysis of PD-1hiCXCR5+ GC TFH populations gated on SMARTA GFP+CD4+ T cells from host mice adoptively transferred with EV, Mettl3-WT, or Mettl3-Mut retrovirus-introduced SMARTA cells. Summary of the frequency and cell numbers of GC TFH cells are shown in h (n = 3 per group). i Quantitative RT-PCR analysis of mRNA abundance of TFH cell-related genes in CD44+CXCR5+ TFH cells ectopically expressed with EV, Mettl3-WT, or Mettl3-Mut, relative expression was normalized to Ctrl cells transduced with EV retrovirus (n = 6 for Ctrl EV, Mettl3fl/flCd4-Cre-EV, and Mettl3fl/flCd4-Cre-Mettl3 WT group; n = 5 for Mettl3fl/flCd4-Cre-Mettl3 Mut group). Data are representative of two independent experiments. Error bars indicate standard error of the mean. P value was calculated by one-way ANOVA, followed by unpaired two-tailed Student’s t test for indicated pairwise comparisons.
m6A modifies Tcf7 mRNA in 3′ UTR to control its stability
To examine how m6A methylation modulates TFH transcriptional program, we performed m6A-miCLIP-SMARTer-seq to map global m6A landscape in Ctrl or Mettl3fl/flCd4-Cre SMARTA CD4+ T cells primed in vivo. Bioinformatic analysis revealed that m6A peaks were significantly abundant in 3′ UTR and near the stop codon of mRNAs (Fig. 6a, b). Approximately 45.3% methylated mRNAs contained three or more peaks (Fig. 6c). Biological duplicates of m6A-miCLIP-SMARTer-seq yielded about 4939 transcripts (called ‘m6A-modified transcripts’ hereafter; Fig. 6d). By stratifying m6A-modified transcripts with differentially expressed genes, we found that 208 differentially expressed transcripts were potentially regulated by m6A methylation (Fig. 6d). GO term analysis showed these transcripts were enriched for functions related to defense response to the virus, T-cell activation and differentiation (Fig. 6e). Among these potential m6A methylated target genes, a set of TFH cell-relevant genes were directly marked by m6A, such as Cxcr5, Icos, Il6ra, and Il6st (Supplementary Fig. 6a). Interestingly, the 3′ UTR of Tcf7 mRNA had highly enriched m6A peaks, whereas Bcl6 and Prdm1 mRNAs were not tagged by m6A (Fig. 6f and Supplementary Fig. 6a). Accordingly, RNA immunoprecipitation (RIP) assay suggested METTL3 directly binds to Tcf7 mRNA (Fig. 6g). The m6A-RIP-qPCR results further indicated Tcf7 mRNA was tagged by m6A methylation and the m6A levels on Tcf7 mRNA were substantially decreased in METTL3-deficient cells (Fig. 6f, h). These data jointly indicated Tcf7 is a bona fide m6A target. Accordingly, both the Tcf7 mRNA and the TCF-1 protein were significantly decreased in METTL3-deficient TFH cells compared with those in wild-type TFH cells (Fig. 4e and Fig. 6i). To further validate METTL3 regulation of Tcf7 expression in an m6A-dependent manner, we identified a high-confidence m6A site (GGA1011CT, a conserved m6A methylation motif) in the Tcf7 3′ UTR region (Fig. 6f). We generated a minigene vector placing the Tcf7 m6A site to the 3′ end of luciferase reporter cDNA. Then the ‘GGACT’ consensus motif in the Tcf7 m6A site was mutated into ‘GGTCT’ to abrogate m6A modification (Fig. 6j). Wild-type Tcf7 3′ UTR exhibited increased luciferase activity compared to that of the empty vector (pGL4.23) in the presence of overexpressed METTL3; notably, mutation of Tcf7 3′ UTR almost completely compromised the increase (Fig. 6k). These results revealed that METTL3 regulates Tcf7 mRNA levels via m6A modifications in the 3′ UTR region (m6A1011). Given m6A modification regulates gene expression by multiple approaches, including mRNA splicing, stability, and translation17,38, alternative splicing assay was applied and exhibited no significant difference in the alternative splicing of Tcf7 mRNA between Ctrl and METTL3-deficient TFH cells (Supplementary Fig. 6b). We then performed RNA decay assay via actinomycin D (ActD) treatment to detect the stability of Tcf7 mRNA, and found that Tcf7 mRNA exhibited substantially accelerated decline in METTL3-deficient cells compared with Ctrl cells at checkpoints after treatment (Fig. 6l, m). Taken together, these data suggested that METTL3 enhances Tcf7 mRNA stability via catalyzing m6A methylation at its 3′ UTR, to ensure TCF-1 expression in promoting TFH differentiation.

a Metagene profiles of m6A site distribution along a normalized transcript containing three rescaled non-overlapping segments: 5′ UTR, CDS, and 3′ UTR in Ctrl and Mettl3fl/flCd4-Cre SMARTA CD4+ T cells. b Pie chart showing the distribution of m6A sites in five regions of Ctrl and Mettl3fl/flCd4-Cre SMARTA CD4+ T cells. c Bar chart depicting percentage of mRNAs with different internal m6A abundance. d Venn diagram showing the overlapping differentially expressed genes from RNA-seq and m6A-modified transcripts from m6A-miCLIP-SMARTer-seq. Heatmap of 208 differentially expressed genes with m6A modification is shown on the right. e Representative GO terms of the biological process categories enriched in differentially expressed transcripts with m6A peaks (Red: upregulated genes; blue: downregulated genes; gray: GO terms). f Integrative Genomics Viewer (IGV) tracks displaying RNA-seq (top panel) and m6A-miCLIP-SMARTer-seq (bottom panel) reads distribution of Tcf7 gene. The high-confidence m6A site is marked as a triangle. g RIP-qPCR analysis showing enrichment of METTL3 on Tcf7 mRNA in SMARTA CD4+ T cells. The Tcf7 mRNA enrichment is presented as IP/input and normalized to IgG group (n = 4 per group). h m6A-RIP-qPCR analysis of m6A enrichment on Tcf7 mRNA of Ctrl and Mettl3fl/flCd4-Cre SMARTA CD4+ T cells (n = 4 per group). i Flow cytometry analysis of expression level of TCF-1 on TFH cells on 8 dpi. Quantification of gMFI of TCF-1 is shown on the right (n = 5 per group). j Constructions of plasmids with wild-type or m6A site mutant in Tcf7 3′ UTR. k Luciferase reporter assay. Results were normalized to the luciferase activity of cells co-transfected with the pGL4.23 empty plasmid and EV-Myc plasmid (n = 6 per group). l RNA decay assay. The half-live of Tcf7 mRNA was detected by quantitative RT-PCR. The remaining mRNAs were normalized to t = 0 (n = 4 per group). m Quantitative RT-PCR analysis of Tcf7 mRNA abundance as in l, relative expression was normalized to t = 0 of Ctrl cells (n = 4 per group). Data are from one experiment with duplicate (a–e) or representative of at least three independent experiments (i–h, k–m). Error bars indicate standard error of the mean. P value was calculated by unpaired two-tailed Student’s t test (g–i) or two-way ANOVA coupled with multiple comparisons (k, m).
Forced expression of TCF-1 restores defective TFH differentiation in METTL3-deficient mice
To determine the functional link between METTL3 and Tcf7 transcript stability in TFH differentiation, we next examined the impact of ectopic expression of TCF-1 in METTL3-deficient cells. Upon transducing in vivo primed SMARTA CD4+ T cells with TCF-1 (full-length CDS of P45 isoform without 3′ untranslated region39) retrovirus (Fig. 7a), on day 8 post LCMV-Armstrong infection, we observed a significant increase in TCF-1 expression in METTL3-deficient SMARTA cells (Fig. 7b). We then analyzed TFH populations from transduced SMARTA CD4+ T cells in recipient mice. Compared with Ctrl SMARTA CD4+ T cells infected with EV retrovirus, the EV-infected Mettl3fl/flCd4-Cre SMARTA CD4+ T cells exhibited defects in CD44+CXCR5+ TFH differentiation; whereas TCF-1 retrovirus could largely rectify the ability of Mettl3fl/flCd4-Cre SMARTA CD4+ T cells to differentiate into TFH cells (Fig. 7c). In addition, TCF-1 overexpression also elevated the cell numbers of Mettl3fl/flCd4-Cre TFH cells, but not TH1 cells (Fig. 7d). Consistently, PD-1hiCXCR5+ GC TFH cells could also be rescued with TCF-1 overexpression (Fig. 7e, f). Meanwhile, forced expression of TCF-1 could largely restore the expression levels of TCF-1, CXCR5, PD-1, ICOS, and Bcl-6 on Mettl3fl/flCd4-Cre TFH cells (Fig. 7g, h). These data collectively demonstrated that METTL3 stabilizes Tcf7 mRNA expression to promote TFH differentiation.

a Scheme of TCF-1 rescue experiment. SMARTA cells were transduced with TCF-1 structure (full-length CDS of P45 isoform without 3′ UTR region) by using a retrovirus transduction system. Then, the transduced cells were adoptively transferred into congenic CD45.1+ wild-type mice followed by LCMV-Armstrong infection, and analyzed on day 8 post viral infection. b Flow cytometry analysis of TCF-1 gMFI in SMARTA GFP+CD4+ cells from recipient mice on 8 dpi. Quantitation of TCF-1 gMFI is shown on the right (n = 3 per group). c, d Flow cytometry analysis of CD44+CXCR5+ TFH populations and CD44+CXCR5– TH1 subsets gated on SMARTA GFP+CD4+ T cells from different host mice adoptively transferred with empty vector (EV) or TCF-1 retrovirus-introduced SMARTA cells at 8 days post infection. Summary of the frequency and cell numbers of TFH cells and TH1 cells are shown in d (n = 3 per group). e, f Flow cytometry analysis of PD-1hiCXCR5+ GC TFH populations gated on SMARTA GFP+CD4+ T cells from different host mice adoptively transferred with EV or TCF-1 retrovirus-introduced SMARTA cells. Summary of the frequency and cell numbers of GC TFH cells are shown in f (n = 3 per group). g, h Flow cytometry analysis of gMFIs of TCF-1, CXCR5, PD-1, ICOS, and Bcl-6 on CD44+CXCR5+ TFH cells transduced with EV or TCF-1 retrovirus. Quantification of the gMFIs is shown in h (n = 3 per group). Data are representative of three independent experiments. Error bars indicate standard error of the mean. P value was calculated by one-way ANOVA, followed by unpaired two-tailed Student’s t test for indicated pairwise comparisons.
Discussion
N6-methyladenosine (m6A) accounts for the prevalent mRNA modifications and has recently emerged as a critical epitranscriptomic regulator to affect the translation and stability of the modified transcripts. Here, we showed that ablation of METTL3 leads to substantial defects in both TFH and TH1 differentiation upon LCMV-Armstrong and KLH challenges. Based on our results, a more severe phenotype was exhibited in TFH cells than that of TH1 cells from METTL3-deficient mice, which strongly supports the notion that the METTL3 acts as an intrinsic modulator of TFH cells.
During acute viral infection, bifurcation of effector CD4+ T cells into TFH cells or TH1 cells can be observed as early as the second to third division40. A recent report demonstrated that METTL3-null CD4+ T cells remained naive state and exhibited defective proliferation when tested both in vivo and in vitro25. Using an adoptive transfer model, we found that METTL3-deficient CD4+ T cells showed a delayed differentiation and a slower proliferation upon acute viral infection, although they were activated normally as shown by expression of the surface markers via both in vivo and in vitro assay. Meanwhile, at 72 h post infection, the activated CD4+ T cells were mostly in the fifth and sixth divisions, whereas METTL3-deficient CD4+ T cells were dominant in third and fourth divisions. These METTL3-deficient CD4+ T cells also showed defects in differentiation into TFH cells and exhibited elevated apoptosis. Further, METTL3 deficiency also resulted in a decreased expression of TFH-defining genes in these nascent TFH cells. Strikingly, the abundances of Tcf7 and Bcl6 mRNAs, both essential for the early commitment of TFH cells41, were profoundly decreased in early METTL3-null TFH cells. Correspondingly, the protein levels of TCF-1 and Bcl-6 were also notably reduced. These findings revealed that METTL3 is required for early TFH commitment, proliferation, and survival by maintaining the key TFH gene expression.
As an RNA binding protein, METTL3 is the essential catalytic component of the conserved heterodimeric m6A writer complex15. Does METTL3-mediated TFH differentiation also directly depend on its m6A catalytic activity? Our results showed that forced expression of METTL3 with a mutated catalytic domain failed to rectify the defects in TFH differentiation of Mettl3fl/flCd4-Cre CD4+ T cells, while wild-type METTL3 did. We thus concluded that METTL3 promotes TFH generation in an m6A catalytic activity-dependent manner. Giving m6A methylation is essential for gene expression regulation42, we hence focused on the methylation status of those differentially expressed genes. The expression patterns of a series of TFH-defining genes were altered at both early and mature stages, including Tcf7, Cxcr5, Bcl6, Pdcd1, and Icos. Through m6A-miCLIP-SMARTer-seq, we found Tcf7 mRNA was tagged by m6A in the 3′ UTR, whereas the other key TFH cell regulator Bcl6 mRNA was not modified. Further study indicated that METTL3-mediated m6A modification regulates the stability of Tcf7 mRNA, ultimately maintains TCF-1 level. Moreover, forced expression of TCF-1 could restore the TFH differentiation in the absence of METTL3. These data collectively suggested that METTL3 directly modulates the Tcf7 expression level in an m6A-dependent manner to promote TFH differentiation.
The functional outcome of m6A modification is determined by an expanding list of m6A reader proteins in a cell type and cellular context-dependent fashion14,43. Although early evidence demonstrated that m6A deposition destabilizes mRNAs, resulting in their faster decay, a recent study has reported that the insulin-like growth factor 2 mRNA binding proteins (IGF2BP1-3), as a class of distinct m6A readers, promote m6A-modified mRNA stability and translation20. Similarly, a more recent study reported Prrc2a as a novel m6A reader that regulates oligodendrocyte specification and myelination by stabilizing target mRNA44. Our data also indicated m6A modification in the 3′ UTR of Tcf7 enhances its mRNA stability to promote TFH differentiation. However, the currently known IGF2BPs family readers are expressed at extremely low levels to detect in CD4+ T cells (Supplementary Fig. 8), implying the possibility of other unidentified proteins might be responsible for deciphering the m6A-modified transcripts in TFH cells. Therefore, it will be of great interest to identify new RNA-binding proteins virtually involved in TFH cells via post-transcriptional networks in future studies.
In METTL3-deficient cells, destabilization of Tcf7 mRNA impacted TFH cells at least in two major aspects. The first aspect is that loss of TCF-1 expression results in massive apoptosis in T cell45, which is coordinated with the decreased TFH cell numbers in the absence of METTL3. The other point lies in that TCF-1 directly regulates Bcl6 mRNA expression11,12,13, and two of them thus collectively affect TFH differentiation. This post-transcriptional regulation of Tcf7 mRNA represents a distinct mechanism that drives TFH differentiation. It should be noted that other post-transcriptional regulators such as RNA-binding proteins and miRNAs also contribute to modulating TFH differentiation program46,47,48. Hence, exploring TFH fate determination on the layer of post-transcriptional level might be a fruitful effort in future investigations.
A recent study referred that induced GAPDH protein by VHL deficiency reduced Icos expression through METTL3/METTL14-catalyzed m6A modification on Icos mRNA, implying that elevated m6A modification on Icos mRNA in VHL-deficient cells decreases Icos expression which is associated with attenuated TFH differentiation49. By analyzing high-throughput data, we also observed m6A modification in the 3′ UTR of Icos mRNA, and the m6A level on Icos mRNA was decreased in the absence of METTL3. However, we found both the mRNA and protein level of ICOS were blunted in METTL3-deficient TFH cells, indicating that loss of m6A modification impairs Icos expression. In addition, Zhu et al. reported that knockdown of METTL3 expression with short hairpin RNA (shRNA) in CD4+ T cells could promote TFH differentiation49, which differs in phenotypes from our genetic knockdown mice model. The varies may be contributed by the distinct experimental approaches and the divergent viewpoints from two studies also reflect the complex regulatory mechanism of m6A modification, which needs to be further disclosed.
In summary, our study uncovers a critical role of METTL3-dependent m6A methylation in directing TFH lineage differentiation. Conditional ablation of m6A ‘writer’ METTL3 in CD4+ T cells intrinsically impaired the TFH differentiation, proliferation, and survival. Consequently, the GC reactions were significantly compromised in METTL3-deficient mice in response to acute viral infection. Our data indicated that METTL3 directs m6A modification in 3′ UTR of Tcf7 mRNA to stabilize the transcript and hence sustain TCF-1 protein expression (Fig. 8). Thus, m6A functions as an important modulator of the METTL3-TCF-1 axis to initiate and secure the differentiation of TFH cells post-transcriptionally.

During acute infection, METTL3-sufficient CD4+ T cells were activated. With m6A machinery, Tcf7 mRNA was m6A modified and stabilized, allowing normal production of TCF-1 protein. TCF-1 in turn regulates expressions of TFH cell regulators, which ultimately program TFH commitment, proliferation, survival, and functional maturation.
Methods
Mice
Mettl3fl/fl mice were kindly provided by Drs. Qi Zhou and Wei Li (Institute of Zoology, Chinese Academy of Sciences). SMARTA mice50 (expressing MHC II I-Ab-restricted TCR specific for LCMV glycoprotein amino acids 66–77) were generously provided by Dr. Rafi Ahmed (Emory University). Cd4-Cre, ERT2-Cre, and C57BL/6J (CD45.2 and CD45.1) mice were purchased from the Jackson Laboratory. All mouse strains used in this study are on a fully C57BL/6J background. All mice were kept in group housing (3–5 mice per cage) in a specific pathogen-free facility with controlled environmental conditions of humidity (50 ± 10%), lighting (a 12-h light/dark cycle), and temperature (21 ± 1 °C) at China Agricultural University. All animal experiments were performed in accordance with the protocol of the Institutional Animal Care and Use Committee of China Agricultural University.
LCMV infection
LCMV-Armstrong strain was grown in BHK-21 cells and titers were determined as described before51. Mettl3fl/flCd4-Cre mice and their wild-type littermates were intraperitoneally infected with 2 × 105 plaque-forming units (pfu) LCMV-Armstrong strain. In adoptive transfer experiments, recipient mice were infected 1 day after cell transfer. For bone marrow chimeric mice, LCMV infection was performed after 8 weeks reconstitution.
Immunization
Mettl3fl/flCd4-Cre mice and their wild-type littermates were intraperitoneally immunized with 100 μg of KLH (Sigma-Aldrich) emulsified in CFA (Sigma-Aldrich). Eight days later, splenic T cells were analyzed.
Flow cytometry and antibodies
Single-cell suspensions of spleens were used for flow cytometry analysis or cell sorting. Surface staining was performed in PBS containing 1% FBS. The antibodies and reagents used for flow cytometry staining are listed as: anti-CD19 (1D3; 1:100), anti-CD25 (PC61.5; 1:100), anti-CD4 (RM4-5; 1:100), anti-CD44 (IM7; 1:100), anti-CD45.1 (A20; 1:100), anti-CD45.2 (104; 1:100), anti-CD62L (MEL-14; 1:100), anti-CD69 (H1.2F3; 1:100), anti-CD8a (53-6.7; 1:100), anti-B220 (RA3-6B2; 1:100), anti-GITR (DTA-1; 1:100), anti-GL7 (GL7; 1:100), anti-PD-1 (J43; 1:100), anti-TCR Vα2 (B20.1; 1:100) (from Thermo Fisher Scientific); anti-CD138 (281-2; 1:100), anti-Fas (Jo2; 1:100) (from BD Biosciences); anti-SLAM (TC15-12F12.2; 1:100), anti-ICOS (C398.4A; 1:100) (from BioLegend), and peanut agglutinin (PNA; Cat. no. FL-1071; 1:500; Vector laboratories). CXCR5 staining was performed with a three steps staining protocol as described before52. Briefly, single-cell suspensions were first stained with purified anti-CXCR5 (2G8; 1:100; BD Biosciences) for 1 h, followed by biotin-conjugated goat anti-rat IgG (Cat. no. 111-066-144; 1:1,000; Jackson ImmunoResearch) for 30 min, and then by APC-eFluor 780-, or eFluor 450-labeled streptavidin (1:500; Thermo Fisher Scientific) at 4 °C for 30 min in PBS supplemented with 2% normal mouse serum (Cat. no. 015-000-120; Jackson ImmunoResearch), 2% FCS, and 0.5% BSA. For detection of cytokines, the splenocytes from KLH-immunized mice were cultured in vitro for 5 h in 2 μg/mL of PMA (Cat. no. P8139; Sigma-Aldrich), 2 μg/mL of Ionomycin (Cat. no. I0634; Sigma-Aldrich), GolgiStop (BD Biosciences), and GolgiPlug (BD Biosciences). Intracellular staining for cytokines was performed with monoclonal antibody against to IL-4 (11B11; 1:100; Thermo Fisher Scientific) and IL-17a (TC11-18H10; 1:100; BD Biosciences), using the Fixation/Permeabilization buffer kit (BD Biosciences). For intracellular staining of Bcl-6 (K112-91; 1:20; BD Biosciences), Foxp3 (FJK-16s; 1:100; Thermo Fisher Scientific), GATA3 (TWAJ; 1:100; Thermo Fisher Scientific), RORγt (AFKJS-9; 1:100; Thermo Fisher Scientific), TCF-1 (C63D9; 1:100; Cell Signaling Technology), and METTL3 (Cat. no. ab195352; 1:100; Abcam), Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) was used following the manufacturer’s instructions. Active Caspase-3 was detected using the CaspGLOW™ Fluorescein Active Caspase-3 Staining Kit (Cat. no. 88-7004-42; 1:200; Thermo Fisher Scientific). All data were collected on a FACSVerse (BD Biosciences) with FACSSuite software (v1.0.5) or an LSRFortessa (BD Biosciences) with FACSDiva software (v8.0.2) and were analyzed with FlowJo software (v10; Treestar). The gating strategies for flow cytometry data analysis are illustrated in Supplementary Fig. 7.
Adoptive transfer
To characterization of cell division at early TFH differentiation, SMARTA CD4+ T cells were labeled with 5 μM of CTV (Invitrogen), and 2 × 106 of labeled Vα2+ SMARTA CD4+ cells were transferred followed by intravenously infected with 2 × 106 pfu of LCMV-Armstrong. To investigate the intrinsic effects with adoptive transfer model, 5 × 106 wild-type SMARTA cells were transferred into Ctrl and Mettl3fl/flCd4-Cre host mice, followed by 2 × 105 pfu LCMV-Armstrong infection intraperitoneally.
Bone marrow chimeric mice
To generate bone marrow chimeric mice, lethally irradiated B6.SJL (CD45.1+) mice were transferred intravenously with a 1:1 mixture of 2.5 × 106 Mettl3fl/flCd4-Cre (CD45.2+) and 2.5 × 106 B6.SJL (CD45.1+) bone marrow cells. After 8 weeks reconstitution, recipient mice were infected with LCMV-Armstrong strain.
ELISA
Analysis of the LCMV-specific antibody in serum was performed as previously described53. Briefly, lysates of LCMV-infected BHK-21 cells were used as substrate and LCMV-specific antibody (IgG) was titrated in serial dilutions of serum using HPR-conjugated goat-anti mouse IgG antibodies (Cat. no. A90-131P-39; 1:5,000; Bethyl laboratories).
Immunofluorescence staining
Tissue specimens submerged in OCT compound were quickly frozen in liquid nitrogen and cut into 10 μm thickness. Frozen tissue sections were then fixed in cold acetone for 30 min at −20 °C, blocked with 1% BSA and Fc-blocker (2.4G2; 1:100; BD Biosciences) in PBS. The tissue sections were then stained with biotinylated PNA (Cat. no. BA-0074; 1:20; Vector laboratories), followed by staining with BV510-labeled anti-CD4 (RM4-5; 1:100; BD Biosciences), APC-labeled anti-IgD (11-26c; 1:100; Thermo Fisher Scientific), and AF488-conjugated streptavidin (1:500; Invitrogen). Then the slides were washed at least three times with PBS. Coverslips were mounted on slides using an antifade kit (Beyotime Biotechnology) and then examined using an Andor Dragonfly confocal microscope. The images were processed with Imaris (v8.1; Bitplane) and Image J (v1.52 g; NIH).
Retroviral vectors, transduction, and cell transfer
Tcf7 (full-length CDS region of P45 isoform without 3′ UTR region39) and Mettl3 (wild-type and catalytic domain dead) coding sequences were amplified and cloned into the pMIG-R1 vector (MSCV-IRES-GFP). Retrovirus was packaged by transfection of HEK293T cells (Cat. no. CRL-3216; ATCC) with the retroviral vectors along with pCLeco plasmid. SMARTA cells were activated in vivo by injection of 200 μg of LCMV GP61-80 (GLNGPDIYKGVYQFKSVEFD) peptide into Mettl3fl/flCd4-Cre SMARTA mice or their littermate wild-type transgenic mice. After 16~18 h, activated SMARTA cells were isolated, purified, and ‘spin-infected’ for 120 min at 37 °C by centrifugation (1000 × g) with freshly harvested retroviral supernatants supplemented with 8 μg/mL of polybrene (Sigma-Aldrich), then cultured overnight in the presence of 20 ng/mL of IL-2 (Peprotech), and 250 nM of LCMV GP61-80. The spinofection was repeated the next day, and a total of 0.5~1 × 106 retroviral infected SMARTA CD4+ T cells were then adoptively transferred into recipient mice, followed by infection of the hosts with 2 × 105 pfu LCMV-Armstrong within 24 h.
RNA-seq and data analysis
For isolation of TH1 cells and TFH cells, splenocytes from Mettl3fl/flCd4-Cre mice and their control littermates on day 8 post viral infection were subjected to depletion of cells positive for lineages markers by using biotin-conjugated antibodies (anti-B220 (RA3-6B2; 1:100), anti-CD8 (53-6.7; 1:100), anti-Gr.1 (RB6-8C5; 1:100), anti-CD11b (M1/70; 1:100), anti-CD11c (N418; 1:100), anti-TER119 (TER-119; 1:100), and anti-CD49b (DX5; 1:100); all from Thermo Fisher Scientific) coupled to Dynabeads M-280 Streptavidin (Invitrogen), followed by surface stained. CD44+SLAMhi TH1 cells and CD44+SLAMlo TFH cells were sorted with a FACSAria II cell sorter (BD Biosciences) with FACSDiva software (v7.0) and subsequently lysed with TRIzol Reagent (Life technologies). Total RNAs were extracted and then subjected to Annoroad (Beijing, China) for library construction and RNA sequencing. The qualities of clean reads were assessed by FastQC (v0.11.5). Then the reads were mapped to mouse genome mm10 (version M17) using TopHat (v2.1.1). The read counts of all genes were estimated by HTseq (v0.6.1) and differentially expressed genes were identified by DESeq2 (v1.18.1). TPM, FPKM, and RPKM were calculated, and upregulated or downregulated genes in Mettl3fl/flCd4-Cre TFH or TH1 cells were identified by expression changes ≥ 2-fold and FDR < 0.01.
Quantitative RT-PCR
Total RNAs were extracted from sorted cells using RNeasy Mini Kit (Qiagen) followed by cDNA synthesis with FastQuant RT Kit (Tiangen). Quantitative RT-PCR was carried out with SuperReal PreMix Plus SYBR Green (Tiangen) on a CFX96 ConnectTM Real-Time System (Bio-Rad). Results were processed by Microsoft Excel and then were normalized to the expression of Hprt1 transcripts. Fold differences in expression levels were calculated according to the 2−ΔΔCT method. All primers used are listed in Supplementary Table 1.
Gene set enrichment analysis
GSEA was performed with GSEA desktop software (v4.1.0) from the Broad Institute. The TFH gene set11, GC TFH gene set11, TH1 gene set12, TH2 gene set34, TH17 gene set35, and Treg gene set34 have been described before. The ‘TCF1-activated genes in TFH cells’ gene set contains 569 genes that are downregulated by ≥1.5-fold in Tcf7fl/flCd4-Cre TFH cells; The ‘TCF1-suppressed genes in TFH cells’ gene set contains 513 genes that are upregulated by ≥1.5-fold in Tcf7fl/flCd4-Cre TFH cells (GSE65693).
Luciferase reporter assay
Tcf7 3′ UTR segment (~200 nt) was amplified from a mouse double-positive (DP) thymocytes cDNA library by PCR and inserted into the pGL4.23 vector (Promega) by using XbaI and FseI restriction sites. The Tcf7 3′ UTR mutation plasmids were generations from site-directed mutagenesis. All plasmids and mutations were verified by sequencing. HEK293T cells were seeded into 24-well plates in triplicate to allow 80% confluency in the next day. A total of 200 ng of reporter plasmids (Fluc) and 20 ng of Renilla luciferase (Rluc) control plasmids (pRL-TK) were co-transfected using Lipofectamine 2000 reagent (Invitrogen) under METTL3 overexpressing. Fluc and Rluc activities were measured 24 h later with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. The relative luciferase activity was calculated by dividing Fluc by Rluc and normalized to pGL4.23 empty vector for each assay.
RNA decay assay
CD4+ T cells were purified from Mettl3fl/flERT2-Cre mice and their control wild-type mice as described above. 5 × 105 purified CD4+ T cells in RPMI 1640 medium supplemented with 10% fetal bovine serum, 20 ng/mL of IL-2, 10 ng/mL of IL-7, and 5 μM of 4-Hydroxytamoxifen (Sigma-Aldrich) were seeded into 48-well plates. After 48 h, actinomycin D (MedChemExpress) was added to a final concentration of 5 μM, and cells were harvested at t = 0, 1, 2 h after actinomycin D treatment. Total RNAs were extracted and subjected to RT-qPCR analysis. Results were processed by Microsoft Excel and then normalized to the expression of Gapdh transcript. Fold differences in expression levels were calculated according to the 2–ΔΔCT method. All primers used are listed in Supplementary Table 1.
m6A-miCLIP-SMARTer-seq and data processing
Total RNAs from CD4+ SMARTA cells sorted from LCMV GP61-80-primed Mettl3fl/flCd4-Cre SMARTA or Ctrl SMARTA mice were extracted with TRIzol Reagent (Life Technologies). mRNAs were further isolated from total RNAs using Dynabeads mRNA purification kit (Ambion). The procedures of m6A-miCLIP-SMARTer-seq were according to the previously reported methods with some modifications54. Briefly, 100 ng of mRNAs were fragmented to ~100 nt by using the fragmentation reagent (Life Technologies) and incubated with 5 μg of specific antibody against m6A (Abcam) in 500 μL of immunoprecipitation buffer (50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 0.05% NP-40) with gentle rotation at 4 °C for 2 h. The mixture was then transferred into a clear flat-bottom 96-well plate (Corning) on ice and irradiated three times with 0.15 J/cm−2 at 254 nm in a CL-1000 Ultraviolet Crosslinker (UVP). The irradiated mixture was then transferred to a new tube and incubated with 50 μL of pre-washed Dynabeads Protein A (Life Technologies) at 4 °C for 2 h. After extensive washing twice with high-salt wash buffer (50 mM Tris-HCl (pH 7.4), 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS) and twice with immunoprecipitation buffer, the mixture on beads was subjected to dephosphorylation with T4 PNK (NEB) for 20 min at 37 °C. After extensive washing, the RNA was eluted from the beads by proteinase K (Sigma-Aldrich) digestion at 55 °C for 1 h, followed by phenol-chloroform extraction and ethanol precipitation. The purified RNA was subjected to library construction using a SMARTer smRNA-Seq Kit for Illumina (Clontech Laboratories) according to the manufacturer’s instructions. Sequencing was carried out on an Illumina X-ten platform. m6A-miCLIP-SMARTer-seq data (paired-end) were analyzed as previously described55. Adapter sequences at the 3′ ends were removed first. R2 reads were then transformed to reverse complementary sequences and merged with R1 reads. PolyA tails from the library were trimmed by Cutadapt (v1.17) and fastq2collapse (v1.1.3) was used to remove duplicated reads; afterward, the barcodes of reads were removed. Low-quality bases were discarded, and only reads longer than 18 nt were retained. The remaining reads were mapped to the reference genome (mm10) using BWA (v0.7.17) with the parameter: -n 0.06. The mutation information was extracted and PCR duplicates according to BWA’s results were removed separately by parseAlignment.pl (–map-qual 1 –min-len 18) and tag2collapse.pl (-EM 30 –seq-error-model alignment). CIMS.pl was used to calculate the coverage (n) of the mutation site and transition number (m). Mutation sites with parameters, m ≥ 3, k/m ≥ 0.01, and k/m ≤ 0.5, were kept. The remaining sites within the RRACH motif sequence were considered as m6A.
m6A-RIP-qPCR
Purified mRNAs of CD4+ SMARTA cells sorted from Mettl3fl/flCd4-Cre SMARTA or Ctrl SMARTA mice were prepared and fragmented into ~100 nt by RNA fragmentation reagents (Life Technologies). Immunoprecipitation was performed using anti-m6A antibody (Abcam) as described above. The enrichment of m6A was measured with quantitative RT-PCR. Primers for m6A-RIP-qPCR are listed in Supplementary Table 1.
RIP-qPCR
5 × 106 CD4+ SMARTA T cells were isolated and lysed with 1 mL cell lysis buffer (150 mM KCl, 10 mM HEPES (pH 7.6), 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1:100 proteinase inhibitor cocktail, and 0.4 U/µL RNasin) at 4 °C for 30 min. After centrifugation, the supernatant (10% of which was kept as input) was subjected to RNA immunoprecipitation with anti-METTL3 (Abcam) coupled with Dynabeads Protein A (Life Technologies). RNA was isolated from the beads and input samples for RT-qPCR. Primers for RIP-qPCR are listed in Supplementary Table 1.
Statistical analysis
Statistical analysis was performed with Prism 8.0 (GraphPad). An unpaired two-tailed Student’s t test with a 95% confidence interval, one-way ANOVA, or two-way ANOVA analysis was used to calculate P values.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA-seq and m6A-miCLIP-SMARTer-seq datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE129650. All data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011).
Ueno, H., Banchereau, J. & Vinuesa, C. G. Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol. 16, 142–152 (2015).
Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542 (2014).
Tangye, S. G., Ma, C. S., Brink, R. & Deenick, E. K. The good, the bad and the ugly - TFH cells in human health and disease. Nat. Rev. Immunol. 13, 412–426 (2013).
Nutt, S. L. & Tarlinton, D. M. Germinal center B and follicular helper T cells: siblings, cousins or just good friends? Nat. Immunol. 12, 472–477 (2011).
Vinuesa, C. G., Linterman, M. A., Yu, D. & MacLennan, I. C. Follicular helper T cells. Annu. Rev. Immunol. 34, 335–368 (2016).
Yu, D. et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468 (2009).
Nurieva, R. I. et al. Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005 (2009).
Johnston, R. J. et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009).
Choi, Y. S., Yang, J. A. & Crotty, S. Dynamic regulation of Bcl6 in follicular helper CD4 T (Tfh) cells. Curr. Opin. Immunol. 25, 366–372 (2013).
Choi, Y. S. et al. LEF-1 and TCF-1 orchestrate TFH differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat. Immunol. 16, 980–990 (2015).
Wu, T. et al. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep. 12, 2099–2110 (2015).
Xu, L. et al. The transcription factor TCF-1 initiates the differentiation of TFH cells during acute viral infection. Nat. Immunol. 16, 991–999 (2015).
Shi, H., Wei, J. & He, C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019).
Wang, P., Doxtader, K. A. & Nam, Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63, 306–317 (2016).
Wang, X. et al. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 542, 260 (2017).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Du, H. et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016).
Huang, H. et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624 (2019).
Zhang, C. et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276 (2017).
Lee, H. et al. Stage-specific requirement for Mettl3-dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nat. Cell Biol. 21, 700–709 (2019).
Vu, L. P., Cheng, Y. & Kharas, M. G. The biology of m6A RNA methylation in normal and malignant hematopoiesis. Cancer Discov. 9, 25–33 (2019).
Li, H. B. et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342 (2017).
Tong, J. et al. m6A mRNA methylation sustains Treg suppressive functions. Cell Res. 28, 253–256 (2018).
Winkler, R. et al. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 20, 173–182 (2019).
Zheng, Q., Hou, J., Zhou, Y., Li, Z. & Cao, X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 18, 1094–1103 (2017).
Szabo, S. J. et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000).
Choi, Y. S. et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34, 932–946 (2011).
Chen, Y. P. & Yu, D. TCF-1 at the Tfh and Th1 divergence. Trends Immunol. 36, 758–760 (2015).
Shaw, L. A. et al. Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat. Immunol. 17, 834–843 (2016).
Hale, J. S. et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38, 805–817 (2013).
Tibbitt, C. A. et al. Single-cell RNA sequencing of the T helper cell response to house dust mites defines a distinct gene expression signature in airway Th2 cells. Immunity 51, 169–184.e165 (2019).
Ciofani, M. et al. A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012).
Oltvai, Z. N., Milliman, C. L. & Korsmeyer, S. J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609–619 (1993).
O’Connor, L. et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17, 384–395 (1998).
Frye, M., Harada, B. T., Behm, M. & He, C. RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018).
Ioannidis, V., Beermann, F., Clevers, H. & Held, W. The β-catenin-TCF-1 pathway ensures CD4+CD8+ thymocyte survival. Nat. Immunol. 2, 691–697 (2001).
Vinuesa, C. G. & Cyster, J. G. How T cells earn the follicular rite of passage. Immunity 35, 671–680 (2011).
Crotty, S. T follicular helper cell biology: a decade of discovery and diseases. Immunity 50, 1132–1148 (2019).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Huang, H., Weng, H. & Chen, J. The biogenesis and precise control of RNA m6A methylation. Trends Genet. 36, 44–52 (2020).
Wu, R. et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 29, 23–41 (2019).
Kovalovsky, D. et al. Beta-catenin/Tcf determines the outcome of thymic selection in response to αβTCR signaling. J. Immunol. 183, 3873–3884 (2009).
Pratama, A. et al. Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity 38, 669–680 (2013).
Vogel, K. U. et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity 38, 655–668 (2013).
Hu, R. et al. miR-155 promotes T follicular helper cell accumulation during chronic, low-grade inflammation. Immunity 41, 605–619 (2014).
Zhu, Y. et al. The E3 ligase VHL promotes follicular helper T cell differentiation via glycolytic-epigenetic control. J. Exp. Med. 216, 1664–1681 (2019).
Oxenius, A., Bachmann, M. F., Zinkernagel, R. M. & Hengartner, H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 28, 390–400 (1998).
Welsh, R. M. & Seedhom, M. O. Lymphocytic choriomeningitis virus (LCMV): propagation, quantitation, and storage. Curr. Protoc. Microbiol. Chapter 15, Unit 15A.1 (2008).
Yao, Y. et al. Long noncoding RNA Malat1 is not essential for T cell development and response to LCMV infection. RNA Biol. 15, 1477–1486 (2018).
Ahmed, R., Salmi, A., Butler, L. D., Chiller, J. M. & Oldstone, M. B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160, 521–540 (1984).
Zhang, Z. et al. METTL3-mediated N6-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 28, 1050–1061 (2018).
Moore, M. J. et al. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc. 9, 263–293 (2014).
Acknowledgements
We thank Drs. Qi Zhou and Wei Li (Institute of Zoology, Chinese Academy of Sciences) for Mettl3fl/fl mice; Dr. Rafi Ahmed (Emory University) for SMARTA mice; Dr. Hai-Hui Xue (Hackensack University Medical Center) for constructive advice on the manuscript; Dr. Xinyuan Zhou (Third Military Medical University) for retroviral vectors; and members of Drs. Ye, Yang and Yu Laboratories for their technical advice and help. This work was supported in part by grants National Key Research and Development Program of China (2017YFA0104401 to S.Y.), National Natural Scientific Foundation of China (31970831, 31630038, 31571522, & 31422037 to S.Y.; 31825011 to L.Y.; 31430022 to Y.-G.Y.), the Project for Extramural Scientists supported by State Key Laboratory of Agrobiotechnology of China Agricultural University (2018SKLAB6-30, 2019SKLAB6-6, & 2019SKLAB6-7 to S.Y.), and the Youth Innovation Promotion Association (CAS 2018133 to Y.-G.Y.).
Author information
Authors and Affiliations
Contributions
Y. Yao and W.G. performed the overall experiments with Y. Yang and L.X. M.Y., Z.S., X.C., G.Y., Z.Q., Jingjing L., and T.Z. helped to carry out experiments and interpreted data. Juanjuan L. helped to breed B6.SJL mice. Y. Yang executed m6A-miCLIP-SMARTer-seq experiments. M.Y., Y.Z., and F.W. analyzed the RNA-seq and m6A-miCLIP-SMARTer-seq data. Y. Yao, W.G., and S.Y. were responsible for analysis of overall data. Y. Yao and S.Y. wrote the manuscript with the revision by all authors. S.Y., L.Y., and Y.-G.Y. conceived experiments and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yao, Y., Yang, Y., Guo, W. et al. METTL3-dependent m6A modification programs T follicular helper cell differentiation. Nat Commun 12, 1333 (2021). https://doi.org/10.1038/s41467-021-21594-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-21594-6
This article is cited by
-
Regulation of inflammatory diseases via the control of mRNA decay
Inflammation and Regeneration (2024)
-
METTL3-mediated m6A methylation regulates ovarian cancer progression by recruiting myeloid-derived suppressor cells
Cell & Bioscience (2023)
-
m6A modification patterns are associated with copy number burden and tumor immune landscape in thyroid cancer
BMC Endocrine Disorders (2023)
-
RNA N6-methyladenosine modification in female reproductive biology and pathophysiology
Cell Communication and Signaling (2023)
-
m6A methylation: a process reshaping the tumour immune microenvironment and regulating immune evasion
Molecular Cancer (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
