
Abstract
Burial of organic material in marine sediments represents a dominant natural mechanism of long-term carbon sequestration globally, but critical aspects of this carbon sink remain unresolved. Investigation of surface sediments led to the proposition that on average 10-20% of sedimentary organic carbon is stabilised and physically protected against microbial degradation through binding to reactive metal (e.g. iron and manganese) oxides. Here we examine the long-term efficiency of this rusty carbon sink by analysing the chemical composition of sediments and pore waters from four locations in the Barents Sea. Our findings show that the carbon-iron coupling persists below the uppermost, oxygenated sediment layer over thousands of years. We further propose that authigenic coprecipitation is not the dominant factor of the carbon-iron bounding in these Arctic shelf sediments and that a substantial fraction of the organic carbon is already bound to reactive iron prior deposition on the seafloor.
Similar content being viewed by others


Introduction
Organic carbon (OC) burial in shelf sediments plays an important role in the global carbon cycle as ∼87% of the estimated 169 × 106 tons of OC deposited at the seafloor were buried in these shallow parts of the ocean each year1. However, the fundamental, physical, biological, and chemical processes that control OC preservation, including sedimentation rate2,3, presence and absence of oxygen4,5,6, selective preservation of biochemically unreactive compounds7,8, and protection of organic matter through interactions with a mineral matrix9,10,11 are complex and not fully understood. Even though a possible chemical association between iron and OC in soils were identified more than half a century ago12, clay minerals were historically viewed as the primary controller for mineral hosted sedimentary carbon. The potentially global importance of reactive iron oxides (FeR; nanoparticulate and amorphous phases of ferric (oxyhydroxides) for OC preservation in marine sediments has only recently been recognised13.
At the Fe2+/Fe3+ redox boundary, typically in the upper centimetres of a shelf sediment profile, oxidation and precipitation of upward diffusing Fe2+ (liberated to the pore waters by dissimilatory iron reduction) leads to an enrichment of sedimentary FeR14. The OC has a strong affinity to these freshly precipitating Fe(III) phases (e.g., ferrihydrite) and the resultant iron/OC association, through coprecipitation of OC within or sorption to reactive iron phases is assumed to promote long-term stabilisation of sedimentary organic matter13,15,16,17. Thus, reactive iron phases may serve as an efficient shuttle to promote carbon burial as OC associated with these Fe(III) phases should be protected against microbial degradation, allowing it to bypass the efficient early diagenetic degradation regime18 and to be buried into anoxic sediments, where the OC preservation potential is much higher.
To date, the mechanisms stabilising OC with FeR in marine sediments have mainly been studied in surface sediments13,19,20,21,22,23. These studies show that the fraction of the total OC bound to FeR (fOC-FeR) is on average 10–20%, with values ranging from ~0.5 to 40%. A series of factors, such as binding mechanisms of OC to FeR, sediment mineralogy, organic matter composition, and iron redox cycling were invoked to explain the wide variations of fOC-FeR. However, the term “surface sediment” is ill-defined and the depth below the seafloor of the investigated sediments is not consistent between different studies (ranging from 0.5 to 3 cm or even unspecified). Moreover, information about the position of studied sediment samples relative to the Fe2+/Fe3+ redox interface is usually absent. However, this information is critical, as Fe(III) phases making up the FeR pool are highly redox-sensitive and under anoxic conditions deeper in the sediment, dissimilatory iron reduction24 may affect the stability of the OC-FeR bonding. The stabilisation of OC by FeR may therefore be transient and only stabilise OC in the oxic surface sediment layer. Hence, while iron redox cycling has been proposed as a controlling factor of fOC-FeR, current findings based on surface sediment investigations might be biased by differences in the depth of oxygen penetration and the Fe2+/Fe3+ redox interface at the different locations. Moreover, downcore investigations of OC-FeR will not only provide a better understanding of the role of early diagenesis in OC-FeR generation and stability, they will also help to reveal the source of the OC-FeR (allochthonous vs. autochthonous) and allow to identify the relative contributions of OC-FeR that was formed on land during the transport process, or at the sediment-water interface.
Besides Fe(III) phases, Mn(III/IV) (oxyhydroxides) (MnR) also strongly interact with OC in marine sediments25,26,27. However, similar to the OC-FeR coupling, OC-MnR in marine sediments has so far only been investigated in surface sediments and a paucity of information remains on the abundance of carbon associated with manganese oxides and their potential role in stabilising OC over longer timescales. It is therefore unclear if manganese oxides help to transfer OC from the sediment surface carbon cycle to the geological carbon cycle or if MnR plays a minor role in OC stabilisation compared to FeR25,26.
To better understand the effect of sedimentary degradation processes on the formation and stability of the OC-FeR and OC-MnR association over long (millennial) timescales, we chemically analysed pore water and sediment samples retrieved at four coring sites along a south-north transect across the Arctic Barents Sea shelf area (Fig. 1). Iron and manganese (oxyhydroxide) reduction play an important role in organic matter degradation in this region28,29 and it is therefore a suitable location to study the combined diagenetic fate of OC, iron, and manganese. Moreover, the Barents Sea region currently experiences the greatest warming in the Arctic, a dramatic loss of sea ice30 and the highest increase of primary productivity across the Arctic ocean31. Thus, the transformation from an icy-land into an open ocean force the entire Barents Sea ecosystem to adapt and restructure, which affects the Arctic carbon cycle through changes in atmospheric CO2 uptake, pelagic-benthic coupling, organic matter sedimentation, and long-term sequestration32,33,34,35,36,37. Understanding the mechanisms responsible for enhancing the stability and long-term storage of OC worldwide and especially in the Arctic is important for predicting how the global carbon cycle will respond to climate change.

The Barents Sea is the largest pan-Arctic shelf sea covering an area of 1.6 million km2 with an average water depth of 230 m67. There are several extensive overviews and reviews about the modern climate setting and ecosystem of the Barents Sea and we refer to these references for a detailed description of the physical and ecological conditions34,68,69,70,71,72. The general oceanic circulation pattern of the western Barents Sea is dominated by the relatively warm northward flowing North Atlantic Current which enters the Barents Sea from the south-west and the southward flowing cold Arctic currents entering the Barents Sea from the north-east. The relatively sharp boundary between these water masses forms the oceanographic Polar Front (golden line)44, which is mainly determined by the bathymetry and is, therefore, relatively stable from year to year73. The northern Barents Sea is seasonally ice covered with maximum and minimum ice coverage in March–April and August–September, respectively. The heat content of the Atlantic water keeps the southern Barents Sea permanently ice-free. River runoff into the Barents Sea is limited. Rivers on Svalbard and in Norway are small and often drain into fjords. Thus, sediment discharge through river inflow is low and the main processes responsible for the Barents Sea surface sediment distribution are re-deposition by winnowing from shallow banks into troughs and depressions and deposition from sea ice. Hence, sedimentation rates are generally low, 0.04–2.1 mm/y since the last glacial period48. The brown arrow indicates the proposed transport of iron-rich sediments from Nordaustlandet (I) into the central Barents Sea. The map was created using the IBCAO V. 3.0 dataset74.
Here, we examine (I) the persistence of fOC-FeR below the oxygenated part of the sediment over millennial time scales, (II) the effect of the Fe2+/Fe3+ redox boundary on OC-FeR binding mechanisms, (III) the importance of different Fe(III) phases for binding OC in competition with other chemical species such as arsenic, (IV) the role of manganese oxides in stabilising OC in marine sediments on longer timescales, and (V) potential allochthonous OC-FeR contribution.
Results and discussion
Reactive and total iron sources
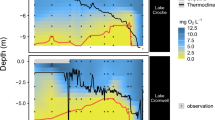
Our results show that at all four study sites (B13–B16), the Fe/Al is highly correlated to the sedimentary FeR contents (r ≥ 0.95; Supplementary Fig. S1), the reactive iron fraction of total iron (fFeR) shows the same downcore pattern as FeR and both parameters are closely related to the total sedimentary iron contents (Figs. 2, 3 and Supplementary Fig. S2). The close relationship between Fe/Al, total iron, and FeR could lead to the conclusion of a common source, e.g. ref. 38, i.e., terrigenous influx from Svalbard (Fig. 1). However, as expected and revealed by the comparison of our Fe2+ pore water and fFeR profiles, early diagenesis plays an important role in the generation of the observed fFeR patterns over depth (Fig. 2). At the Fe2+/Fe3+ redox boundary, oxidation and precipitation of upward diffusing Fe2+ leads to an enrichment of Fe/Al and reactive Fe(III) phases, which results in an accompanying increase of fFeR at all four sites. At stations B13, B14, and B16, the iron redox interface occurs in the top 5 cm at each site. At station B15, oxygen penetrates deeper into the sediment39 and the iron redox interface is located between ~5 and 12 cm. Therefore, diagenetic processes have a strong impact on fFeR patterns at the study location. Seasonal and annual primary productivity changes, for example, through the differences in sea ice cover during the three consecutive sampling years possibly caused variations in the depth of the redox boundary. However, early diagenesis variability had probably only a minor effect on our fFeR results as the iron redox interface was remarkably stable in all investigated cores during our sampling campaigns in summer 2017, 2018, and 2019 (Supplementary Fig. S4). Moreover, the stable redox interface also indicates only minor disturbance of the sediment column through bioturbation, which is in accordance with a recently reported very shallow mean bioturbation depth (<1 cm) at all investigated stations40.

The pore water iron content represents the average iron concentration from three different cores at the same coring location. Black squares are uncalibrated radiocarbon dates in kyr BP. The shaded grey area at station B15 marks the location of a reddish/pink sediment layer.

The three iron phases from the sequential iron extraction, shown by black circles, triangles and squares, are: Fe bound to carbonates or very labile iron oxides (FeOx-1); poorly crystalline Fe (oxyhydr)oxides like ferrihydrite (FeOx-2) and crystalline Fe (oxyhydr)oxides such as hematite and goethite (FeOx-3). Blue bars indicate the sum of FeOx1–3 which shows that the dithionite extraction of FeR (orange dots) represents the entire reactive iron pool. The grey hatched area (B15) indicates a pink, iron-rich sediment layer, which is enriched in more crystalline iron phases (FeOx-3).
At stations B13, B14, and B16, we found no indication of significant changes in the external input of FeR phases to the seafloor over time. Sediment cores from these sites show surface enrichments of fFeR with maxima (~20–30%) in the top 5 cm, which are related to the precipitation of authigenic Fe(III) phases and relatively stable values (~10%) to the core bottom (Fig. 2). In comparison to the other stations, fFeR at station B15 is much higher and shows a distinct peak of up to 51% between 9 and 15 cm. This peak corresponds to high total iron contents of up to 8% and a reddish/pink, fine-grained sediment layer. Such reddish sediment layers are known in sediments north and west of Svalbard and probably originate from iron-rich Devonian sandstones in central Svalbard41. To our knowledge, no such pink/reddish sediment layers have previously been reported from the central Barents Sea. But modern glacial discharge at the eastern side of Svalbard (Fig. 1; Nordaustlanded) creates reddish meltwater plumes42, and sea ice covered with reddish sediment (Supplementary Fig. S3) has been observed during our and other expeditions around eastern Svalbard. The Barents Sea current system transports the fine-grained components incorporated in the meltwater plume south-westward and into the central Barents Sea (Fig. 1; refs. 43,44,45), towards the location of station B15. Hence, we suggest that the high iron content at station B15 is related to north-eastern glacial sediment discharges on Nordaustlanded.
Reactive iron content and OC-FeR bonding
The large downcore variations in the fFeR profiles raise the question of whether the FeR content has a substantial control on the fraction of OC bound to FeR. Due to the strong affinity of OC to reactive Fe(III) phases (e.g., ref. 46), we would expect that an increase in FeR results in an overall higher OC-FeR, presuming sufficient OC is present. However, in our downcore records and agreement with data from the beach and marine surface sediments47,48, OC-FeR, as well as the fraction of total OC bound to FeR (fOC-FeR), do not seem to be controlled by the amount of available sedimentary FeR. At stations B13, B14, and B16, fOC-FeR shows a slight gradual decrease from the core top to the bottom and does not seem to follow the fFeR profile. And fOC-FeR at station B15 shows principally no difference between the fFeR-rich (up to 51%) reddish sediment layer at 9–15 cm depth and the sediments above (Fig. 2). This indicates that increased terrigenous iron and FeR input does not necessarily result in higher fOC-FeR, and therefore, sedimentary iron and FeR contents are not the exclusive controlling factors for the association of OC with FeR. A possible methodical explanation for this result, however, could be the extraction of iron phases with reduced surface reactivities, especially in very iron-rich sediment layers. To deconvolve the reactive iron species in more detail, we conducted a sequential iron extraction on selected samples from our sediment cores (Fig. 3), which reveals that the dominant reactive iron fraction in all sediment cores and in particular in the reddish sediment layer, is attributed to more crystalline iron oxide phases such as haematite and goethite (Fig. 3). The maturation and crystallisation of FeR from more or less fresh phases like ferrihydrite to goethite/haematite decreases its surface area, reactivity, and solubility49. Thus, a large fraction of more crystalline oxides in the dithionite extractable FeR pool may lead to an overestimation of fFeR. Moreover, the gradual decrease of fOC-FeR and OC-FeR (Fig. 4) with increasing depth could be related to the predominance of more crystalline iron oxide phases in the FeR pool below the surface sediments (Fig. 3). Thus, the loss of OC-FeR may be caused by the maturation of these reactive iron phases and an accompanying release of the bound OC50. However, the OC-FeR association not only protects the OC from degradation but is also believed to have a stabilising effect on the iron oxides and, therefore, helps to prevent the transformation to more crystalline phases, e.g. ref. 51. Furthermore, OC associated with less reactive iron phases (e.g., goethite and haematite), probably via mono- or multi-layer sorption, is possibly more accessible for microbial degradation52. Further investigations are required to quantify the role of the different Fe(III) phases within the reactive iron pool in stabilising OC in natural sediments. Nevertheless, the decreasing trends of OC-FeR and fOC-FeR are accompanied by overall declining total sedimentary OC content with increasing depth at all stations (Fig. 4) and we cannot rule out that downcore variability in the fOC-FeR has been affected by processes completely independent from iron. In fact, we fully acknowledge that the downcore patterns in the amounts of OC bound to FeR may have been affected by various processes. These include the remineralisation of iron-bound OC over time, but also a combination of chemical, physical, and biological processes that affect sedimentary OC records, including a variable fraction of OC being bound to clay minerals or variable amounts of non-bound OC being degraded53,54. Nonetheless, the fact that on average 19.2% of the total organic carbon remains bound to FeR below the oxygenated surface sediment layer still highlights the important role that this OC-FeR association plays in long-term carbon storage, despite the variance in environmental parameters over time. Moreover, as none of the presented iron metrics (fFeR, FeR, Fe/Al) shows a consistent connection with either OC-FeR or fOC-FeR across all study sites our findings, reveal that the fOC-FeR is not generally controlled by FeR availability and a substantial increase of terrigenous iron and FeR input does not necessarily have a direct effect on fOC-FeR. This raises the question of how much of the OC-FeR is allochthonous, i.e., formed in the water column, in sea ice, on land, and how much is autochthonous, i.e., formed by biogeochemical processes within the sediments.

The gradual downcore decrease in all three parameters may indicate that sedimentary OC degradation also affects the OC bound to FeR, even though fOC-FeR values are still relatively high in the lower part of these Arctic sediment cores. Other factors than diagenetic processes, such as environmental change, OC, and allochthonous OC-FeR input changes, probably also play a role during the time span investigated here. Note the different scale of the x-axis (sediment core depth) and y-axis (TOC) between each core.
Suspended and bed sediments from the river and glacial systems show a clear link between OC and FeR55, and substantial amounts of OC-FeR in marine sediments may originate from land19, especially at locations or during periods with high clastic sedimentation rates. The molar ratio of iron-bound OC to reactive iron (OC:Fe) has been interpreted as an indicator for the bonding mechanism between reactive iron and OC13, with low values (<1) indicating the major OC-FeR bonding form to be simple mono-layer sorption, while coprecipitation results in higher ratios52. As coprecipitation occurs at the oxic/anoxic interface in marine sediments, low OC:Fe ratios, for example, found in Mississippi river delta deposits were, therefore, interpreted as evidence for terrigenous OC-FeR contribution56. Hence, low OC:Fe ratios at stations B15 and B16 could be interpreted such that sorption of OC is the dominant form of OC-FeR association (Fig. 2). This suggests that at least part of the OC-FeR in these sediments was formed on land, during the transport process, or at the sediment-water interface before burial57. This assumption is further supported by the high fOC-FeR values (>10%) at the sediment surface at all four locations, above the Fe2+/Fe3+ redox boundary, where coprecipitation of OC with Fe(III) phases would occur. Such coprecipitation of OC and FeR at the Fe2+/Fe3+ redox boundary has been proposed as the dominant mechanism behind the formation of OC-FeR e.g. (refs. 13,17). Our comparison of iron pore water profiles, FeR contents, and fOC-FeR provides the first real indication that coprecipitation at the iron redox boundary in marine sediments is less important for OC-FeR bonding than previously assumed13,17 and other controlling factors seem to be more important for the sedimentary fOC-FeR contents.
Based on pore water data and the molar ratio of OC:Fe as a proxy for mono-layer sorption vs. coprecipitation, our data could indicate that at B14 and B15, coprecipitation may occur below the zone of FeR precipitation (Fig. 2) and while OC:Fe at station B14 increases with increasing fOC-FeR, e.g., due to coprecipitation or multi-layer sorption, fOC-FeR concentration at B15 decreases with increasing OC:Fe ratios. Even though OC:Fe and fOC-FeR show similar trends at station B16, an increase in OC:Fe at the Fe2+/Fe3+ redox boundary does not result in higher fOC-FeR. Sediments from stations B14 and B13 show much higher OC:Fe values compared to B15 and B16, and fOC-FeR and OC:Fe are clearly related in these two cores. It thus emerges from our data that the processes generating the OC-FeR coupling at the different study sites are not exclusively related to the coprecipitation of OC and FeR at the Fe2+/Fe3+ redox boundary. Several factors may be responsible for the different relationship between OC:Fe and fOC-FeR at stations B13/14 vs. B15/16. For example, experimental laboratory studies showed that the organic matter composition can influence the OC:Fe ratio regardless of the adsorption and coprecipitation bonding mechanism15,58,59. Moreover, as our sediment cores represent a time span of several thousands of years, microbial degradation might have, over time, selectively modified the adsorbed or coprecipitated OC-FeR content60,61. Additionally, the determination of FeR via chemical extraction yields an operationally defined reactive iron pool, including FeR that is not associated with OC. The molar ratio of OC:Fe might, therefore, be biased and especially low OC:Fe ratios, as in core B15 and B16, should be interpreted with care62. Finally, crystalline iron (oxyhydroxides) in natural sediments are impure, which influences the dynamics of the OC to FeR association. In a competition to OC, phosphate, arsenic, and other heavy metals have a strong affinity to iron (oxyhydroxide) surfaces and can, therefore, influence the OC:Fe ratio as well as fOC-FeR16. Arsenic contents in the Barents Sea surface sediments are strongly related to FeR contents (r = 0.9)48. Our downcore data show similar results with a clear relationship between As/Al, Fe/Al, and FeR, especially at the Fe2+/Fe3+ redox boundary (Supplementary Fig. S5). Hence, we speculate that the strong relationship between arsenic and FeR indicates that arsenic sorption could affect the mineral surface properties and reactivities of the Fe(III) phases and, therefore, their capacity to bind to OC16.
The role of reactive manganese in OC stabilisation
Manganese is another element that potentially interacts with OC in marine sediments and might have biased existing fOC-FeR estimates23,26,27. Besides Fe(III) phases, Mn(III/IV) (oxyhydroxides) (MnR) also strongly interact with OC in marine sediments25, but their effect on carbon stabilisation in natural sediments is almost completely unconstrained. In marine sediments, MnR is typically reductively dissolved below the depth of oxygen penetration14. Thus, the OC-MnR association might not be stable over longer timescales due to the strong effect of the oxic-anoxic redox interface on MnR stability27. Our data from the Barents Sea show that MnR is strongly related to total manganese contents and a rapid increase of pore water Mn2+ is accompanied by a decrease in MnR and total manganese close to the sediment surface. Below the top few cm of our cores, MnR is virtually absent at station B14 and very low at stations B13 (<38 ppm) and B16 (<55 ppm; Supplementary Table S3 and Fig. S6). Therefore, we propose that MnR in marine sediments is not important or plays only a very minor role, in the stabilisation of OC on longer timescales.
Synthesis and implications
Based on our investigation of the effect of sedimentary degradation processes on the formation and stability of the OC-FeR association in the Arctic marine sediments, we posit that an increased influx of iron or a higher fraction of FeR does not necessarily enhance fOC-FeR values and that iron redox cycling and associated authigenic FeR formation are less important for the stabilisation of OC in marine sediments than so far assumed, e.g. (refs. 17,23,46,57). We show that significant amounts (>10%) of fOC-FeR are present above the iron redox zone, before OC-FeR coprecipitation presumably occurs, which suggests that at least some of the OC-FeR binding forms on land, during transport to the seafloor, or at the sediment-water interface prior to deposition. Moreover, the association between OC and FeR via coprecipitation does not necessarily result in higher fOC-FeR and is, therefore, not the dominant factor controlling fOC-FeR in the Barents Sea sediments. Other factors such as organic matter composition15,58,59, inconsistent effects of different binding mechanisms on organic matter loadings60,61, and changes in Fe(III) phase reactivity due to incorporation and adsorption of other elements with a high affinity to FeR16, likely all play a (combined) role in natural environments.
A recent investigation of the Barents sea surface sediment samples found that the spatial distribution of the fOC-FeR content seems to be unrelated to sea ice cover, Atlantic water inflow, proximity to land, grain size distribution, or sediment composition48. Although more work is needed to elucidate the impact of climate and environmental changes on the fOC-FeR in marine sediments, the finding of this study could indicate that future Arctic warming might neither enhance nor decrease average carbon burial through the adsorption to iron oxides as, even though fOC-FeR profiles at all stations show some degree of variability, total fOC-FeR values averaged over all depths of all four sediment cores are surprisingly similar (B13: 18.1 ± 7.3%, B14: 17.7 ± 3.6%, B15: 22.5 ± 6.4%, B16: 17.9 ± 5.6% (mean ± s.d.)). These values are in agreement with published estimates for the Barents Sea (21.0 ± 8.3%) as well as global marine surface sediments (21.8 ± 8.6%)13,48. Thus, while highlighting new and important complexities in the coupling of OC with iron in marine sediments, our results clearly underline the importance of reactive iron phases for OC burial. In particular, we show that despite clear evidence for the iron reduction in the studied deposits, on average 19.2% of the total OC remains bound to FeR below the oxygenated surface sediment layer over thousands of years in the Arctic marine sediments. This shows that the rusty carbon sink is not disabled by diagenetic processes affecting both OC and Fe(III) phases and data from surface sediments can be used for meaningful estimates of OC-FeR burial rates.
Methods
Sediment cores: sampling and preparation
In July 2017, 2018, and 2019, sediment cores were collected by using a multi-corer at the same four stations (Supplementary Table S1) along a south-north gradient in the western Barents Sea (Fig. 1). One tube per multi-corer deployment at each station visited in 2017 was sliced at 0.5 cm intervals until 2 cm core depth, and at 1 cm intervals thereafter. Samples were stored in plastic bags at –20 °C immediately after recovery on-board the Royal Research Ship James Clark Ross. Prior to any chemical sediment analysis, all samples were freeze-dried and homogenised by gentle grinding using an agate mortar and pestle. For pore water analysis, four sediment cores were collected in all three years from three (two at B14 in 2017) sequential multi-corer deployments at each station (including the coring site for sediment sampling), with about 20–50 m distance between each deployment to account for spatial variability in sediment properties. Pore water was extracted with Rhizon samplers63 inserted into pre-drilled Perspex core liners (D = 110 mm). Following extraction, pore water samples from the same sediment depths from four core tubes per multi-corer deployment were combined into acid-washed and MilliQ-rinsed vials to reach maximum pore water volumes for individual sediment layers. Pore water splits of 3 mL for cation analysis were acidified with 10 µL concentrated ROMIL-UpA™ HCl and stored at 4 °C.
Sediment and pore water analysis
Sedimentary bulk iron, manganese, aluminium, and arsenic contents were determined by wavelength dispersive X-ray fluorescence (XRF; Supplementary Table S4). A sample split of 700 mg was mixed with 4200 mg di-lithiumtetraborate (Li2B4O7, Spectromelt A10), preoxidized at 500 °C with 1.0 g NH4NO3 (p.a.), and fused to homogenous glass beads. The glass beads were analysed using a Philips PW-2400 WD-XRF spectrometer calibrated with 53 geostandards at the University of Oldenburg. Analytical precision and accuracy were better than 5% as checked by in-house and international reference materials. Pore water concentrations of iron and manganese were determined by inductively coupled plasma optical emission spectrometry (Thermo Scientific iCAP 7400 Radial ICP-OES) at the University of Leeds. Analytical precision was ±3.5% and results were provided in the Supplementary Table S2.
OC, iron and manganese extraction, and analysis
To quantify the amount of OC bound to iron and manganese (oxyhydroxides) in our samples, we applied a method described in detail by Lalonde et al.13 and Salvadó et al.19. Briefly, 0.25 g of each sample was transferred into 30 ml centrifuge tubes. Fifteen millilitres of a solution containing 0.27 M trisodium citrate (Na3C6H5O7·H2O) and 0.11 M sodium bicarbonate (NaHCO3) was added, mixed and heated up to 80 °C in a water bath. 0.1 M sodium dithionite (Na2S2O4; 0.25 g) was added to the mixture, the temperature was maintained at 80 °C, and the tube was shaken every five minutes. After 15 min, the mixture was centrifuged for 10 min at 3360 g, the supernatant was decanted, and 200 μl of HCl were added to prevent Fe(III) precipitation. The remaining sediment samples were rinsed three times with artificial seawater and then freeze-dried. To quantify the OC loss unrelated to metal oxide dissolution, a control experiment was conducted: A 0.25 g aliquot of each sample was treated the same way as for the reduction experiment, but the complexing and reducing agents (sodium citrate and sodium dithionite) were replaced with sodium chloride to reach a solution of the same ionic strength. All samples were weighed after the experiment to account for mass loss. Dissolved iron and manganese in the supernatant and rinse water of the control and reduction experiments were analysed using a Thermo Scientific iCE3000 Atomic Absorption Spectrometer (AAS) at the University of Leeds, UK. Results are shown in the Supplementary Table S3 and the relative error of the analysis, based on eleven sample replicates, was ±3.2% for iron and 9.9% for manganese. Unlike iron, the dithionite extraction method is not well established for manganese. As manganese concentrations in all control experiments were below the detection limit, we assume that dithionite extracted manganese represents only reactive manganese phases64.
We also performed a three-step sequential iron extraction on a subset of sediment samples (n = 18) following the procedure of Poulton and Canfield65, and März et al.66. The extracted iron fractions were operationally defined but usually constitute Fe bound to carbonates or very labile iron (oxyhydroxides) (FeOx-1), poorly crystalline Fe (oxyhydroxides) like ferrihydrite (FeOx-2) and crystalline Fe oxides such as haematite and goethite (FeOx-3). FeOx-1 was extracted using 1 M Na-acetate solution (pH 4.5) for 24 h; for FeOx-2 a solution comprised of 50 g Na-citrate, 50 g Na-bicarbonate, and 20 g ascorbic acid (per litre) at pH 8 for 24 h was used; Fe-S3 was extracted in a solution of 0.2 M Na-citrate and 0.28 M Na-dithionite buffered to pH 4.8 with 0.35 M acetic acid for 2 h. To ensure reproducibility, one sample was extracted in triplicate at all three stages, and the relative error was <2.9%. The iron fractions FeOx-1-3 were analysed using a Thermo Scientific iCE3000 Atomic Absorption Spectrometer (AAS) at Leeds University, UK.
The OC content of the bulk sediment before and after the reduction and control experiments was analysed on decarbonated samples using 10% (vol.) HCl, rinsed three times and dried overnight at 50 °C. OC content was determined with a LECO SC-144DR combustion analyser at the University of Leeds, UK (Supplementary Table S3). The certified reference material LECO 502-062 and blanks were included in every batch, and results are given in weight percentage. The relative error of the OC analysis was ±1.7%. To account for the mass loss during the extraction experiment we applied the mass balance calculation of Salvadó et al.19 (Supplementary information). Note that, liquid-HCl decarbonation of the bulk sediment samples may also dissolve reactive iron phases in addition to carbonates. This could potentially liberate some iron-bound OC, which would bias our bulk OC results to lower values and thus bias our OC-FeR results upwards (Supplementary information).
Chronology
Nine radiocarbon ages (Fig. 2) were obtained from benthic foraminifera using a Mini Carbon Dating System (MICADAS) at the Bristol Radiocarbon Accelerator Mass Spectrometry facility. Uncalibrated carbon-14 ages are shown in the Supplementary Table S3.
Data availability
The authors declare that all data supporting the findings of this study are available within the Supplementary Data.
References
Smith, R. W., Bianchi, T. S., Allison, M., Savage, C. & Galy, V. High rates of organic carbon burial in fjord sediments globally. Nat. Geosci. 8, 450–453 (2015).
Müller, P. J. & Suess, E. Productivity, sedimentation rate, and sedimentary organic matter in the oceans - I. Organic carbon preservation. Deep Sea Res. Part a Oceanographic Res. Pap. 26, 1347–1362 (1979).
Ingall, E. D. & Vancappellen, P. Relation between sedimentation-rate and burial of organic phosphorus and organic-carbon in marine-sediments. Geochimica Et. Cosmochimica Acta. 54, 373–386 (1990).
Canfield, D. E. Factors influencing organic carbon preservation in marine sediments. Chem. Geol. 114, 315–329 (1994).
Pedersen, T. & Calvert, S. E. Anoxia vs. Productivity: what controls the formation of organic-carbon-rich sediments and sedimentary rocks? AAPG Bull. 74, 454–466 (1990).
Hartnett, H. E., Keil, R. G., Hedges, J. I. & Devol, A. H. Influence of oxygen exposure time on organic carbon preservation in continental margin sediments. Nature 391, 572–574 (1998).
Burdige, D. J. Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem. Rev. 107, 467–485 (2007).
Hatcher, P. G., Spiker, E. C., Szeverenyi, N. M. & Maciel, G. E. Selective preservation and origin of petroleum-forming aquatic kerogen. Nature 305, 498–501 (1983).
Hedges, J. I. & Keil, R. G. Sedimentary organic matter preservation: an assessment and speculative synthesis. Mar. Chem. 49, 81–115 (1995).
Mayer, L. M. Relationships between mineral surfaces and organic-carbon concentrations in soils and sediments. Chem. Geol. 114, 347–363 (1994).
Hemingway, J. D. et al. Mineral protection regulates long-term global preservation of natural organic carbon. Nature 570, 228–231 (2019).
Beutelspacher, H. Wechselwirkung zwischen anorganischen und organischen Kolloiden des Bodens. Z. f.ür. Pflanzenernährung, D.üngung, Bodenkd. 69, 108–115 (1955).
Lalonde, K., Mucci, A., Ouellet, A. & Gelinas, Y. Preservation of organic matter in sediments promoted by iron. Nature 483, 198–200 (2012).
Froelich, P. N. et al. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochimica et. Cosmochimica Acta. 43, 1075–1090 (1978).
Chen, C., Dynes, J. J., Wang, J. & Sparks, D. L. Properties of Fe-organic matter associations via coprecipitation versus adsorption. Environ. Sci. Technol. 48, 13751–13759 (2014).
Chen, C. & Sparks, D. L. Fe(II)-induced mineral transformation of ferrihydrite–organic matter adsorption and coprecipitation complexes in the absence and presence of As(III). ACS Earth Space Chem. 2, 1095–1101 (2018).
Riedel, T., Zak, D., Biester, H. & Dittmar, T. Iron traps terrestrially derived dissolved organic matter at redox interfaces. Proc. Natl Acad. Sci. USA 110, 10101–10105 (2013).
Henrichs, S. M. Early diagenesis of organic matter in marine sediments: progress and perplexity. Mar. Chem. 39, 119–149 (1992).
Salvadó, J. A. et al. Organic carbon remobilized from thawing permafrost is resequestered by reactive iron on the Eurasian Arctic Shelf. Geophys. Res. Lett. 42, 8122–8130 (2015).
Zhao, B. et al. The role of reactive iron in the preservation of terrestrial organic carbon in estuarine sediments. J. Geophys. Res.: Biogeosciences 123, 3556–3569 (2018).
Wang, D., Zhu, M. X., Yang, G. P. & Ma, W. W. Reactive iron and iron‐bound organic carbon in surface sediments of the river‐dominated Bohai Sea (China) Versus the Southern Yellow Sea. J. Geophys. Res.: Biogeosciences 124, 79–98 (2019).
Faust, J. C. et al. Does Arctic warming reduce preservation of organic matter in Barents Sea sediments? Philos. Trans. A Math. Phys. Eng. Sci. 378, 20190364 (2020).
Ma, W.-W., Zhu, M.-X., Yang, G.-P. & Li, T. Iron geochemistry and organic carbon preservation by iron (oxyhydr)oxides in surface sediments of the East China Sea and the South Yellow Sea. J. Mar. Syst. 178, 62–74 (2018).
Lovley, D. R. Dissimilatory Fe(III) and Mn(IV) reduction. Microbiological Rev. 55, 259–287 (1991).
Roy, M. et al. Reactive iron and manganese distributions in seabed sediments near small mountainous rivers off Oregon and California (USA). Continental Shelf Res. 54, 67–79 (2013).
Estes, E. R., Andeer, P. F., Nordlund, D., Wankel, S. D. & Hansel, C. M. Biogenic manganese oxides as reservoirs of organic carbon and proteins in terrestrial and marine environments. Geobiology 15, 158–172 (2017).
Johnson, K. et al. Towards a mechanistic understanding of carbon stabilization in manganese oxides. Nat. Commun. 6, 7628 (2015).
Nickel, M., Vandieken, V., Brüchert, V. & Jørgensen, B. B. Microbial Mn(IV) and Fe(III) reduction in northern Barents Sea sediments under different conditions of ice cover and organic carbon deposition. Deep Sea Res. Part II: Topical Stud. Oceanogr. 55, 2390–2398 (2008).
Vandieken, V., Nickel, M. & Jorgensen, B. B. Carbon mineralization in Arctic sediments northeast of Svalbard: Mn(IV) and Fe(III) reduction as principal anaerobic respiratory pathways. Mar. Ecol. Prog. Ser. 322, 15–27 (2006).
Screen, J. A. & Simmonds, I. Increasing fall-winter energy loss from the Arctic Ocean and its role in Arctic temperature amplification. Geophys. Res. Lett. 37, L16707 (2010).
Lewis, K. M., van Dijken, G. L. & Arrigo, K. R. Changes in phytoplankton concentration now drive increased Arctic Ocean primary production. Science 369, 198–202 (2020).
Post, E. et al. Ecological consequences of sea-ice decline. Science 341, 519–524 (2013).
Piepenburg, D. Recent research on Arctic benthos: common notions need to be revised. Polar Biol. 28, 733–755 (2005).
Dalpadado, P. et al. Productivity in the barents sea–response to recent climate variability. PLoS ONE 9, e95273 (2014).
Wassmann, P. Arctic marine ecosystems in an era of rapid climate change. Prog. Oceanogr. 90, 1–17 (2011).
Wassmann, P., Carroll, J. & Bellerby, R. G. J. Carbon flux and ecosystem feedback in the northern Barents Sea in an era of climate change: an introduction. Deep Sea Res. Part II: Topical Stud. Oceanogr. 55, 2143–2153 (2008).
Arrigo, K. R. & van Dijken, G. L. Secular trends in Arctic Ocean net primary production. J. Geophys. Res. 116, C09011 (2011).
Sun, C.-H. et al. Examining bulk and iron-associated organic carbon through depth in margin sea sediments (China) under contrasting depositional settings: chemical and NEXAFS spectral characterization. J. Mar. Syst. 207, 103344 (2020).
Freitas, F. S. et al. Benthic-pelagic coupling in the Barents Sea: an integrated data-model framework. Philos. Trans. A Math. Phys. Eng. Sci. 378, 20190359 (2020).
Solan, M. et al. Climate-driven benthic invertebrate activity and biogeochemical functioning across the Barents Sea polar front. Philos. Trans. A Math. Phys. Eng. Sci. 378, 20190365 (2020).
Rasmussen, T. L. & Thomsen, E. Pink marine sediments reveal rapid ice melt and Arctic meltwater discharge during Dansgaard–Oeschger warmings. Nat. Commun. 4, 2849 (2013).
Dowdeswell, J. A. et al. Sediment-rich meltwater plumes and ice-proximal fans at the margins of modern and ancient tidewater glaciers: observations and modelling. Sedimentology 62, 1665–1692 (2015).
Loeng, H. Features of the physical oceanographic conditions of the Barents Sea. Polar Res. 10, 5–18 (1991).
Harris, C. L., Plueddemann, A. J. & Gawarkiewicz, G. G. Water mass distribution and polar front structure in the western Barents Sea. J. Geophys. Res.: Oceans 103, 2905–2917 (1998).
Pfirman, S. L. & Solheim, A. Subglacial meltwater discharge in the open-marine tidewater glacier environment—observations from Nordaustlandet, Svalbard Archipelago. Mar. Geol. 86, 265–281 (1989).
Linkhorst, A., Dittmar, T. & Waska, H. Molecular fractionation of dissolved organic matter in a shallow subterranean estuary: the role of the iron curtain. Environ. Sci. Technol. 51, 1312–1320 (2017).
Sirois, M., Couturier, M., Barber, A., Gélinas, Y. & Chaillou, G. Interactions between iron and organic carbon in a sandy beach subterranean estuary. Mar. Chem. 202, 86–96 (2018).
Faust, J. C. et al. Does Arctic warming reduce preservation of organic matter in Barents Sea sediments? Philos. Trans. A Math. Phys. Eng. Sci. 378, 20190364 (2020).
Cornell, R. M. & Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses (John Wiley & Sons, 2003).
Jelavić, S., Mitchell, A. C. & Sand, K. K. Fate of organic compounds during transformation of ferrihydrite in iron formations. Geochem. Perspect. Lett. 15, 25–29 (2020).
van der Zee, C., Roberts, D. R., Rancourt, D. G. & Slomp, C. P. Nanogoethite is the dominant reactive oxyhydroxide phase in lake and marine sediments. Geology 31, 993–996 (2003).
Wagai, R. & Mayer, L. M. Sorptive stabilization of organic matter in soils by hydrous iron oxides. Geochimica et. Cosmochimica Acta. 71, 25–35 (2007).
Arndt, S. et al. Quantifying the degradation of organic matter in marine sediments: a review and synthesis. Earth-Sci. Rev. 123, 53–86 (2013).
LaRowe, D. E. et al. The fate of organic carbon in marine sediments—New insights from recent data and analysis. Earth-Sci. Rev. 204, 103146 (2020).
Poulton, S. W. The low-temperature geochemical cycle of iron: from continental fluxes to marine sediment deposition. Am. J. Sci. 302, 774–805 (2002).
Shields, M. R., Bianchi, T. S., Gélinas, Y., Allison, M. A. & Twilley, R. R. Enhanced terrestrial carbon preservation promoted by reactive iron in deltaic sediments. Geophys. Res. Lett. 43, 1149–1157 (2016).
Barber, A., Lalonde, K., Mucci, A. & Gélinas, Y. The role of iron in the diagenesis of organic carbon and nitrogen in sediments: a long-term incubation experiment. Mar. Chem. 162, 1–9 (2014).
Mikutta, C. X-ray absorption spectroscopy study on the effect of hydroxybenzoic acids on the formation and structure of ferrihydrite. Geochimica et. Cosmochimica Acta. 75, 5122–5139 (2011).
Eusterhues, K. et al. Fractionation of organic matter due to reaction with ferrihydrite: coprecipitation versus adsorption. Environ. Sci. Technol. 45, 527–533 (2011).
Eusterhues, K. et al. Reduction of ferrihydrite with adsorbed and coprecipitated organic matter: microbial reduction by Geobacter bremensis vs. abiotic reduction by Na-dithionite. Biogeosciences 11, 4953–4966 (2014).
Cooper, R. E., Eusterhues, K., Wegner, C.-E., Totsche, K. U. & Küsel, K. Ferrihydrite-associated organic matter (OM) stimulates reduction by Shewanella oneidensis MR-1 and a complex microbial consortia. Biogeosciences 14, 5171–5188 (2017).
Barber, A. et al. Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron. Sci. Rep. 7, 366 (2017).
Seeberg-Elverfeldt, J., Schlüter, M., Feseker, T. & Kölling, M. Rhizon sampling of porewaters near the sediment-water interface of aquatic systems. Limnol. Oceanogr.: Methods 3, 361–371 (2005).
McManus, J. et al. Benthic manganese fluxes along the Oregon–California continental shelf and slope. Continental Shelf Res. 43, 71–85 (2012).
Poulton, S. W. & Canfield, D. E. Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221 (2005).
März, C., Poulton, S. W., Brumsack, H. J. & Wagner, T. Climate-controlled variability of iron deposition in the Central Arctic Ocean (southern Mendeleev Ridge) over the last 130,000 years. Chem. Geol. 330–331, 116–126 (2012).
Carmack, E. et al. Climate variability and physical forcing of the food webs and the carbon budget on panarctic shelves. Prog. Oceanogr. 71, 145–181 (2006).
Wassmann, P. et al. Food webs and carbon flux in the Barents Sea. Prog. Oceanogr. 71, 232–287 (2006).
Smedsrud, L. H. et al. The role of the Barents Sea in the Arctic climate system. Rev. Geophysics. 51, 415–449 (2013).
Jørgensen, L. L. et al. Distribution of benthic megafauna in the Barents Sea: baseline for an ecosystem approach to management. ICES J. Mar. Sci. 72, 595–613 (2015).
Loeng, H., Ozhigin, V. & Adlandsvik, B. Water fluxes through the Barents Sea. Ices J. Mar. Sci. 54, 310–317 (1997).
Jakobsen, T. & Ozhigin, V. K. The Barents Sea—Ecosystem, Resources, Management. Half a Century of Russian—Norwegian Cooperation (Tapir Akademisk Forlag, 2011).
Drinkwater, K. F. The influence of climate variability and change on the ecosystems of the Barents Sea and adjacent waters: review and synthesis of recent studies from the NESSAS Project. Prog. Oceanogr. 90, 47–61 (2011).
Jakobsson, M. et al. The International Bathymetric Chart of the Arctic Ocean (IBCAO) Version 3.0. Geophys. Res. Lett. 39, L12609 (2012).
Poulton, S. W. & Canfield, D. E. Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221 (2005).
Acknowledgements
We thank the crew of the RRS James Clark Ross for their professional support during our expedition. Further, we would like to express our gratitude to Andy Connelly, Andrew Hobson, Fiona Keay, Carola Lehners, Corinna Mori, and Bernhard Schnetger for their help with the laboratory work at the University of Leeds and at the ICBM Oldenburg, as well as Heather Birch and Timothy Knowles for training in foraminiferal taxonomy and MICADAS analyses at the University of Bristol. This work resulted from the ChAOS project (NE/P006493/1), part of the Changing Arctic Ocean programme, jointly funded by the UKRI Natural Environment Research Council (NERC) and the German Federal Ministry of Education and Research (BMBF).
Author information
Authors and Affiliations
Contributions
J.C.F. was the lead author, wrote the manuscript, and created all figures. J.C.F., M.Z., A.T., K.D., and C.M. conducted fieldwork/sampling together and compiled datasets. J.C.F., S. P., B.F., and K.H. conducted all the required analytical work. All authors contributed early ideas, revised the initial manuscript and provided a lively discussion.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Thomas Blattmann, Jordon Hemingway, and Michael Shields for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Faust, J.C., Tessin, A., Fisher, B.J. et al. Millennial scale persistence of organic carbon bound to iron in Arctic marine sediments. Nat Commun 12, 275 (2021). https://doi.org/10.1038/s41467-020-20550-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-20550-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
