
Abstract
Respiratory electron transport complexes are organized as individual entities or combined as large supercomplexes (SC). Gram-negative bacteria deploy a mitochondrial-like cytochrome (cyt) bc1 (Complex III, CIII2), and may have specific cbb3-type cyt c oxidases (Complex IV, CIV) instead of the canonical aa3-type CIV. Electron transfer between these complexes is mediated by soluble (c2) and membrane-anchored (cy) cyts. Here, we report the structure of an engineered bc1-cbb3 type SC (CIII2CIV, 5.2 Å resolution) and three conformers of native CIII2 (3.3 Å resolution). The SC is active in vivo and in vitro, contains all catalytic subunits and cofactors, and two extra transmembrane helices attributed to cyt cy and the assembly factor CcoH. The cyt cy is integral to SC, its cyt domain is mobile and it conveys electrons to CIV differently than cyt c2. The successful production of a native-like functional SC and determination of its structure illustrate the characteristics of membrane-confined and membrane-external respiratory electron transport pathways in Gram-negative bacteria.
Similar content being viewed by others


Introduction
Respiratory chains couple exergonic electron transport from nutrients to the terminal acceptor oxygen (O2) and generate a proton motive force used for ATP synthesis. Complex I (NADH dehydrogenase) and Complex II (succinate dehydrogenase) are the entry points into the chain of reducing equivalents, reducing quinone (Q) to hydroquinone (QH2). Complex III (cytochrome (cyt) bc1 or CIII2) oxidizes QH2 to reduce cyt c, which in turn is oxidized by Complex IV (cyt c oxidase or CIV) converting oxygen to water1 (Fig. 1a).

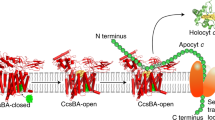
a Bifurcated electron transfer reaction conveys one electron from QH2 oxidation to the FeS protein (FeS, yellow) [2Fe-2S] cluster and another electron to cyt b (periwinkle) hemes bL and bH. Reduced [2Fe-2S] cluster transfers the electron to cyt c1 (green) heme c1 via the movement of the FeS protein from near heme bL to near heme c1. The subsequent electron transfer step from heme bH to Q from the pool is not shown for clarity. A cyt c (c2 or cy) carries electrons from heme c1 to CIV, where electrons reach the heme-Cu (Cu-b3) site, where O2 is reduced to H2O, via the CcoP (P, light blue) hemes cp1 and cp2, CcoO (O, dark green) heme co, and CcoN (N, purple) heme b. b R. capsulatus petABC encodes the structural genes of the bc1-type CIII2 subunits, the FeS protein (petA, yellow), cyt b (B, periwinkle), and cyt c1 (C, green), and ccoNOQP encodes those of the cbb3-type CIV subunits, the CcoN (ccoN, purple), CcoO (O, dark green), CcoQ (Q, gray), and CcoP (P, light blue). cycY gene (red) encodes cyt cy, and its 30-residue transmembrane helix (TMH), 69-residue linker (L), and 100-residue cyt c (cyt c) domain are indicated. Heme cofactors of b- and c-type cyts are indicated by black and white asterisks, respectively, whereas diamond and dot designate the [2Fe-2S] cluster and Cu atom, respectively. c Plasmid-borne genetic fusions. The bipartite fusion (left) is formed by in-frame linking the 3′-end of petC to the 5′-end of ccoP and the tripartite fusion (right) by adding the linker and cyt c domain of cycY to the 3′-end of ccoP. Colors and cofactors are as in a, and the his (H) and flag (F) affinity tags (dark purple) of the bipartite and tripartite fusion subunits are shown. d Schematic depiction of bipartite (left) and tripartite (right) supercomplexes (SC). The bipartite SC encodes a CIII2 dimer fused on each side to a His-tagged CIV. The tripartite SC also contains the Flag-tagged cyt domain of cy (red) at the end of CcoP (blue).
Respiratory complexes are evolutionarily conserved, and bacterial enzymes are structurally simple, consisting mainly of the catalytic subunits. CIII2 is a dimer with each monomer comprised of three subunits: the Rieske FeS (FeS) protein with a [2Fe-2S] cluster, cyt b with hemes bH and bL, and cyt c1 with heme c1 cofactors (Fig. 1a, b). The FeS protein external domain (FeS-ED) is mobile between the b (close to heme bL) and c (close to heme c1) positions2,3. Some bacterial species such as Rhodobacter capsulatus4 and pathogens like Helicobacter pylori, Campylobacter jejuni5, and Neisseria6 contain only a high oxygen affinity cbb3-type CIV to support their micro-aerophilic growth. The cbb3-type CIV is a monomer comprised of four subunits: CcoN with heme b and heme b3-Cu binuclear center, CcoO with heme co, CcoQ, and CcoP with hemes cp1 and cp2 cofactors7 (Fig. 1a, b).
Many Gram-negative bacteria contain soluble (e.g., cyt c2) and membrane-anchored (via transmembrane domains or fatty acids) cyts c (e.g., R. capsulatus cyt cy8, Paracoccus denitrificans cyt c5529, and Helicobacterium gestii cyt c55310) as electron carriers. Conversely, Gram-positive bacteria have no freely diffusing electron carrier but contain cyt c domains fused to their CIII2 like in Mycobacterium smegmatis11, or CIV like in Bacillus subtilis12. In R. capsulatus, both the diffusible cyt c2 and the membrane-anchored cyt cy carry electrons from CIII2 to CIV in respiration13.
In recent years, the co-occurrence of individual complexes together with multi-enzyme supercomplexes (SCs) in energy-transducing membranes has become evident14,15,16,17. SCs may enhance catalytic efficiency through substrate/product channeling or minimize production of harmful reactive oxygen species to decrease cellular distress18. The structures of mitochondrial SCs, such as CICIII2CIV or its smaller variants CICIII2 and CIII2CIV19 are well known20,21. Some bacterial SCs (e.g., P. denitrificans22 or C. glutamicum23) have also been characterized biochemically, but only the structure of the Gram-positive M. smegmatis SC (CIII2CIV2) is available24,25.
As of yet, no respiratory SC structure has been determined for Gram-negative bacteria, the evolutionary precursors of mitochondria. Furthermore, SCs containing cbb3-type ancient forms of CIV with primordial respiratory features remain unknown26. Such structural studies have been hampered due to unstable interactions between CIII2 and CIV, yielding trace amounts of “native” SCs. We have overcome this hurdle using a genetic approach and obtained large amounts of engineered SCs from the Gram-negative facultative phototroph R. capsulatus. Here we report the cryo-electron microscopic (cryo-EM) structure of this functional bc1-cbb3-type SC (CIII2CIV, at 5.2 Å resolution), together with three structural conformers of native CIII2 (at 3.3–4.2 Å resolution). The structures show that the membrane-bound cyt cy and the diffusible cyt c2 interact differently with CIV for electron transfer. This work illustrates the structural and functional features of a native-like CIII2CIV and its distinct membrane-confined and membrane-peripheral respiratory electron transport pathways in Gram-negative bacteria.
Results
Isolation and characterization of engineered functional SCs
Earlier studies have indicated that in R. capsulatus CIII2, CIV, and cyt cy are in close proximity to each other27. Analyses of native membranes of this species indicated barely detectable entities around ~450 kDa Mr, larger than the masses of CIV monomers or CIII2 dimers (Supplementary Fig. 1a, b). Similar entities were seen with dodecyl-maltoside or digitonin used to disperse wild-type membrane, but they were of low abundance and unstable, rendering their isolation and study difficult.
Earlier works of the assembly processes of CIII2 and CIV showed that cyt c1 interacts with cyt b to form a cyt b-c1 subcomplex28, and similarly, CcoP associates with CcoNOQ subcomplex to yield an active CIV29. Thus we hypothesize that translationally fusing the C-terminus (C-ter) of cyt c1 to the N-terminus (N-ter) of CcoP (on the inner (n) side of the membrane) as a cyt c1-CcoP fusion might produce a bipartite bc1-cbb3-type SC that may form a native-like SC and enrich possible SC assembly factors during protein isolation for biochemical and structural studies (left panels of Fig. 1c, d). Further, adding the 69-residue linker (L) and the 100-residue cyt domain of cy to the C-ter of cyt c1-CcoP (on the outer (p) side of the membrane) as a cyt c1-CcoP-cy fusion might yield a tripartite SC with its attached electron carrier (right panels of Fig. 1c, d). This approach (see “Methods” and Supplementary Table 1) was successful and yielded high amounts of fused SCs of native-like electrophoretic mobilities (Supplementary Fig. 1a, b; a was overstained for IGA as compared to b). Indeed, these fused native-like SCs were functional in vivo and complemented a mutant lacking both CIII2 and CIV for photosynthesis proficiency, which requires a functional CIII2, and colonies producing them exhibited CIV activity in situ (Supplementary Fig. 1c). Thus the functional properties of the native-like SCs were like those of native SCs.
The His-tagged bipartite and Flag-tagged tripartite SCs were purified by tag-affinity and size exclusion chromatography (SEC; “Methods”; Supplementary Fig. 1d, e). Analyses of isolated proteins showed the presence of large entities of Mr ~450 kDa range (Supplementary Fig. 1d, e fractions A-1 and B-1 in insets). These fractions contained the cyt c1-CcoP (~65 kDa) or cyt c1-CcoP-cy fusions (~80 kDa) (Supplementary Fig. 1f), and the protein bands were identified by mass spectrometry (MS) as subunits of CIII2 and CIV (Supplementary Table 2). The fusion proteins contained covalently attached heme cofactor(s) as revealed by a cyt c-specific staining (Supplementary Fig. 1g). CcoQ (Mr ~7 kDa) of CIV was absent in the SC preparations.
Purified SCs were characterized for their cyts b and c contents. The optical redox difference spectra were distinct from those of CIII230 or CIV31, and the tripartite SC contained more c-type cyt than the bipartite SC, due to the additional cyt domain of cy (Supplementary Fig. 1h). The samples had both QH2:cyt c reductase (specific to CIII2) and cyt c oxidase (specific to CIV) activities (Supplementary Fig. 1i, j). Thus the native-like SCs were enzymatically active in vitro. Moreover, the tripartite SC also exhibited QH2:O2 reductase (coupled CIII2 + CIV) activity without addition of any electron carrier (e.g., horse heart cyt c or R. capsulatus cyt c2; Supplementary Fig. 1k), suggesting that the cyt domain of cy fused to cyt c1-CcoP transferred electrons from CIII2 to CIV.
Structures of the tripartite SCs
Cryo-EM analysis of the tripartite SC preparations (Supplementary Fig. 1e, fraction B-1) showed that the initial three-dimensional (3D) classes were mainly of two different sizes (Supplementary Fig. 2, Box 1, left). The size (~180 Å length) and shape of the smaller particles suggested that these may correspond to a dimeric CIII2 associated with a single CIV. Focused classification and processing of the subclass containing ~62,000 particles with the highest initial resolution and best discernable features yielded a tripartite CIII2CIV map (SC-1A, EMD-22228) at 6.1 Å resolution (Supplementary Fig. 2a, “Methods”), while another dataset produced a map (SC-1B, EMD-22230) at 7.2 Å resolution (Supplementary Fig. 2b and Supplementary Table 3). The larger particles (~250 Å length, Supplementary Fig. 2, Box 1, left) represented a dimeric CIII2 flanked by two CIV, as expected based on two c1-CcoP-cy subunits per CIII2. However, these particles were rare (~5000) and their map (SC-1C) could not be refined beyond ~10 Å resolution (Supplementary Fig. 2c).
A homology model of R. capsulatus cbb3-type CIV was built using as a template the highly homologous Pseudomonas stutzeri structure (PDB: 3MK7; 3.2 Å resolution) (“Methods,” Supplementary Table 3). The available CIII2 model (PDB: 1ZRT; 3.5 Å resolution) was further refined (PDB: 6XI0; 3.3 Å resolution) based on the cryo-EM data obtained in this work (Supplementary Table 3). These models were fitted as rigid bodies into the maps SC-1A and SC-1B (Supplementary Fig. 3a), with correlation coefficients CCbox of 0.75 and CCbox of 0.71, respectively. The [2Fe-2S] clusters of the FeS proteins of CIII2 could be recognized closer to b (heme bL) than to c (heme c1) positions but had lower occupancy and resolution likely due to conformational heterogeneity. The heterogeneity of the FeS-ED of CIII2 in monomer B (away from CIV) was more pronounced than that in monomer A (adjacent to CIV) (Supplementary Fig. 3b). Lower resolutions of the CIII2 FeS-ED portions were anticipated due to their mobility2,3.
Superimposition of the CIII2 portions of SC-1A and SC-1B maps showed that CIV was in different orientations in different maps (Supplementary Fig. 3c). The two extreme locations of CIV with respect to CIII2 were displaced from each other by a translation of ~3 Å and a rotation of ~37° (Supplementary Fig. 3d, e; SC-1A in red and SC-1B in blue). Other subclasses identified in 3D classifications showed CIV in various orientations between those seen in SC-1A and SC-1B maps. This CIII2CIV interface flexibility is attributed to the limited interaction between the CcoP (N-ter transmembrane helix (TMH)) of CIV and the cyt b (TMH7) of CIII2 (Fig. 2, top view). In the interface regions of SC-1A and SC-1B maps, additional weaker features that are not readily attributable to CIII2 and CIV structures were observed. However, no membrane-external feature that might correspond to cyt domain of cy, which is an integral part of the cyt c1-CcoP-cy subunit, could be discerned in these maps.

a The structures of CIII2 (PDB: 6XI0) and the homology model of R. capsulatus cbb3-type CIV were fitted into the cryo-EM map SC-2A (gray), depicted as a side view. All subunits are colored and labeled as in Fig. 1, and the arrow at the edge of CcoN of CIV is for the extra N-ter TMH (TMH0, light purple) specific to R. capsulatus. b Representative regions of the cryo-EM map showing map quality and model fitting. The TMH2 and TMH10 of CcoN show heme b and some bulky side chains. The protein backbone and hemes bL and bH are resolved between cyt b TMH2–TMH4 (compare to Fig. 3b, CIII2 map at 3.3 Å). Large side chains are visible between the TMH4s of CIII2 monomers A and B (4A and 4B). c Top view of CIII2CIV with TMHs depicted as cylinders and colored as in a. The TMHs of cyt b (only CIII2 monomer A) and CcoN of CIV are numbered, and the TMHs of the FeS protein (yellow), cyt c1 (green), CcoO (dark green), CcoP (light blue), CcoH/cy (blue/red with an arrow), and CcoN TMH0 (light purple) are shown. d 180° rotated view for the back view of CIII2CIV interface. The two extra TMHs at the interface are those of CcoH (blue) and cyt cy (red). e Enlarged view of CIII2CIV interface. The view is slightly rotated relative to d for better visibility of CcoP TMH in the background (light blue). CcoN TMH9 is next to CcoH and cyt cy TMHs. The fusion region between cyt c1 and CcoP is indicated at the bottom, with cyt c1 C-ter (green) and CcoP N-ter (light blue) with their respective end residues (Lys257c1 and Thr13CcoP) (only resolved portions). The 12 N-ter CcoP residues connecting these two chains (dashed line) are not resolved. f Enlarged view showing close interaction between CcoH and cyt cy TMHs. Characteristic features of cyt cy TMH (NH2-Gly11xxxPhe15xxxxxTyr21-COOH) are used to determine the registration, with the helix break induced by Gly11, and bulky side chains for Phe15 and Tyr21 clearly visible.
Structure of bipartite SC supplemented with cyt c y
The bipartite SC preparations (Supplementary Fig. 1d, fraction A-1) were supplemented with either purified full-length cyt cy or its soluble variant lacking the TMH (i.e., cyt S-cy)32 to yield the bipartite SC + cy and SC + S-cy samples. Following SEC, the elution fractions showed that only the intact cyt cy, but not the cyt S-cy, remained associated with the SC (Supplementary Fig. 4a). Thus the cyt domain of cy does not bind tightly to, and its TMH is required for its association with, the SC.
The cryo-EM analyses of the bipartite SC + cy yielded a map (SC-2A, EMD-22227) at 5.2 Å resolution (Supplementary Fig. 5a, b), with local resolutions ranging from 4.3 to 8.0 Å (Supplementary Fig. 6a, c). The homology model of CIV and the refined model of CIII2 (PDB: 6XI0) were fitted as rigid bodies into SC-2A (Fig. 2a), with a correlation coefficient CCbox of 0.74. Comparison of the maps SC-2A (bipartite CIII2CIV) with SC-1A (tripartite CIII2CIV) showed that they were similar, with root-mean-square deviation of 1.6 Å. Hence, they are collectively referred to as CIII2CIV, irrespective of their bipartite or tripartite origins.
The dimensions (~155 × 60 × 90 Å, L × W × H) of the slightly curved CIII2CIV structure were consistent with a CIII2 dimer associated with one CIV. On SC-2A map (5.2 Å resolution), some large aromatic side chains could be readily discerned (Fig. 2b). Of the TMHs seen, 34 were accounted for by two FeS proteins, two cyts b, and two cyts c1 (2, 16, and 2 TMHs per dimer, respectively) and single CcoN, CcoO, and CcoP (12, 1, and 1 TMHs, respectively) (Fig. 2c). The features corresponding to the heme cofactors of CIII2CIV were attributed to hemes bH and bL of cyt b, heme c1 of cyt c1, and to hemes b and b3 of CcoN, heme co of CcoO, and hemes cp1 and cp2 of CcoP proteins. Like the tripartite maps, in this case also the FeS-EDs of CIII2 had lower resolution and the [2Fe-2S] clusters were located closer to heme bL (b position). An additional TMH was observed at the distal end of CIV (Fig. 2a, rotated 180° in Fig. 2d) close to CcoN TMH3 and TMH4 (Fig. 2c). Due to its location, this TMH (depicted in Fig. 2 as an ab initio model of CcoN residues Arg25 to Leu48) was tentatively attributed to the extra N-ter TMH (TMH0) of CcoN, specific to R. capsulatus.
The interface of CIII2CIV is delimited by CcoN TMH8 and TMH9, CcoP TMH, cyt b TMH5 and TMH7, and cyt c1 TMH of monomer A, with the closest interaction being between CcoP TMH and cyt b TMH7 (Fig. 2a, c). Two interacting TMHs of unknown identities (Fig. 2d, in red and blue) and an inter-complex connection are seen at the interface. This connection linking CIII2 to CIV is at the n face of the membrane, near the TMHs of cyt c1 and CcoP (Fig. 2e, Lys257c1 and Thr13CcoP). It is tentatively attributed to the 13 amino acid residues spanning the fusion joint between the C-end of PetC and N-end of CcoP (with no extra amino acid inserted in between) that are not clearly observed.
Assembly factor CcoH and cyt c y TMHs are located at CIII2CIV interface
A co-evolution-based approach predicting the residue–residue contacts in protein–protein interactions, RaptorX-ComplexContact33, was used to identify the TMHs at CIII2CIV interface. The single TMH containing CIV-related proteins (i.e., CcoQ subunit, CcoS and CcoH assembly factors29, and cyt cy8) were analyzed against the subunits of CIII2 and CIV. Significant predictions of interacting residue pairs (confidence value >0.5) were found only between CcoN (primarily TMH9) and the putative N-term TMH of CcoH (Supplementary Fig. 7a, b). An ab initio model of CcoH TMH was docked via PatchDock34 onto CIV using the predicted residue–residue contacts as distance restraints (15 Å threshold; “Methods”). The top scoring 50 models converged to a single cluster at the location of the unknown TMH, next to CcoN TMH9 (Supplementary Fig. 7c). Examination of the interactions between CcoH TMH and CcoN TMH9 showed multiple co-evolutionarily conserved residues in close contacts (Supplementary Fig. 7b, d). Earlier studies had indicated that CcoH can be cross-linked by disuccinimidyl suberate (spacer length ~11 Å) to CcoP and CcoN35. Thus the TMH close to CcoN TMH9 (Fig. 2e, blue TMH) is tentatively assigned to the assembly factor CcoH.
A clear difference between the maps SC-2A (bipartite CIII2CIV + cy) and SC-1A (tripartite CIII2CIV) was seen at the interface. The unidentified TMHs were barely visible in SC-1A but highly enhanced in SC-2A (Fig. 2e), indicating higher occupancy. The observation that only the native cyt cy binds to bipartite SC via its TMH suggested that the TMH (red in Fig. 2e) next to CcoH TMH (blue in Fig. 2e) corresponds to the membrane anchor of cyt cy. This explanation is supported by fact that the bipartite CIII2CIV + cy is supplemented with full-length cyt cy while the tripartite SC contains only the fused cyt domain of cy. Indeed, landmark densities corresponding to the helix-breaking Gly11 and the correctly spaced bulky side chains of Phe15 and Tyr21 of cyt cy TMH (NH2-xxxGly11xxxPhe15xxxxxTyr21-COOH) were discerned (Fig. 2f).
We noted that some CIII2CIV + cy subclasses exhibited a weak feature on the p side of the membrane that may reflect the cyt domain of cy (Supplementary Fig. 5g, SC-2B). However, this feature could not be refined to high resolution, consistent with the weak binding of cyt domain of cy to CIII2CIV. Moreover, upon addition of cyt cy, the predominant conformation of CIV in the bipartite CIII2CIV + cy (Supplementary Fig. 5a, b, SC-2A) shifted toward that seen in map SC-1A of tripartite SC (Supplementary Fig. 2a), with no major class corresponding to SC-1B (Supplementary Fig. 2b). This suggested that local interactions between the CcoH and cyt cy TMHs and CIV decreased the interface flexibility of CIII2CIV (Fig. 2e).
Cryo-EM structures of R. capsulatus native CIII2
The bipartite SC + cy samples also contained smaller particles (~110 Å length) (Supplementary Fig. 2, Box 2) that were the size of CIII2 Supplementary (Fig. 5c, d). Analyses of these particles using C2 symmetry led to the map CIII2 at 3.3 Å resolution for native CIII2 (Supplementary Fig. 5e), with local resolutions ranging from 3.0 to 4.0 Å (Supplementary Fig. 6b, d and Supplementary Table 3). The FeS-ED parts showed lower occupancy and resolution compared to the rest of the map, indicating conformational heterogeneity. When similar analyses were carried out without imposing C2 symmetry, three distinct maps were obtained for CIII2 (CIII2 c-c, CIII2 b-c, and CIII2 b-b at 3.8, 4.2, and 3.5 Å resolutions, respectively; Supplementary Fig. 5f). These maps were superimposable with respect to cyt b and cyt c1 subunits, except for the FeS-ED portions. The CIII2 structures depicted by the CIII2 b-b (Fig. 3a–c) and CIII2 c-c (Fig. 3d) maps exhibited overall C2 symmetry, but in the former the FeS-EDs were located in b, whereas in the latter in c position3. Notably, the third structure (CIII2 b-c) was asymmetric, with the FeS-ED of one monomer being in c and the other one in b positions (Fig. 3e). Such asymmetric structures of native CIII2 have not been observed frequently, although they are proposed to occur during QH2 oxidation36,37,38.

a Cryo-EM map CIII2 b-b (EMD-22226) with both FeS proteins in b position. b Representative region of CIII2 b-b demonstrating map quality and model fitting. TMH2 and TMH4 of cyt b with hemes bL and bH are shown. c–e Maps and models showing different conformations of the FeS proteins. The left panels show the CIII2 structure fitted into the different maps with the subunit colorings (cyt b in periwinkle, cyt c1 in green, and the FeS protein in yellow) as in Fig. 2. The right panels show the top half of the models with the membrane-external domain of cyt c1 omitted to better visualize the b-b, c-c, and c-b positions of FeS-EDs. The [2Fe-2S] clusters are shown as yellow–red spheres and indicated by arrows. c Structure of native CIII2 with both FeS-EDs in b position (map CIII2 b-b, EMD-22226; PDB: 6XKV). d Structure of native CIII2 with both FeS-EDs in c position (map CIII2 c-c, EMD-22224; PDB: 6XKT). e Structure of native CIII2 with one FeS-ED in c and one in b position (map CIII2 b-c, EMD-22225; PDB: 6XKU).
Interactions of cyt c 2 and cyt c y with CIII2CIV
As the tripartite or bipartite SC maps contained no feature that could clearly represent cyt domain of cy, the interaction interfaces between SC and its electron carrier remained unknown. Thus a docking approach combined with crosslinking mass spectrometry (XL-MS) was used to characterize these protein-protein interactions. XL-MS has been used successfully in vivo with whole cells39,40,41,42 or in vitro with purified materials43,44, and PatchDock34 is an efficient rigid docking method that maximizes geometric shape complementarity. First, the co-crystal structure (PDB: 3CX5) of yeast CIII2 with its electron carrier iso-1 cyt c45 was used as a template (homology between yeast cyt c1 and R. capsulatus cyt c1: 31% identity and 58% similarity; iso-cyt c and cyt c2: 25% identity and 56% similarity) to model the binding of cyt c2 on bacterial CIII2. The R. capsulatus cyt c1 (PDB: 6XI0) and cyt c2 (PDB: 1C2N) structures were superimposed with their yeast counterparts on the co-crystal structure, and a model with a single cyt c2 docked to a monomer of CIII2 was generated (Supplementary Fig. 4e). This docking model was then experimentally validated using the protein crosslinker 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM). The crosslinks (XLs) were identified using MeroX44,46 search engine, and among them those of high confidence were cross-validated using FindXL43,47. The intra-subunit XLs within CIII2CIV provided controls (Supplementary Fig. 4c, d, “Methods”). The three XLs between cyt c1 and cyt c2 supplied distance restraints for docking cyt c2 to CIII2 using PatchDock (Supplementary Table 4 and Supplementary Fig. 4f), and the docking models (top 50) clustered at a single region per monomer of CIII2 (Fig. 4a, right). This region overlapped with the binding site of cyt c2 defined by the model obtained by alignment to the yeast co-crystal structure (Supplementary Fig. 4g). The distance from cyt c2 heme-Fe to cyt c1 heme-Fe was ~16.8 Å for the co-crystal-derived model, while comparable distances were between ~13.8 and 20.4 Å with the docking models. Thus docking via PatchDock integrating XL-MS-mediated distance restraints defined reliably, but with limited accuracy, the interaction region of cyt c2 on CIII2.

The binding regions were defined by XL-MS-guided docking, and the subunits of CIII2CIV are colored as in Fig. 2 except the monomer B of CIII2 shown in light gray. Only the binding regions on monomer A are shown. a Cyt c2 (PDB: 1C2N) was docked onto CIII2 and CIV using PatchDock guided by DMTMM-generated XLs as distance restraints. This yielded one cluster of models on CIV and one per monomer of CIII2. b A model of cyt domain of cy, generated using P. denitrificans cyt c552 structure (PDB: 3M97) as a template (RMSD between template and model: 0.2 Å) was docked on CIII2 as in a, except that both DMTMM- and DSBU-generated XLs provided distance restraints. Two binding clusters for cyt domain of cy per monomer of CIII2 were found. The two clusters are located behind each other on a side view but are clearly visible on top views (Supplementary Fig. 4h–j, labeled 1 and 2). Only cluster 1 that is closer to cyt c1 and overlapping with the binding region of cyt c2 is shown. In all cases, ten representative models are shown to visualize the clusters of binding models. No binding region for cyt cy on CIV could be defined since no XL was found between the proteins.
No information about the binding of cyt c2 and cbb3-type CIV was previously available. First, this binding was shown biochemically (Supplementary Fig. 4b) and then subjected to XL-MS using DMTMM. The XLs between the proteins (1 between cyt c2 and CcoP and 8 between cyt c2 and CcoO) provided distance restraints for docking cyt c2 to CIV (Supplementary Table 4). The docking models (top 50) clustered in a single region of CIV (Fig. 4a, left), closer to heme cp2 (c2 heme-Fe to cp2 heme-Fe: ~15.2–35.6 Å) than heme cp1 (c2 heme-Fe to cp1 heme-Fe: ~23.0–42.0 Å) of CcoP (Fig. 5). The two cyt c2-binding regions on CIII2CIV are highly distant from each other (closest c2 heme-Fe on CIII2 to that on CIV is ~69 Å; Fig. 5a).

a The cofactors are shown inside the transparent map SC-2A of CIII2CIV with the same subunit colors as in Fig. 2: hemes bL and bH (periwinkle), heme c1 (green), hemes cp1 and cp2 (light blue), heme co (dark green), hemes b and b3 (purple). The [2Fe-2S] clusters are shown as yellow–red spheres. In all cases, the distances (heme-Fe to heme-Fe) between the heme cofactors are indicated. The positions of docked cyt c2 and cyt domain of cy are indicated as orange (heme c2) and red (heme cy) spheres, respectively, representing their heme-Fe atoms. All heme-Fe atoms corresponding to the top 50 docking positions for cyt c2 on CIV are shown as solid (<25 Å) or transparent (>25 Å) spheres, depending on their distances to heme cp2. In the case of CIII2, only the docking positions of cyt c2 and cyt cy on monomer A and between the monomers A and B are shown, omitting those located on monomer B. The TMH of cyt cy is shown in red at CIII2CIV interface. b The heme-Fe atoms of all 50 cyt c2 models docked onto CIV are plotted as function of their distances from heme cp1 and heme cp2, with the Fe atoms within 25 Å shown as solid spheres and those beyond 25 Å as transparent spheres. The vast majority of heme-Fe atoms of docked cyt c2 models are closer to heme cp2 than heme cp1 of CIV. c Top view of the map shown in a is presented to better visualize the distribution of the docked cyt domain of cy on monomer A and between the monomers A and B. In all cases, the heme-Fe atoms are depicted by spheres and colored as indicated above and on the figure.
Next, the binding interactions between cyt domain of cy and CIII2CIV were sought using the longer, primary amine reactive disuccinimidyl dibutyric urea (DSBU) together with the shorter, carboxylic acid reactive DMTMM. The use of two crosslinkers with different chemical properties and spacer lengths provided better coverage and tighter distance restraints, as the SC interface and the FeS-ED of CIII2 were, and the cyt domain of cy was expected to be, flexible (see below). Similar to DMTMM, DSBU yielded multiple intra-subunit XLs within the subunits of CIII2CIV as experimental controls (Supplementary Table 4 and Supplementary Fig. 4c, d). Six XLs (five cyt cy to cyt c1 and one cyt cy to FeS protein) with DMTMM and four XLs (only cyt cy to FeS protein) with DSBU were found (Supplementary Table 4). No XL was observed between cyt cy and CIV, suggesting that cyt c domain of cy is closer to CIII2 in CIII2CIV. In this case, the models (top 50) generated by PatchDock clustered in two binding regions for cyt domain of cy on each CIII2 monomer of SC. One of the clusters was on top of cyt c1 and overlapped with cyt c2 cluster (Fig. 4b), whereas the other one was between cyt c1 and the FeS-ED near CIII2 inter-monomer space (Supplementary Fig. 4h–j). These two clusters are best visible in a top view (Supplementary Fig. 4j), and the distances between cyt cy heme-Fe and cyt c1 heme-Fe of CIII2 monomer A are between 13.8 and 47.1 Å (Fig. 5). To further support these binding locations, we searched in our cryo-EM datasets 3D classes that contained extra features attributable to cyt domain of cy, and a minor class (~18,000 particles; Supplementary Fig. 5g) showing such an extra feature between CIV and CIII was found (Supplementary Fig. 4h, i).
To better define the clusters of cyt domain of cy, the docking analyses were repeated using the three different conformers of native CIII2 (Fig. 3c–e, CIII2 b-b, c-c, and b-c). Interestingly, when the FeS-EDs are in c position (CIII2 c-c), the docking models gathered as a single cluster on cyt c1, slightly displaced toward the FeS-ED of the same monomer (Supplementary Fig. 8a–c). However, when the FeS-EDs are in b position (CIII2 b-b), such models were more spread out (Supplementary Fig. 8d–f). The third model with one FeS-ED in c and the other in b positions yielded the expected clustering pattern depending on the local FeS-ED conformation. In CIII2CIV both FeS-EDs being in the b position, we assumed that cyt domain of cy docks similarly to that seen with CIII2 b-b and that spreading of docking originates from the variable conformations of the FeS-EDs in SC (Supplementary Fig. 8). Heme c1 and not the FeS protein being the electron exit site of CIII248,49, the cluster on cyt c1 was taken as the productive binding region of cyt domain of cy.
Examination of all pertinent distances between the cofactors of CIII2CIV (Fig. 5a) indicates that the binding region of cyt domain of cy near heme c1 is far from CIV. The large distance (~50.8 Å) separating cyt c1 heme-Fe of CIII2 monomer A from CcoP cp1 heme-Fe (the closest compared with heme cp2) makes it impossible to define a position for cyt cy close enough to heme c1 reducing it, and heme cp1 oxidizing it, while transferring electrons from CIII2 to CIV. The distance constraint, the inability to resolve the cyt domain of cyt cy by cryo-EM, and the more frequent XLs to CIII2 led us to conclude that cyt cy oscillates within CIII2CIV during QH2:O2 oxidoreduction (Fig. 6).

The likely binding regions of cyt c2 and cyt c domains of cy (orange and red ellipsoids, respectively), defined by XL-MS-guided docking, are depicted by the distributions of their heme-Fe atoms on the transparent map SC-2A of CIII2CIV. Only the positions that are within 25 Å of heme c1 of CIII2 or heme cp2 of CIV are indicated. The cofactors together with the TMH of cyt cy are shown as in Fig. 5. The linker region (indicated by dotted or dashed lines) between the TMH and the cyt domain of cy is not resolved in the cryo-EM map. The proposed electron transport pathways are shown by thicker black arrows: upon QH2 oxidation by CIII2, cyt cy that is integral to CIII2CIV receives an electron from heme c1. It then moves (double-headed dashed red arrow) to an undefined binding region (dashed oval with cy?) on CIV, where it delivers the electron to the nearest heme cp1 of CIV. Similarly, cyt c2 that is peripheral to CIII2CIV also receives an electron from heme c1, diffuses away to reach CIV, and conveys it to heme cp2. The canonical electron transfers occurring from QH2 to heme c1 in CIII2, and from heme cp1 to O2 in CIV, are indicated by thinner arrows. The double headed dashed black arrow depicts the movement of the [2Fe-2S] of FeS protein from the b position (b-pos, black, solid spheres) to the c position (c-pos, gray, transparent spheres) in CIII2 during QH2 oxidation. A possible electron equilibration between the two heme bL of CIII2 is indicated by double arrows, and the electron transfer steps subsequent to heme bH reduction are not shown.
Discussion
Prior to this work, no information was available neither on the occurrence or structure of any cbb3-type CIV containing SC nor on its interactions with physiological electron carriers. Here we describe the cryo-EM structures of engineered native-like active bc1-cbb3-type respiratory SCs, together with three structural conformers of native CIII2 at high resolution, from the Gram-negative, facultative phototroph R. capsulatus. Although the 3D structures of bacterial bc1-type CIII2 are available, native CIII2 heterodimers have not been reported. Similarly, only one structure was available for cbb3-type CIV50. Members of this subfamily of heme-Cu:O2 reductases are widespread among bacteria and essential for microaerobic processes, including anaerobic photosynthesis and bacterial infection4.
The facultative CIII2CIV of R. capsulatus is more flexible, possibly due to the involvement of CIII2 into multiple metabolic pathways, unlike the highly rigid obligate CIII2CIV2 SC of Actinobacteria dedicated to respiration24,25. In species like R. capsulatus, the dual function of CIII2, interacting with the photochemical reaction center during photosynthesis and with CIV during respiration, may need this natural flexibility to allow metabolic adaptations for growth fitness. The native-like SCs, obtained by genetically fusing a subunit of CIII2 to another one of CIV, are fully active both in vivo and in vitro and exhibit increased stability. These SCs are produced in cells without any harmful effect, mature properly, and support growth of mutants lacking either CIII2 or CIV or both. Moreover, detergent-dispersed membranes and affinity-purified SCs exhibit both CIII2 and CIV, as well as CIII2-CIV coupled, enzymatic activities. Clearly this innovative approach, although yielded heterogeneous preparations, containing mixtures of CIII2CIV2, CIII2CIV, and CIII2 particles, allowed resolution of their structures and defined the likely binding regions of cyt c2 and cyt cy. Insertion of different spacers at the cyt c1-CcoP fusion junction and overexpression of the subunits and related assembly components could not further improve this heterogeneity (“Methods”). Certainly, it remains unknown to what extent the structural and functional properties of the native-like SCs described here are identical to their “native” counterparts, which are currently unavailable.
The structures of the tripartite CIII2CIV, which contains a fused cyt c domain39, and the bipartite CIII2CIV + cy, which has a bound native cyt cy, were highly similar at sub-nanometer resolution (~7.2–5.2 Å, respectively). Their flexibility was due to limited protein–protein interactions at their interface, where the TMHs of cyt cy and CcoH are located (Fig. 2). Previously, neither the exact location nor the mobility of cyt cy between CIII2 and CIV were known. The SC structure shows that the N-terminal TMH of cyt cy is locked at the interface, allowing mobility of its cyt domain within CIII2CIV (Fig. 6). The linker region of cyt cy remains unresolved but is long enough to reach both CIII2 and CIV. Early studies with R. capsulatus cyt cy had shown that a full-length linker (69 residues) is needed for rapid (~<50 μs) electron transfer from CIII2 to the photosynthetic reaction center51, whereas a shorter linker (~45 residues) is proficient for respiratory electron transfer to CIV52.
In native CIII2 conformers, different positions of the [2Fe-2S] cluster bearing FeS-EDs were observed. Crystallographic structures have often depicted bacterial CIII2 as symmetrical homodimers3,53,54. These structures were obtained in the presence of inhibitors constraining FeS-EDs near heme bL or used mutants stabilizing it on cyt b surface. Alternatively, they contained crystal contacts restricting the FeS-ED movement55. To our knowledge, no native heterodimeric CIII2 structure of bacterial origin with different conformations of its FeS-EDs has been reported. Only recent cryo-EM structures of mitochondrial SCs have shown different maps for CIII2 FeS-EDs17,56. Thus native CIII2 is not always a symmetric homodimer, and the FeS-ED of each monomer is free to move independently from the other. The Q-cycle models describe the mechanism of CIII2 catalysis by two turnovers of a given monomer, with the different positions of the FeS-ED protein attributed to different catalytic steps3,48,49. Emerging asymmetric structures of bacterial and mitochondrial native CIII2 obtained by cryo-EM in the absence of inhibitors or mutations, combined with the inter-monomer electron transfer between the hemes bL of the monomers57,58, now provide a glimpse into plausible “heterodimeric Q-cycle” mechanism(s)37,38. Accordingly, the FeS-EDs of CIII2 monomers might cycle between homo- and hetero-dimeric conformations during the Q-cycle.
This work defined hitherto unknown structural interactions between the cyt c2 or cyt cy and native-like CIII2CIV (Fig. 6). The structures indicate that the distances separating heme c1 of CIII2 monomer A and hemes cp1 or cp2 of CIV are too large (Fig. 5) for direct microsecond time scale electronic communication59 to sustain the turnover rate of CIII2CIV. Thus, even when CIII2 and CIV form a SC, an electron carrier is required for QH2:O2 oxidoreduction. The binding location of cyt c2 on CIV defined here, the redox midpoint potentials (Em) of the cofactors, and the distances separating them (Fig. 5a) suggest that cyt c2 would confer electrons to the closer heme cp2, rather than the more distant heme cp1, of CcoP. This will then initiate canonical electron transfer via heme cp1, heme co, and heme b to heme b3-CuB site for O2 reduction60,61 (Fig. 6). For purified R. capsulatus proteins, the Em values of cyt c2, cyt c130, and heme co of CcoO are known31. The Em values of R. capsulatus CcoP hemes cp1 and cp2 are unknown, but based on similar Em values of heme co for Bradyrhizobium japonicum and R. capsulatus, they are expected to be similar62.
The likely interaction region of cyt cy on cyt c1 of CIII2, which is close to that of cyt c2, is now known but that on CIV remains less well defined. The distance separating the redox centers is a major factor that controls the rate of electron transfer59. The binding region of cyt domain of cy on CIII2 suggests that reduced cyt cy, upon its movement to CIV, might preferentially convey electrons to the closer heme cp1 than heme cp2 of CcoP (Fig. 6). If so, under physiological conditions, heme cp1 would be the primary receiver of electrons derived from QH2 oxidation by CIII2, forming a fully membrane-confined electronic wiring within CIII2CIV. In contrast, cyt c2 carries electrons from heme c1 to heme cp2 via free diffusion. This membrane-external pathway might accommodate electrons not only from QH2 but also from other donors distinct from CIII2. As such, reduction of cyt c2 during methylamine oxidation63 or degradation of sulfur containing amino acids, converting toxic sulfite (SO32−) to sulfate (SO42−) by sulfate oxidase64, might provide electrons to CIV, contributing to energy production.
In summary, the architecture of an engineered native-like CIII2CIV along with its interactions with its physiological redox partners establishes salient structural and functional features of two distinct respiratory electron transport pathways (membrane-confined and membrane-external) that operate between CIII2 and CIV in Gram-negative bacteria.
Methods
Bacterial strains, growth conditions, and primers used
Bacterial strains, plasmids, and primers used are listed in Supplementary Table 1. LB medium supplemented as appropriate with ampicillin, gentamicin, kanamycin, or tetracycline at 100, 12, 50, or 12.5 μg/mL, respectively, was used for growing Escherichia coli strains at 37 °C. R. capsulatus strains were grown chemoheterotrophically under semi-aerobic/dark conditions at 35 °C on enriched (MPYE) medium, supplemented as needed with gentamicin, kanamycin, spectinomycin, or tetracycline at 3, 10, 10, or 2.5 μg/mL, respectively. Colonies were stained for cbb3-type CIV activity by incubating plates with a 1:1 (v/v) mixture of 35 mM 1-naphtol and 30 mM N,N-dimethyl-1,4-phenylenediamine (NADI staining)65.
Construction of petABC::ccoP-His8 (cyt bc 1-CcoP) fusion
Using the primers Fw-ccoP (StuI) and Rv-ccoP (HindIII) (Supplementary Table 1), petABC::ccoP-His8 fusion was constructed by ligating in-frame the PCR-amplified 1.13 kb StuI-HindIII fragment containing ccoP (with a C-terminal His8 tag) to the 3′ end of petABC, after elimination of petC stop codon and ccoP start codon on StuI-HindIII digested plasmid pMTSI, to yield pYO60 (Supplementary Table 1). The 3.85 kb BamHI fragment of pYO60 carrying petABC::ccoP fusion was transferred to the plasmids pBSII and pRK415 using the same sites, yielding pYO63 and pYO76, respectively. The linker-spaced versions of petABC::ccoP fusion (petABC::L2-ccoP, petABC::L3-ccoP, and petABC::L4-ccoP) were also constructed by exchanging the StuI-HindIII fragment of pYO63 carrying petABC-ccoP with the PCR-amplified linker added versions, yielding pYO77, pYO78, and pYO80, respectively. L2, L3, and L4 linkers were introduced via the primers F-L2, F-L3, and F-L4 (Supplementary Table 1) and contained the amino acid sequences of NH2-GGSGGGSG-COOH, NH2-GGSGGGSGGGSG-COOH, and NH2-ASIAGGRTASGP-COOH, respectively. The 3.9 kb KpnI-XbaI fragments carrying these linker added versions were cloned to pRK415, yielding pYO81, pYO82, and pYO83, respectively. All constructs were subsequently verified by DNA sequencing. As the protein yields and enzymatic activities of all constructs were similar, only pYO76, which has no linker (i.e., native-like fusion), was used for subsequent work.
Construction of petABC::ccoP::cycY-Flag (cyt bc 1-CcoP-cyt c y-Flag)
Using the primers Fw-cp-cy (BalI) and Rv-cp-cy (BglII + HinIII), the C-terminally Flag-tagged cycY lacking its N-terminal TMH anchor (i.e., amino acid residues 1–30) was PCR amplified. The 619-bp-long PCR fragment containing cycY-Flag without its anchor was digested with BalI-HinIII and exchanged with its counterpart that encompasses the 3′ end of ccoP on pYO63 to yield the petABC::ccoP::cycY-Flag fusion carried by pYO91. The 4.5 kb KpnI-XbaI fragment of pYO91 containing this fusion was cloned into pRK415 using the same sites to yield pYO92 (Supplementary Table 1).
Inactivation of CIII2 (cyt bc 1) and CIV (cbb 3-type Cox) structural genes
The Δ(petABC::gm) deletion–insertion allele carried by pYO3432 was transferred using the gene transfer agent into the chromosome of R. capsulatus strain MG1 (ccoP::kan) to yield YO12, a mutant background lacking both CIII2 and the CcoP subunit of CIV. A similar strain, M7G-CBC1 that lacks both CIII2 and CcoP but overproducing CcoN and CcoO of CIV31, was also used.
Construction of strains producing bipartite and tripartite fusion SCs
The plasmids pYO76 and pYO92 encoding the bipartite and tripartite SCs, respectively, were conjugated into YO12 and M7G-CBC1 using triparental mating to yield pYO76/YO12 and pYO92/M7G-CBC1 strains as in ref. 65 for protein purification.
Purification of fusion SCs
R. capsulatus cells (~25–30 g from 8 L media) were resuspended in a final volume of 35–40 mL of buffer A (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 20 mM EDTA, 1 mM aminocaproic acid, and 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) supplemented with Pierce Protease Inhibitor (Thermo Scientific, 1 mini tablet per 30 mL). Cells were disrupted by three passages through a French Pressure Cell (SLM Aminco) at 13,000 psi in the presence of 5–10 mg of DNase I (GoldBio), and cell debris was removed by centrifugation at 27,000 × g for 30 min31. Chromatophore membranes were sedimented by centrifugation at 190,000 × g for 2 h and resuspended in buffer A. The protein concentration was determined using the Pierce Protein BCA Assay Kit (Thermo Scientific), the suspension was supplemented with 10% glycerol and adjusted to a protein concentration of 10 mg/mL with buffer A. Membrane proteins were solubilized with 2% n-dodecyl-β-d-maltoside (DDM; Anatrace) for 15 min on ice, and non-solubilized materials were sedimented by centrifugation at 100,000 × g for 15 min. The supernatant was subjected to chromatography, as appropriate.
The His-tagged bipartite SC was purified by anion exchange chromatography on Bio-Gel A-50 (Bio-Rad), followed by affinity chromatography on Ni-Sepharose High Performance (GE Healthcare). Solubilized proteins were loaded onto a 40 mL Bio-Gel column equilibrated with Tris buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1% glycerol, and 0.01% DDM) and washed with three column volumes (CVs) of the buffer. After another washing step with 3 CVs of Tris buffer containing 150 mM NaCl to eliminate weakly bound proteins, elution was carried out with a linear, five CV gradient from 150 to 400 mM NaCl in Tris buffer. Fractions containing both cyt c reductase and cyt c oxidase activities were combined and loaded onto a 7-mL Ni-Sepharose column equilibrated with the binding buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1% glycerol, and 0.01% DDM). After washing with 30 mL of binding buffer, the column was eluted with a linear 100 mL gradient from 20 to 260 mM imidazole.
The Flag-tagged tripartite SC was purified by affinity chromatography using the Anti-Flag affinity gel (Bimake). The supernatant containing solubilized proteins was loaded onto a 1 mL column equilibrated with TBS buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.01% DDM, and 1 mM AEBSF), washed with 10 ml TBS, and eluted with 5 mL of Flag (DYKDDDDK) peptide (100 μg/mL) (Sigma) in TBS buffer.
Eluted proteins were concentrated (Amicon Ultra-15, 30 kDa molecular weight cut-off (MWCO)) (Millipore) to a final volume of 150–200 μL and loaded onto a Superose 6 Increase 10/300 GL (GE Healthcare) size exclusion column equilibrated with TBS buffer and eluted with the same buffer at a flow rate of 0.4 mL/min. Peak fractions were combined and concentrated, and PD MiniTrap G-25 columns (GE Healthcare) were used for buffer exchanges, as needed.
Biochemical characterization of fusion SCs
Native polyacrylamide gel electrophoresis (PAGE) was performed using 4–13% gels66, and sodium dodecyl sulfate (SDS)-PAGE used 12.5 or 18% gels67. Gels were stained with Coomassie Brilliant Blue (Bio-Rad) or colloidal silver68. Immunoblot analyses were performed using polyclonal antibodies specific of R. capsulatus cyt b28 and monoclonal anti-rabbit-IgG-alkaline phosphatase (Sigma) used at 1:10,000 dilution. CIV in-gel activity was revealed by incubating native gels with 0.5 mg/mL 3,3′-diaminobenzidine in 50 mM Na-phosphate, pH 7.269. The c-type cyts were revealed by their heme peroxidase activity, using 3,3′,5,5′-tetramethylbenzidine (TMBZ) and hydrogen peroxide (H2O2)70. Briefly, gels were washed with 0.25 M Na-acetate buffer (pH 5) and incubated with 6.3 mM TMBZ in 30% methanol and 0.25 M Na-acetate, pH 5.0. After 1-h incubation, 0.4% H2O2 were added to reveal the peroxidase activity of c-type cyts.
Reduced minus oxidized spectra were recorded in 50 mM MOPS, pH 7.0 using a Cary 60 UV-Vis spectrophotometer (Agilent). Usually, 50 μg of purified protein was fully oxidized by adding a grain of K3[Fe(CN)6] and then gradually reduced with a few grains of sodium ascorbate or sodium dithionite, as appropriate. Protein concentrations were determined with a NanoDrop 2000c (Thermo Scientific) using the A280 method71.
Cyt c reductase (cyt bc1) activity was determined as described72. Accordingly, 10 mM stock solution of 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone (DB) (Sigma) in dimethyl sulfoxide was reduced to DBH2 with a few grains of sodium borohydride and excess borohydride was quenched by adding HCl to a final pH of 6.0. For assays, 40 μM DBH2 were added to 25 μM horse heart cyt c (Sigma) in 500 μL assay buffer (40 mM sodium phosphate, pH 7.4, 20 mM sodium malonate, 0.5 mM EDTA, 0.5 mM KCN, and 0.01% DDM) in a stirred cuvette, and the non-enzymatic rate of cyt c reduction was recorded at 550 nm for 1 min (Cary 60 UV-Vis spectrophotometer, Agilent). The reaction was initiated by adding 1–2 μg of purified protein and cyt c reduction was monitored. The enzymatic activity was inhibited by adding 1 μM stigmatellin (Fluka), inhibitor of cyt bc1-type CIII2.
The CIV (cyt cbb3-Cox) activity was determined as described73. Reduced horse heart cyt c (Sigma) was prepared by incubating a 1–2 mM stock solution with 10 mM sodium dithionite for 15 min at room temperature, excess dithionite was removed by gel filtration using a PD-10 desalting column (GE Healthcare), and the final concentration of reduced cyt c was calculated based on its absorption at 550 nm and an ε = 20 mM−1 cm−1. In all, 20 μM reduced cyt c in 500 μL assay buffer (10 mM Tris-HCl, pH 7.0, 100 mM KCl) were prepared in a stirred cuvette (Cary 60 UV-Vis spectrophotometer), and the baseline was recorded for 1 min. The reaction was started by adding 1–2 μg of purified protein, and cyt c oxidation monitored at 550 nm. The enzymatic activity was inhibited by addition of 250 μM KCN as an inhibitor of cbb3-Cox.
DBH2-dependent oxygen consumption was monitored using a mini Clark-type oxygen electrode (Instech Laboratories, PA) at 25 °C. The assay was performed in the same buffer as that used for cyt c reductase activity (40 mM sodium phosphate, pH 7.4, 20 mM sodium malonate, 0.5 mM EDTA, and 0.01% DDM) but without KCN. A baseline was recorded after adding 100 μM DBH2 to 1 mL of assay buffer, and the reaction was started by adding 30 μg of purified protein. As needed, purified protein was pre-incubated with a 1:1 (w/w) mixture of E. coli polar lipids (Avanti), which improved the activity. In all, 20 μM horse heart cyt c (Sigma), 250 μM KCN, or 1 μM of stigmatellin were used as specificity controls. For all measurements, three biological and three technical repeats are used.
Identification of protein subunits by MS
Protein subunits were identified by MS using appropriate gel bands reduced (dithiothreitol (DTT); Sigma), alkylated (iodoacetamide; Bio‐Rad) and subjected to in‐gel trypsin digestion (Promega, Sequencing Grade Modified Trypsin) overnight at 37 °C. Peptides eluted from the gel samples were dried, desalted using ZipTips (Millipore U‐C18 P10, Millipore), lyophilized, and stored at −80 °C. They were resuspended in 10 μL 5% acetonitrile (ACN)/0.1% formic acid prior to MS, using either a LCQ Deca XP + ion trap, or a Q-Exactive Quadrupole-Orbitrap mass spectrometer (both from Thermo Fisher Scientific)74. The LCQ Deca XP+ mass spectrometer was coupled to a Thermo-Dionex LC Packings Ultimate Nano HPLC system controlled by the Thermo Xcalibur version 2.0 software. Peptides were separated on a 15 cm C18 nanocolumn (Thermo-Dionex, NAN-75-15-03-PM) using a 45‐min linear gradient from 4 to 40% buffer B (100% ACN, 0.1% formic acid), followed by a 7‐min gradient from 40 to 80% buffer B and 8‐min wash with 80% B (constant flow rate 150 nL/min). MS/MS data were acquired in data-dependent analysis mode with dynamic exclusion enabled (repeat count: 3, exclusion duration: 3 min). Full MS survey scans (mass range 300–2000 m/z) were followed by MS/MS fragmentation (normalized collision energy 35) of the top 3 most intense ions. The Q‐Exactive Quadrupole‐Orbitrap mass spectrometer was coupled to an Easy‐nLC™1000 nano HPLC (Thermo Fisher Scientific), and samples were loaded in buffer A (0.1% formic acid) onto a 20‐cm‐long fused silica capillary column (75 μm ID), packed with reversed‐phase Repro‐Sil Pur C18‐AQ 3 μm resin (Dr. Maisch GmbH, Ammerbuch, Germany). Peptides were eluted using a 45‐min linear gradient from 4 to 40% buffer B (100% ACN, 0.1% formic acid), followed by a 7‐min gradient from 40 to 80% buffer B and 8‐min wash with 80% B (constant flow rate 300 nL min). The Q‐Exactive was operated in data‐dependent acquisition mode with dynamic exclusion enabled (repeat count: 1, exclusion duration: 20 s). Full MS survey scans (mass range 300–1600 m/z) at high resolution (70 000 at 200 m/z) were followed by MS/MS fragmentation of the top 15 most intense ions with higher energy collisional dissociation at a normalized collision energy of 22 (resolution 17 500 at 200 m/z). Dual lock mass calibration was enabled with 371.101233 and 445.120024 m/z background ions.
MS spectra were searched against the R. capsulatus protein database (https://www.uniprot.org, last modified 01/15/2020; R. capsulatus (strain ATCC BAA-309/NBRC 16581/SB1003)) using Proteome Discoverer 1.4 (Thermo Fisher Scientific) with Sequest‐HT search engine. Search parameters were set to full trypsin digestion, with maxima of three missed cleavages and three modifications per peptide. Oxidation of methionine (+16 Da) and carbamidomethylation of cysteine (+57 Da) were selected as dynamic modifications. Precursor and fragment ion tolerances were set to 2 and 1 Da, respectively, for the LCQ DecaXP+, and 10 ppm and 0.6 Da, respectively, for the Q-Exactive data. False discovery rates (FDRs) by target‐decoy search were set to 0.01 and Xcorr filter based on charge (z) were: >2 for z = 2, >2.5 for z = 3, and >2.6 for z = 474. All identifications are listed in Supplementary Table 2.
Binding of cyt c y and soluble cyt S-c y to purified proteins
Binding of purified cyt cy75 or a variant of it containing only the soluble cyt domain (cyt S-cy) (residues 99–199) to the bipartite SC was assayed by mixing a 5–10-fold molar excess of purified cyt c with 125 μg of purified SC in a final volume of 150 μL TBS buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.01% DDM). The mixture was separated by chromatography using Superose 6 Increase size exclusion column, and elution fractions were analyzed by SDS-PAGE followed by silver staining. Cyt S-cy was purified from the R. capsulatus strain pYO135/FJ2-R4 (Supplementary Table 1) grown under photosynthetic conditions to maximize its yield32. Cells were washed with 20 mM Tris-HCl, pH 8.0, and resuspended 1:5 (w/v) in the same buffer supplemented with 50 mM NaCl. Polymyxin B sulfate (1 mg/mL) was added, and the cell suspension was incubated for 75 min on ice with gentle stirring. After centrifugation for 20 min at 10,000 × g followed by 3 h at 150,000 × g, the supernatant was collected and concentrated to a final volume of 2 mL (Amicon Ultra-15 3 kDa MWCO) (Millipore). Aliquots of 300 μL were loaded onto Superose 6 Increase (GE Healthcare) equilibrated in 30 mM Tris-HCl, pH 8.0, and eluted in the same buffer. Fractions eluting after 20 mL were combined and concentrated using Amicon Ultra-15 3 kDa MWCO filters (Millipore).
Binding of cyt c 2 to purified proteins
Binding of cyt c2 to cbb3-type CIV was determined by mixing 2-fold molar excess of purified R. capsulatus cyt c276 with 300 μg of purified CIV31 under low-salt conditions (20 mM Tris-HCl, pH 7.4, 1 mM NaCl, and 0.01% DDM). The mixture (250 μL total volume) was loaded onto a Superose 6 Increase (GE Healthcare) sizing column equilibrated with the same buffer to separate the proteins. Elution fractions were concentrated (Amicon Ultra-15, 3 kDa MWCO) (Millipore), and a volume containing 1–5 μg of protein was analyzed by SDS-PAGE followed by silver staining.
Protein crosslinking using chemical crosslinkers
For crosslinking, 180 μg of purified bipartite SC at a concentration of 1 mg/mL in phosphate-buffered saline buffer (50 mM Na-Phosphate, pH 7.4, 150 mM NaCl, and 0.01% DDM) mixed with 5-fold molar excess (90 μg) of purified cyt cy75 were supplemented with 6 mM DSBU (Thermo Fisher Scientific) and incubated on ice for 2 h. The reaction was quenched by adding 50 mM of ammonium bicarbonate, and the mixture was analyzed by MS. In all, 300 μg of purified cbb3-type CIV31 were mixed with a 2-fold molar excess of purified cyt c276 at a final protein concentration of 200 μg/mL under low-salt conditions (20 mM Na-phosphate, pH 7.4, 1 mM NaCl, and 0.01% DDM), and incubated with 20 mM 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) (Sigma) for 1 h at room temperature. Similarly, 300 μg of purified bc1-type CIII2 were mixed with 2-fold molar excess of purified cyt c2 and treated with DMTMM as above. In both cases, the reaction was stopped by removing excess DMTMM with a PD MiniTrap G-25 desalting column (GE Healthcare), and crosslinked proteins were precipitated with 20% (w/v) trichloroacetic acid (TCA, Sigma) at 4 °C for 1 h. Proteins were pelleted by centrifugation at 21,000 × g for 15 min and washed with 10% TCA in 0.1 M Tris-HCl and then with acetone (Fisher). The solvent was discarded, and the pellet was air-dried and then stored at −80 °C for analysis by MS.
MS of crosslinked proteins
Crosslinked proteins were resuspended in an appropriate volume of solution A (2.5% SDS and 50 mM triethylammonium bicarbonate final concentrations) and reduced with 10 mM DTT (US Biological) for 30 min at 30 °C, followed by alkylation with 50 mM iodoacetamide (Sigma Aldrich) for 30 min at 30 °C. The proteins were processed using an S-TrapTM according to the protocol recommended by the supplier (Protifi, C02-mini) and digested with trypsin (Thermo Fisher Scientific) in 1:10 (w/w) enzyme/protein ratio for 1 h at 47 °C. Peptides eluted from this column were vacuum-dried and resuspended with the peptide fractionation–elution buffer for liquid chromatography-MS (LC-MS; (70% (v/v) LC-MS-grade water (Thermo Fisher Scientific), 30% (v/v) ACN (Thermo Fisher Scientific) and 0.1 % (v/v) trifluoroacetic acid (TFA, Thermo Fisher Scientific)]. Peptides were fractionated using AKTA Pure 25 with Superdex 30 Increase 3.2/300 (GE Life Science) at a flow rate of 30 μL/min of the elution buffer, and 100 μL fractions were collected. Based on the elution profile, fractions containing enriched crosslinked peptides of higher molecular masses were vacuum-dried and resuspended with LC-MS-grade water containing 0.1% (v/v) TFA for MS analysis. One half of each fraction was analyzed by a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) coupled to a Dionex Ultimate 3000 UHPLC system (Thermo Fischer Scientific) equipped with an in-house made 15 cm long fused silica capillary column (75 μm ID), packed with reversed‐phase Repro‐Sil Pur C18‐AQ 2.4 μm resin (Dr. Maisch GmbH, Ammerbuch, Germany). Elution was performed using a gradient from 5 to 45% B (90 min), followed by 90% B (5 min), and re-equilibration from 90 to 5% B (5 min) with a flow rate of 400 nL/min (mobile phase A: water with 0.1% formic acid; mobile phase B: 80% ACN with 0.1% formic acid). Data were acquired in data-dependent MS/MS mode. Full scan MS settings were: mass range 300–1800 m/z, resolution 120,000; MS1 AGC target 1E6; MS1 Maximum IT 200. MS/MS settings were: resolution 30,000; AGC target 2E5; MS2 Maximum IT 300 ms; fragmentation was enforced by higher-energy collisional dissociation with stepped collision energy of 25, 27, 30; loop count top 12; isolation window 1.5; fixed first mass 130; MS2 Minimum AGC target 800; charge exclusion: unassigned, 1, 2, 3, 8, and >8; peptide match off; exclude isotope on; dynamic exclusion 45 s43. Raw files were converted to mgf format with TurboRawToMGF 2.0.877.
MeroX 2.0.1.444,46 (for MS-cleavable and non-cleavable crosslinkers, http://www.stavrox.com/) was used as the main search engine for both DMTMM and DSBU. Additionally, FindXL46,47 (for non-cleavable crosslinkers, http://biolchem.huji.ac.il/nirka/software.html) was used to analyze DMTMM spectra, and MassAI 19.0778 (for MS-cleavable and non-cleavable crosslinkers, http://www.massai.dk) was used to analyze DSBU spectra to cross-validate results from MeroX in each case. Only the crosslinked peptides that were cross-validated by two different search engines (i.e., MeroX and FindXL for DMTMM and MeroX and MassAI for DSBU) were used for docking experiments (Supplementary Table 4). MeroX was run in RISEUP mode, with default crosslinker mass and fragmentation parameters for DSBU and in Quadratic mode with default crosslinker mass parameters for DMTMM; precursor mass range, 300–10,000 Da; minimum precursor charge 4; precursor and fragment ion precisions 5.0 and 10.0 ppm, respectively; maximum number of missed cleavages 3; carbamidomethylation of cysteine and oxidation of methionine, as fixed and variable modifications, respectively; results were filtered for score (>10) as well as the MeroX built-in CL spectral matches FDR (<1%). The database consisted of the proteins listed in Supplementary Table 2, supplemented with the assembly factors such as CcoH and the 116 proteins of the cRAP database (www.thegpm.org; contaminant proteins commonly occurring in MS analyses). The latter increased the size of the decoy database to yield more reliable FDRs. FindXL was used to analyze and validate MeroX results for DMTMM CLs. The default FindXL parameters were used as described before47 with the possible CL amino acids adjustments for K, Y, S, or T on one peptide and E or D on the other peptide. MassAI was used to validate CLs identified by MeroX for DSBU with standard settings, except 5 ppm MS accuracy, 0.05 Da MS/MS accuracy, 3 allowed missed cleavages, and carbamidomethylation of cysteine and oxidation of methionine, as fixed and variable modifications, respectively. Additional filters were: # fragments A ≥4, # fragments B ≥4, Intensity ≥1000, Score ≥10. Databases used in FindXL and MassAI consisted of the proteins listed in Supplementary Table 2, supplemented with the assembly factors, such as CcoH. Visualization of the CLs in the structures used Chimera79 with the Xlink Analyzer plug-in80.
Negative staining and cryo-EM sample preparation
For negative staining, purified SCs were diluted to 0.01–0.05 mg/mL concentrations in TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.01% DDM, and 1 mM AEBSF) buffer, 5 μL applied to glow-discharged (20 s, 25 mA, Pelco easiGlow) carbon-coated Cu grids (CF300-CU, EMS), incubated for 1 min, and stained with 2% uranyl acetate. Grids were imaged on FEI Tecnai 12 transmission electron microscope operating at 120 kV, with a CCD camera (Gatan BM-Ultrascan).
For cryo-EM, the tripartite SC was used as purified, whereas the purified bipartite SC was mixed with a 5-fold molar excess of purified cyt cy75. In all, 2.5 μL of the mixture, containing 3–5 mg/mL proteins, were applied to CFlat holey carbon grids (1.2/1.3–400 mesh or 2/2–300 mesh) (EMS), which were glow discharged (2 min, 25 mA, Pelco easiGlow) before sample application. Grids were blotted for 9 s at force 0 (CFlat-1.2/1.3) or for 3 s at force −5 (CFlat-2/2) and flash-frozen in liquid ethane cooled with liquid nitrogen using FEI Vitrobot Mark IV (25 °C, 100% humidity). Plunge freezing conditions were optimized using FEI Tecnai TF20 TEM operating at 200 kV, equipped with an FEI Falcon II camera.
Cryo-EM data acquisition and processing
All cryo-EM grids were imaged using FEI Titan Krios electron microscope operating at 300 kV. Images of the tripartite SC (SC~cy) were recorded using a Gatan K2 Summit direct electron detector, equipped with an energy quantum filter (20 eV), and operated in super-resolution mode at a nominal magnification of ×105,000, resulting in a binned pixel size of 1.32 Å. Images were dose-fractionated to 40 frames with a total exposure time of 10 s, and a total dose of 40 e−/A2. Automated data acquisition was carried out using the Latitude software (Gatan), and nominal underfocus values varied from 1 to 2.5 μm. Movies were motion corrected using MotionCor281 and CTF parameters were determined with CTFFIND 4.182. About 10,000 particles were manually picked and subjected to an initial reference-free two-dimensional (2D) classification using Relion 3.083. Representative classes were selected and used as a template for auto-picking. After sorting and two rounds of 2D classification, ~30,000 particles were retained from the dataset. For 3D classification, an initial model was created using R. capsulatus CIII2 structure (PDB: 1ZRT) and low-pass filtered to 60 Å using EMAN284. A 3D map with a nominal resolution of ~11 Å containing a total of ~12,000 particles (40%) was obtained, showing a CIII2 associated on one side with a single copy of CIV. For further analyses, additional datasets were collected using samples from the same batch and identical imaging conditions. From a total of ~17,000 images, ~1,000,000 particles were automatically picked using the template obtained from the first dataset, and after sorting and two rounds of 2D classification, ~500,000 particles were retained. Various subsets of all images were processed separately and yielded the same overall results with slightly different versions of individual maps. For clarity, only the paths to the best representative of each of the final maps are shown (Supplementary Fig. 2a–c). For 3D classification, the map obtained from the first dataset (Supplementary Fig. 2, Box 3) was low-pass filtered to 60 Å and used as the initial model. Of the five classes obtained, one showed clear features of a CIII2 dimer associated with a single monomer of CIV (Supplementary Fig. 2, class005 in a, class002 in b and c). The remaining classes showed the same overall shape but lacked density or resolution in different parts of the structure. Only two of the five maps contained a second monomer of CIV associated with the other monomer of CIII2 (Supplementary Fig. 2c). These two classes showing this feature were combined, yielding a subset of ~220,000 particles. These particles were further processed by another round of 2D classification and subjected to 3D classification, yielding three classes (Supplementary Fig. 2c). Only one of the maps obtained in this second round of 3D classification clearly showed density for a second monomer of CIV. Aligned particles from this group were subclassified into six classes in another round of 3D classification using a soft mask and no image alignment. The low number of particles (~5,000) per class limited the resolution of the maps (<10 Å) and could not be further improved through 3D refinement.
Refinement of the individual maps (Supplementary Fig. 2a, b) led to relatively low-resolution reconstructions, particularly in the CIV portion of the map, probably due to greater structural heterogeneity in the sample with respect to this portion of the tripartite SC. The highest resolution was obtained with a map containing a single copy of CIV by combining all classes showing well-defined features corresponding to it and subjecting them to a second round of 3D classification, focused on CIV portion of the map. For this purpose, a soft mask around CIV was created by fitting the closely related structure of P. stutzeri (PDB: 3MK7) into the map and low-pass filtering it to 10 Å. All information outside of this mask was subtracted from the aligned particles, and the remaining particles were subjected to a masked classification without image alignment, to yield six classes (Supplementary Fig. 2a). Alternatively, a cylindrical mask was wrapped around the CIV portion of the map and used in a similar procedure (Supplementary Fig. 2b). In each case, classes showing the highest level of details were retained, and the entire unsubtracted particle dataset was subjected to 3D auto-refinement followed by per-particle CTF refinement, Bayesian polishing, and post-processing using Relion 3.0. The two extreme conformations of the orientation of CIV relative to CIII2 were represented by the maps SC-1A and SC-1B (Supplementary Fig. 2a, b), which were refined to 6.1 and 7.2 Å resolutions, respectively (Supplementary Table 3).
Images of the bipartite SC supplemented with cyt cy (SC + cy) were recorded by a Gatan K3 direct electron detector equipped with an energy quantum filter (20 eV) and operated in counting mode at a nominal magnification of ×64,000, corresponding to a pixel size of 1.36 Å. Images were dose-fractionated to 80 frames with a total exposure time of 3.1 s and a total dose of 40 e−/A2. Nominal underfocus values varied from 1 to 2.5 μm. Automated data acquisition was carried out using the Latitude software (Gatan) and image shift (~ 2 μm, 4 images per stage position) was used for accelerated data collection. For this sample, 5480 images were collected in the first session, motion corrected using MotionCor281, and CTF parameters were determined with CTFFIND 4.182. For auto-picking, the template previously obtained with the tripartite SC was used, with this and all subsequent steps done in Relion 3.083. After sorting and two rounds of 2D classification, ~228,000 particles were retained (Supplementary Fig. 5a). The same initial model as for the tripartite SC was used for 3D classification into five classes. Two classes (001 and 002) resembled more to the overall shape of a CIII2 without CIV, while the other three classes showed the same overall shape of a CIII2CIV SC, as seen with the tripartite sample. In all, 37,460 particles corresponding to the SC class with the highest level of detail were combined with SC particles from the second dataset (Supplementary Fig. 5b) to yield the best final map of the bipartite SC. The second dataset consisted of 12,200 images, which were collected and processed under the same conditions as the first dataset. In all, 1.6 million particles were auto-picked, and after 2 rounds of 2D classification, classes were split into ~340,000 large particles likely representing CIII2CIV and ~465,000 particles resembling CIII2 (Supplementary Fig. 5b, d). The former particles were subjected to 3D classification into three classes, and the class most similar to the overall shape of CIII2CIV was identified. It contained ~118.000 particles, but due to its low level of detail, it was subjected to another round of 2D classification. The 34,819 particles thus retained were combined with 37,460 SC particles from the first dataset (Supplementary Fig. 5a). After 3D classification into five classes, the class with the highest level of detail contained 56% of particles and clearly showed the overall shape of CIII2CIV. In all, 41,017 aligned particles corresponding to this class were extracted and subjected to a second round of 3D classification without image alignment using a soft mask. Of the six 3D classes, the one with the highest nominal resolution contained 14,978 particles (36%) and was subjected to 3D auto-refinement and post-processing, followed by per-particle CTF refinement and Bayesian polishing. After a second round of 3D auto-refinement and post processing, the final map (SC-2A) of the bipartite CIII2CIV was obtained at a nominal resolution of 5.18 Å (Supplementary Table 3). This map was very similar to that (SC-1A) of the tripartite CIII2CIV, except for its higher resolution and strongly improved density and resolution of the extra TMHs at the interface. Unlike the tripartite samples, no major class corresponding to the conformation seen in SC-1B was identified.
As the bipartite SC samples contained a significant amount of CIII2 particles without CIV, subsets of smaller 2D classes consistent with a CIII2 dimer were selected and processed separately. Of the ~376,000 total particles retained from the dataset 1, ~267,000 were identified as resembling CIII2. After a second round of 2D classification (Supplementary Fig. 5c), ~213,000 particles were retained and used to generate an ab initio 3D model. This model was low-pass filtered to 60 Å and used as initial model for the 3D classification of ~465,000 CIII2 particles from dataset 2 (Supplementary Fig. 5d). Particles corresponding to the class with the highest resolution and level of detail (~185,000) were combined with the CIII2 particles from dataset 1 to yield ~400,000 particles and subjected to 3D classification. The ~170,000 particles corresponding to the best class were further processed following two different strategies. CIII2 being a homodimer based on X-ray structures, C2 symmetry was applied in the next round of 3D classification and in all subsequent steps (Supplementary Fig. 5e). The best class contained ~120,000 particles (70%), which were extracted and, similar to CIII2CIV, subjected to another round of 3D classification without image alignment using a soft mask. Of the six classes obtained, some showed the ED of both monomers of the FeS protein close to heme bL (“b position”, b-b) while other classes showed both EDs close to heme c1 (“c position”, c-c). Individual classes were subjected to 3D auto-refinement and post-processing, followed by two rounds of per-particle CTF refinement, Bayesian polishing, 3D auto-refinement, and post-processing. Only the final map with the highest nominal resolution (3.30 Å) is shown (Supplementary Fig. 5e, CIII2; Supplementary Table 3). This map contained 37,997 particles and showed both FeS-EDs close to b position but at a lower local resolution and occupancy than the rest of the map, indicating structural flexibility and conformational heterogeneity. A similar approach was used in Supplementary Fig. 5f, except that no C2 symmetry was applied in the two rounds of 3D classifications. Of the three best classes obtained in the second round, one showed both EDs in the b position, one showed both in the c position, and one showed a heterodimeric conformation with one monomer in the b and the other in the c position. Each of the maps was further processed as in Supplementary Fig. 5e, but C2 symmetry was only applied in case of the homodimeric structures. The final maps were CIII2 c-c with both EDs in the c position at 3.8 Å, CIII2 b-b with both ED’s in the b position at 3.5 Å, and CIII2 b-c with one ED in the b and the second in the c position at 4.2 Å (Supplementary Table 3). The nominal resolutions thus obtained were slightly lower than in map CIII2 (Supplementary Fig. 5e). However, by omitting C2 symmetry application to the 3D classifications, the conformational heterogeneity of the FeS protein EDs was resolved, and a subset of particles showing a heterodimeric conformation was identified. The different locations (i.e., b or c positions) of the EDs were clear in the three maps (Supplementary Fig. 5f), but their occupancy and local resolutions was lower than the rest of the structure.
A third dataset was also collected and yielded ~303,000 particles after the first 2D classification. Adding these particles to the first two datasets did not improve the maps shown in Supplementary Fig. 5a, b, but it turned out to be informative. As shown in Supplementary Fig. 5g, particles from dataset 3 were subjected to two rounds of 3D classification. Interestingly, an extra density near the periplasmic domain of CcoP could be seen in some classes. This was not observed in any subclass obtained from the datasets 1 and 2 and tentatively thought to correspond to the cyt domain of cyt cy. Focused classification followed by 3D auto-refinement and post-processing, using wider soft masks around the periplasmic domain of CcoP to avoid cutting off any of the extra density, led to a final map (SC-2B) with limited resolution (10.5 Å) and could not be further improved by sub-classification due to the low number of particles in each subclass.
Refinement of R. capsulatus CIII2 in the cryo-EM maps
The X-ray based structure of R. capsulatus CIII2 (PDB: 1ZRT) was fitted into map CIII2 (EMD-22189), which had the highest nominal resolution of all maps obtained (Supplementary Fig. 5e), and refined using Phenix1.1685. The real space refinement approach included four rounds of global minimization, local grid search, morphing, and simulated annealing, with the final round also including ADP (B-factor) refinement. Each round included five cycles using default settings, and morphing and annealing were performed in each cycle. Secondary structures were determined by Phenix1.16, using default search settings, and restrictions were applied during real space refinement. Due to the low occupancy and limited local resolution, the FeS-ED proteins (residues 50–191) were only subjected to rigid body fitting followed by two rounds of global minimization and local grid search (5 cycles each) but not to morphing and simulated annealing. To ensure the correct cofactor geometry, hemes and [2Fe-2S] clusters including their coordinating residues were copied from the high-resolution X-ray structure of the homologous CIII2 (cyt bc1) from R. sphaeroides (PDB: 6NHH). Validation was performed using MolProbity86 (http://molprobity.biochem.duke.edu/), and outliers (Ramachandran, rotamer, bonds, angles) were manually corrected in Coot87, using real space refinement and regularization. The model that was refined in map CIII2 (EMD-22189) was subsequently used for rigid body fitting into the maps CIII2 b-b, CIII2 c-c, and CIII2 b-c (Supplementary Fig. 5f and Supplementary Table 3). Each chain was treated as one separate body, except the FeS protein, which was split into its TMH (11–49) and ED (50–191) residues. After the procedure, the linker region between the ED and TMH (residues 40–50) was remodeled in Coot using real space refinement and regularization.
Modeling of R. capsulatus cbb 3 -type CIV subunits
R. capsulatus cbb3-type CIV was modeled using P. stutzeri cbb3-type CIV structure (PDB: 5DJQ or 3MK7) (sequence identities for R. capsulatus CcoN: 68%, CcoO: 55%, CcoP: 34%) as a template, and comparative models were computed using MODELLER v9.1888 with defined secondary structure and crosslinking restraints. The secondary structures for the regions without any template coverage (CcoP residues 104–112, 176–178, 184–197, and 204–215; CcoO residues 172–185 and 192–199), due to the insertion sequences that are present only in R. capsulatus CIV, were estimated using PsiPred Protein Sequence Analysis Workbench89,90, and added as restraints to MODELLER. Cross-link distances were added as Gaussian restraints to MODELLER with a mean of 18.0 Å and a standard deviation of 1.0 Å. Problematic loop regions (a total of ten loops longer than four amino acids) were detected by MolProbity86,91 and remodeled using MODELLER “slow” refinement method (Supplementary Table 3). As R. capsulatus CcoN is longer than that of P. stutzeri with a predicted extra N-ter TMH, whereas CcoP is shorter with only one predicted N-ter TMH instead of two, these regions were not included into the model. The regions CcoO 179–214 as well as CcoP 1–12, 53–59, 161–173, and 272–280 without template coverage that were not supported by the cryo-EM maps were omitted.
Modeling of R. capsulatus CcoH and cyt c y
For CcoH (residues Met 1 to Thr35), five ab initio models were obtained using I-TASSER server92. All models were almost identical, and one with the lowest energy score was retained. For R. capsulatus cyt cy, its homologs with known structures were detected using HHpred93, and the structure of cyt domain of cyt c552 from P. denitirificans (PDB: 3M97; sequence identity: 61%) was used as a template for the soluble cyt domain of cy, and comparative models were computed using MODELLER88.
Modeling of the extra N-ter TMH of CcoN (TMH0)
A 29-residue model consisting of CcoN residues Arg25 to Asp53, which includes the predicted transmembrane region (Met30-Leu48, UniProtKB, D5ARP4), was obtained from the I-TASSER server94. The alpha helical region included residues Leu27 to Thr50, and the model including the residues Arg25 to Leu48 was manually fitted into the density map for visualization purpose in Fig. 2. Related coordinates were not deposited in the PDB because the registration could not be determined due to the lack of side chain density.
Modeling of cyt c y and CcoH TMH interactions
The 30-residue-long cyt cy TMH was manually docked into the density map by positioning Phe15 and Tyr21 into the corresponding densities and refined by real-space refinement and regularization using Coot. The I-TASSER model of CcoH was docked into the map by moving it along the corresponding density, retaining its close association with cyt cy TMH. The best fit was found when Ala23 and Val24 of CcoH were located at the interface with cyt cy TMH. The interaction of the two helices was optimized by GalaxyWEB95, and the model with the lowest energy profile was refined in map SC-2A using Phenix 1.17. The refinement strategy included five cycles of global minimization, rigid body fitting (where cyt cy and CcoH chains were treated as separate bodies), local grid search, and ADP (B-factor) refinement with default settings for all steps. Secondary structure restrictions were applied to the α-helical parts of CcoH as predicted by I-TASSER (residues 12–34) and to cyt cy as predicted by GalaxyWEB (residues 2–9 and 12–29). CIII2 and CIV models were present during the refinement to keep the rest of the map occupied, but changes made during this procedure were discarded as these models were refined separately.
Integrative modeling of SC
The entire SC was assembled by an integrative modeling using the cryo-EM map, XL-MS, co-evolutionary analysis, and subunit models described above. CIII2 and CIV models were fitted into the cryo-EM maps in UCSF Chimera79.
Docking of CcoH, cyt c y, and cyt c 2 to bipartite SC
Additional data about the interaction interfaces between the different subunits were obtained using RaptorX-ComplexContact33 for each pair of the SC subunits. Based on co-evolution and machine learning, this method predicted the pairs of residues that are in contact. The PatchDock, which is an efficient rigid docking method that maximizes geometric shape complementarity34, was used to generate docked configurations of CcoH, cyt c2, and cyt cy to CIII2 and CIV, as appropriate, and in all docking analyses the top 50 models were retained. The different subunits were docked in parallel independently from each other.
Docking of CcoH to CIV
A total of 16 contacts predicted by RaptorX-ComplexContact, with probabilities >0.5 were used as distance restraints for protein–protein docking34. The models satisfied 13 of the contacts, and a single cluster of docked models evidenced by the convergence of the top 50 results was obtained (Supplementary Fig. 7). This cluster coincided with the additional feature seen at the SC interface, and CcoH TMH was modeled into this feature.
Docking of cyt c 2 to CIII2 and CIV
R. capsulatus cyt c2 of known structure (PDB: 1C2N) was docked to CIII2 using the three distance restraints between cyt c2 and cyt c1 derived from the protein crosslinking data (Supplementary Table 4). Similarly, cyt c2 was docked to CIV using nine distance restraints provided by XLs (1 CL to CcoP and 8 to CcoO, Supplementary Table 4). The models yielded one main cluster in each case and covered 100 and 89% of the data for CIII2 and CIV, respectively.
Docking of cyt c y to CIII2CIV SC
The model of cyt domain of cy was docked by PatchDock to bipartite CIII2CIV, which has both FeS-EDs of CIII2 in the b position. Ten distance restraints derived from the XL-MS data (6 with DMTMM with max limit of 30 Å and 4 with DSBU with max limit of 35 Å, Supplementary Table 4) were used, top models yielded two main clusters (1 and 2) of the docked models on the p side of CIII2 (Supplementary Fig. 4), and satisfied 100% of the restraints. As the cryo-EM data revealed that CIII2 particles could have their FeS-EDs in the c position, the corresponding CIII2 c-c, CIII2 b-b, and CIII2 b-c models were used for docking via PatchDock the cyt domain of cy onto CIII2.
All the models were ranked using statistically optimized atomic potentials (SOAP)96, and those that have low SOAP scores were retained.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
R. capsulatus database used is: https://www.uniprot.org, last modified 01/15/2020; R. capsulatus strain ATCC BAA-309/NBRC 16581/SB1003. All structural data and corresponding cryo-EM maps that support the findings of this study have been deposited to PDB and EMDB under the accession codes: CIII2 (PDB: 6XI0; EMD-22189); CIII2 c-c (PDB: 6XKT; EMD-22224); CIII2 b-c (PDB: 6XKU; EMD-22225); CIII2 b-b (PDB: 6XKV; EMD-22226); SC-2A (PDB: 6XKW; EMD-22227); SC-1A (PDB: 6XKX; EMD-22228); SC-1B (PDB: 6XKZ; EMD-22230). XL-MS data have been deposited to ProteomeXchange PRIDE repository with the dataset identifier PXD020038. All other relevant data are available upon request from the corresponding authors. Source data are provided with this paper.
References
Nicholls, D. G. & Ferguson, S. J. Bioenergetics 4 (Elsevier, 2013).
Darrouzet, E., Moser, C. C., Dutton, P. L. & Daldal, F. Large scale domain movement in cytochrome bc1: a new device for electron transfer in proteins. Trends Biochem. Sci. 26, 445–451 (2001).
Esser, L. et al. Surface-modulated motion switch: capture and release of iron-sulfur protein in the cytochrome bc1 complex. Proc. Natl Acad. Sci. USA 103, 13045–13050 (2006).
Khalfaoui-Hassani, B. et al. In Cytochrome Complexes: Evolution, Structures, Energy Transduction, and Signaling (eds Cramer, W. & Kallas, T.) 527–555 (Springer, 2016).
Smith, M. A., Finel, M., Korolik, V. & Mendz, G. L. Characteristics of the aerobic respiratory chains of the microaerophiles Campylobacter jejuni and Helicobacter pylori. Arch. Microbiol. 174, 1–10 (2000).
Aspholm, M. et al. Structural alterations in a component of cytochrome c oxidase and molecular evolution of pathogenic Neisseria in humans. PLoS Pathog. 6, e1001055 (2010).
Ekici, S., Pawlik, G., Lohmeyer, E., Koch, H. G. & Daldal, F. Biogenesis of cbb(3)-type cytochrome c oxidase in Rhodobacter capsulatus. Biochim. Biophys. Acta 1817, 898–910 (2012).
Jenney, F. E. Jr. & Daldal, F. A novel membrane-associated c-type cytochrome, cyt cy, can mediate the photosynthetic growth of Rhodobacter capsulatus and Rhodobacter sphaeroides. EMBO J. 12, 1283–1292 (1993).
Turba, A., Jetzek, M. & Ludwig, B. Purification of Paracoccus denitrificans cytochrome c552 and sequence analysis of the gene. Eur. J. Biochem. 231, 259–265 (1995).
Albert, I., Rutherford, A. W., Grav, H., Kellermann, J. & Michel, H. The 18 kDa cytochrome c553 from Heliobacterium gestii: gene sequence and characterization of the mature protein. Biochemistry 37, 9001–9008 (1998).
Kim, M. S. et al. Isolation and characterization of a hybrid respiratory supercomplex consisting of Mycobacterium tuberculosis cytochrome bcc and Mycobacterium smegmatis cytochrome aa3. J. Biol. Chem. 290, 14350–14360 (2015).
Winstedt, L. & von Wachenfeldt, C. Terminal oxidases of Bacillus subtilis strain 168: one quinol oxidase, cytochrome aa3 or cytochrome bd, is required for aerobic growth. J. Bacteriol. 182, 6557–6564 (2000).
Hochkoeppler, A., Jenney, F. E. Jr., Lang, S. E., Zannoni, D. & Daldal, F. Membrane-associated cytochrome cy of Rhodobacter capsulatus is an electron carrier from the cytochrome bc1 complex to the cytochrome c oxidase during respiration. J. Bacteriol. 177, 608–613 (1995).
Enriquez, J. A. Supramolecular organization of respiratory complexes. Annu. Rev. Physiol. 78, 533–561 (2016).
Melo, A. M. & Teixeira, M. Supramolecular organization of bacterial aerobic respiratory chains: from cells and back. Biochim. Biophys. Acta 1857, 190–197 (2016).
Brzezinski, P. New structures reveal interaction dynamics in respiratory supercomplexes. Trends Biochem. Sci. https://doi.org/10.1016/j.tibs.2019.10.011 (2019).
Letts, J. A., Fiedorczuk, K., Degliesposti, G., Skehel, M. & Sazanov, L. A. Structures of respiratory supercomplex I+III2 reveal functional and conformational crosstalk. Mol. Cell 75, 1131–1146 e1136 (2019).
Quintana-Cabrera, R. & Soriano, M. E. ER stress priming of mitochondrial respiratory suPERKomplex assembly. Trends Endocrinol. Metab. 30, 685–687 (2019).
Letts, J. A., Fiedorczuk, K. & Sazanov, L. A. The architecture of respiratory supercomplexes. Nature 537, 644–648 (2016).
Gu, J. et al. The architecture of the mammalian respirasome. Nature 537, 639–643 (2016).
Hartley, A. M. et al. Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc1. Nat. Struct. Mol. Biol. 26, 78–83 (2019).
Berry, E. A. & Trumpower, B. L. Isolation of ubiquinol oxidase from Paracoccus denitrificans and resolution into cytochrome bc1 and cytochrome c-aa3 complexes. J. Biol. Chem. 260, 2458–2467 (1985).
Kao, W. C. et al. The obligate respiratory supercomplex from Actinobacteria. Biochim. Biophys. Acta 1857, 1705–1714 (2016).
Gong, H. et al. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. Science. https://doi.org/10.1126/science.aat8923 (2018).
Wiseman, B. et al. Structure of a functional obligate complex III2IV2 respiratory supercomplex from Mycobacterium smegmatis. Nat. Struct. Mol. Biol. 25, 1128–1136 (2018).
Ducluzeau, A. L., Ouchane, S. & Nitschke, W. The cbb3 oxidases are an ancient innovation of the domain bacteria. Mol. Biol. Evol. 25, 1158–1166 (2008).
Myllykallio, H., Drepper, F., Mathis, P. & Daldal, F. Electron-transfer supercomplexes in photosynthesis and respiration. Trends Microbiol. 8, 493–494 (2000).
Davidson, E., Ohnishi, T., Tokito, M. & Daldal, F. Rhodobacter capsulatus mutants lacking the Rieske FeS protein form a stable cytochrome bc1 subcomplex with an intact quinone reduction site. Biochemistry 31, 3351–3358 (1992).
Kulajta, C., Thumfart, J. O., Haid, S., Daldal, F. & Koch, H. G. Multi-step assembly pathway of the cbb3-type cytochrome c oxidase complex. J. Mol. Biol. 355, 989–1004 (2006).
Valkova-Valchanova, M. B., Saribas, A. S., Gibney, B. R., Dutton, P. L. & Daldal, F. Isolation and characterization of a two-subunit cytochrome b-c1 subcomplex from Rhodobacter capsulatus and reconstitution of its ubihydroquinone oxidation (Qo) site with purified Fe-S protein subunit. Biochemistry 37, 16242–16251 (1998).
Gray, K. A. et al. Rhodobacter capsulatus contains a novel cb-type cytochrome c oxidase without a CuA center. Biochemistry 33, 3120–3127 (1994).
Ozturk, Y. et al. Soluble variants of Rhodobacter capsulatus membrane-anchored cytochrome cy are efficient photosynthetic electron carriers. J. Biol. Chem. 283, 13964–13972 (2008).
Zeng, H. et al. ComplexContact: a web server for inter-protein contact prediction using deep learning. Nucleic Acids Res. 46, W432–W437 (2018).
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. & Wolfson, H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–W367 (2005).
Pawlik, G. et al. The putative assembly factor CcoH is stably associated with the cbb3-type cytochrome oxidase. J. Bacteriol. 192, 6378–6389 (2010).
Covian, R. & Trumpower, B. L. Rapid electron transfer between monomers when the cytochrome bc1 complex dimer is reduced through center N. J. Biol. Chem. 280, 22732–22740 (2005).
Castellani, M. et al. Direct demonstration of half-of-the-sites reactivity in the dimeric cytochrome bc1 complex: enzyme with one inactive monomer is fully active but unable to activate the second ubiquinol oxidation site in response to ligand binding at the ubiquinone reduction site. J. Biol. Chem. 285, 502–510 (2010).
Cooley, J. W., Lee, D. W. & Daldal, F. Across membrane communication between the Qo and Qi active sites of cytochrome bc1. Biochemistry 48, 1888–1899 (2009).
Chavez, J. D. et al. Chemical crosslinking mass spectrometry analysis of protein conformations and supercomplexes in heart tissue. Cell Syst. 6, 136.e5–141.e5 (2018).
Chavez, J. D. & Bruce, J. E. Chemical cross-linking with mass spectrometry: a tool for systems structural biology. Curr. Opin. Chem. Biol. 48, 8–18 (2019).
Larance, M. et al. Global membrane protein interactome analysis using in vivo crosslinking and mass spectrometry-based protein correlation profiling. Mol. Cell. Proteomics 15, 2476–2490 (2016).
Leitner, A., Faini, M., Stengel, F. & Aebersold, R. Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci. 41, 20–32 (2016).
Slavin, M. & Kalisman, N. Structural analysis of protein complexes by cross-linking and mass spectrometry. Methods Mol. Biol. 1764, 173–183 (2018).
Gotze, M. et al. Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom. 26, 83–97 (2015).
Solmaz, S. R. & Hunte, C. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J. Biol. Chem. 283, 17542–17549 (2008).
Iacobucci, C. et al. A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein-protein interactions. Nat. Protoc. 13, 2864–2889 (2018).
Kalisman, N., Adams, C. M. & Levitt, M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl Acad. Sci. USA 109, 2884–2889 (2012).
Osyczka, A., Moser, C. C. & Dutton, P. L. Fixing the Q cycle. Trends Biochem. Sci. 30, 176–182 (2005).
Crofts, A. R. et al. The Q-cycle reviewed: how well does a monomeric mechanism of the bc(1) complex account for the function of a dimeric complex? Biochim. Biophys. Acta 1777, 1001–1019 (2008).
Buschmann, S. et al. The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 329, 327–330 (2010).
Myllykallio, H., Drepper, F., Mathis, P. & Daldal, F. Membrane-anchored cytochrome cy mediated microsecond time range electron transfer from the cytochrome bc1 complex to the reaction center in Rhodobacter capsulatus. Biochemistry 37, 5501–5510 (1998).
Lee, D. W., Ozturk, Y., Osyczka, A., Cooley, J. W. & Daldal, F. Cytochrome bc1-cy fusion complexes reveal the distance constraints for functional electron transfer between photosynthesis components. J. Biol. Chem. 283, 13973–13982 (2008).
Berry, E. A. et al. X-ray structure of Rhodobacter capsulatus Cytochrome bc1: comparison with its mitochondrial and chloroplast counterparts. Photosynth. Res. 81, 251–275 (2004).
Xia, D. et al. The road to the crystal structure of the cytochrome bc1 complex from the anoxigenic, photosynthetic bacterium Rhodobacter sphaeroides. J. Bioenerg. Biomembr. 40, 485–492 (2008).
Esser, L. et al. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. J. Biol. Chem. 283, 2846–2857 (2008).
Sousa, J. S., Mills, D. J., Vonck, J. & Kuhlbrandt, W. Functional asymmetry and electron flow in the bovine respirasome. Elife. https://doi.org/10.7554/eLife.21290 (2016).
Swierczek, M. et al. An electronic bus bar lies in the core of cytochrome bc1. Science 329, 451–454 (2010).
Lanciano, P., Lee, D. W., Yang, H., Darrouzet, E. & Daldal, F. Intermonomer electron transfer between the low-potential b hemes of cytochrome bc1. Biochemistry 50, 1651–1663 (2011).
Moser, C. C., Keske, J. M., Warncke, K., Farid, R. S. & Dutton, P. L. Nature of biological electron transfer. Nature 355, 796–802 (1992).
Wikstrom, M., Krab, K. & Sharma, V. Oxygen activation and energy conservation by cytochrome c oxidase. Chem. Rev. 118, 2469–2490 (2018).
Brzezinski, P. & Gennis, R. B. Cytochrome c oxidase: exciting progress and remaining mysteries. J. Bioenerg. Biomembr. 40, 521–531 (2008).
Verissimo, A. F., Sousa, F. L., Baptista, A. M., Teixeira, M. & Pereira, M. M. Thermodynamic redox behavior of the heme centers of cbb3 heme-copper oxygen reductase from Bradyrhizobium japonicum. Biochemistry 46, 13245–13253 (2007).
Otten, M. F. et al. Cytochromes c550, c552, and c1 in the electron transport network of Paracoccus denitrificans: redundant or subtly different in function? J. Bacteriol. 183, 7017–7026 (2001).
Kappler, U. & Dahl, C. Enzymology and molecular biology of prokaryotic sulfite oxidation. FEMS Microbiol. Lett. 203, 1–9 (2001).
Khalfaoui-Hassani, B. et al. Widespread distribution and functional specificity of the copper importer CcoA: distinct Cu uptake routes for bacterial cytochrome c oxidases. MBio. https://doi.org/10.1128/mBio.00065-18 (2018).
Wittig, I., Braun, H. P. & Schagger, H. Blue native PAGE. Nat. Protoc. 1, 418–428 (2006).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 (1970).
Bartsch, H., Arndt, C., Koristka, S., Cartellieri, M. & Bachmann, M. Silver staining techniques of polyacrylamide gels. Methods Mol. Biol. 869, 481–486 (2012).
Yan, L. J. & Forster, M. J. Resolving mitochondrial protein complexes using nongradient blue native polyacrylamide gel electrophoresis. Anal. Biochem. 389, 143–149 (2009).
Thomas, P. E., Ryan, D. & Levin, W. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal. Biochem. 75, 168–176 (1976).
Gill, S. C. & von Hippel, P. H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 (1989).
Atta-Asafo-Adjei, E. & Daldal, F. Size of the amino acid side chain at position 158 of cytochrome b is critical for an active cytochrome bc1 complex and for photosynthetic growth of Rhodobacter capsulatus. Proc. Natl Acad. Sci. USA 88, 492–496 (1991).
Peters, A., Kulajta, C., Pawlik, G., Daldal, F. & Koch, H. G. Stability of the cbb3-type cytochrome oxidase requires specific CcoQ-CcoP interactions. J. Bacteriol. 190, 5576–5586 (2008).
Selamoglu, N. et al. Comparative differential cuproproteomes of Rhodobacter capsulatus reveal novel copper homeostasis related proteins. Metallomics 12, 572–591 (2020).
Myllykallio, H., Jenney, F. E. Jr., Moomaw, C. R., Slaughter, C. A. & Daldal, F. Cytochrome cy of Rhodobacter capsulatus is attached to the cytoplasmic membrane by an uncleaved signal sequence-like anchor. J. Bacteriol. 179, 2623–2631 (1997).
Holden, H. M., Meyer, T. E., Cusanovich, M. A., Daldal, F. & Rayment, I. Crystallization and preliminary analysis of crystals of cytochrome c2 from Rhodopseudomonas capsulata. J. Mol. Biol. 195, 229–231 (1987).
Sheng, Q. et al. Preprocessing significantly improves the peptide/protein identification sensitivity of high-resolution isobarically labeled tandem mass spectrometry data. Mol. Cell. Proteomics 14, 405–417 (2015).
Rutsdottir, G. et al. Structural model of dodecameric heat-shock protein Hsp21: Flexible N-terminal arms interact with client proteins while C-terminal tails maintain the dodecamer and chaperone activity. J. Biol. Chem. 292, 8103–8121 (2017).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Kosinski, J. et al. Xlink Analyzer: software for analysis and visualization of cross-linking data in the context of three-dimensional structures. J. Struct. Biol. 189, 177–183 (2015).
Zheng, S. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5 6 1–32 (2014).
Buchan, D. W. A. & Jones, D. T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 47, W402–W407 (2019).
Jones, D. T., Tress, M., Bryson, K. & Hadley, C. Successful recognition of protein folds using threading methods biased by sequence similarity and predicted secondary structure. Proteins (Suppl 3), 104–111 (1999).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960 (2005).
Zhang, W. et al. Integration of QUARK and I-TASSER for ab initio protein structure prediction in CASP11. Proteins 84(Suppl 1), 76–86 (2016).
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 40, W294–W297 (2012).
Dong, G. Q., Fan, H., Schneidman-Duhovny, D., Webb, B. & Sali, A. Optimized atomic statistical potentials: assessment of protein interfaces and loops. Bioinformatics 29, 3158–3166 (2013).
Acknowledgements
This work was supported partly by the NIH grants GM 38237 to F.D., GM123233 to K.M., GM110174 and AI118891 to B.A.G., T32-GM008275 to T.V., T32-GM071339 to H.J.K., and partly by the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences of Department of Energy grant DE-FG02-91ER20052 to F.D., by ISF 1466/18, BSF 2016070, and Ministry of Science and Technology 80802 grants to D.S.-D. Y.O. was supported by the grant GRK2202-23577276/RTG from DFG, Germany. Data analysis was partly supported by the NIH grant S10OD023592. This research was supported in part by the NCI, National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. The authors thank Ulrich Baxa, Thomas Edwards, and Adam Wier for their support and helpful discussions. Some cryo-EM data were also obtained at the University of Massachusetts Cryo-EM Core Facility, and we thank Dr. Chen Xu, Dr. KangKang Song, and Dr. Kyounghwan Lee for their support. Cryo-EM sample screening and optimization was performed at the Electron Microscopy Resource Laboratory at the Perelman School of Medicine, University of Pennsylvania, and we thank Dr. Sudheer Molugu for his support. We also thank Dr. S. Saif Hasan, Dr. Brian G. Pierce, and Dr. Christian Presley at the Institute for Bioscience and Biotechnology Research, University of Maryland for insightful discussions and invaluable help they provided during this study. S.S. and F.D. thank Vivian Kitainda for her assistance with protein purification and O2 consumption measurements.
Author information
Authors and Affiliations
Contributions
All authors have given approval to the final version of the manuscript. S.S., T.V., Y.O., H.J.K., M.B., N.S., B.A.G., D.S.-D., K.M., and F.D. all contributed to aspects of the experiments, analyzed data, and wrote and edited the manuscript. S.S. purified, characterized, and prepared samples; S.S. and T.V. processed data and built models; Y.O. performed genetic constructs; M.B. and D.S.-D. conducted computer modeling; H.J.K., N.S., and B.A.G. obtained MS data; and K.M. and F.D. managed the overall project and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Steimle, S., van Eeuwen, T., Ozturk, Y. et al. Cryo-EM structures of engineered active bc1-cbb3 type CIII2CIV super-complexes and electronic communication between the complexes. Nat Commun 12, 929 (2021). https://doi.org/10.1038/s41467-021-21051-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-21051-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
