
Abstract
Catalytic asymmetric conjugate allylation of unsaturated carbonyl compounds is usually difficult to achieve, as 1,2-addition proceeds dominantly and high asymmetric induction is a challenging task. Herein, we disclose a copper(I)-NHC complex catalyzed asymmetric 1,6-conjugate allylation of 2,2-dimethyl-6-alkenyl-4H-1,3-dioxin-4-ones. The phenolic hydroxyl group in NHC ligands is found to be pivotal to obtain the desired products. Both aryl group and alkyl group at δ-position are well tolerated with the corresponding products generated in moderate to high yields and high enantioselectivity. Moreover, both 2-substituted and 3-substituted allylboronates serve as acceptable allylation reagents. At last, the synthetic utility of the products is demonstrated in several transformations by means of the versatile terminal olefin and dioxinone groups.
Similar content being viewed by others

Introduction
Catalytic asymmetric conjugate addition of various metal reagents to unsaturated compounds is identified as one of the most important tools in the construction of carbon-carbon bonds in organic synthesis1,2,3. Among the various carbon-based metal reagents, allyl metal reagents exhibit advantages over other alkyl metal reagents as the olefin moiety is more synthetically versatile. Non-enantioselective methods based on different allyl metal (such as, Si, B, Zn, and Sn) species have been disclosed in the past several decades4. However, the catalytic asymmetric conjugate addition with allyl metal reagents is still in its infancy as such a reaction is not easy to achieve due to the competitive 1,2-addition and the difficulty in the asymmetric induction.
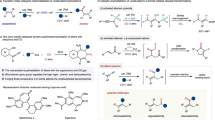
Indeed, catalytic asymmetric allylation of aldehyde, ketone, and imine receives significant research efforts from the chemical community, which leads to the identification of efficient catalytic systems based on Cu5,6,7,8,9,10,11,12,13,14,15,16, Zn17,18, Ag19,20,21,22, and In23,24. The proposed six-membered ring transition state formed by the coordination of the allyl metal species to carbonyl group allows excellent asymmetric induction. Especially, copper(I)-catalysts serve as powerful weapons to enable the highly enantioselective allylation of carbonyl groups and imines5,6,7,8,9,10,11,12,13,14,15,16. However, the affinity of the highly nucleophilic allylcopper(I) species to carbonyl group set up an obstacle on the conjugate allylation. For example, in the presence of 10 mol % copper(I)-(R)-BINAP, 2 equiv allylboronate, and 1 equiv LiOtBu, the allylation of α,β-unsaturated ester produced tertiary alcohol only and the 1,4-conjugate allylation product was not obtained (Fig. 1a). Moreover, without the assistance of the six-membered ring transition state, there is a concern about the enantioselectivity in the conjugate allylation.

a Failed copper(I)-catalyzed asymmetric conjugate allylation. b Reported catalytic asymmetric conjugate allylation. c This work: copper(I)-catalyzed asymmetric conjugate 1,6-allylation.
Several research group made their contributions in the challenging catalytic asymmetric conjugate allylation (Fig. 1b)25,26,27,28,29,30,31,32,33. Snapper reported a Cu(II)-BOX-catalyzed asymmetric conjugate allylation of unsaturated cyclic β-keto-esters with allylsilane25. In 2007, Morken and co-workers disclosed an impressive Ni-catalyzed regioselective conjugate allylation of α,β-α′,β′-di-unsaturated ketones26. Later, the Morken group uncovered two efficient methods to carry out the catalytic asymmetric version in excellent control of the regioselectivity with either a palladium catalyst or a nickel catalyst27,28. The Hoveyda group achieved a three-component reaction of 1,3-butadiene, B2Pin2 and alkylidenemalonates in high yields with excellent enantioselectivity29. However, aliphatic substituents were not well tolerated at the β-position. The same group also succeeded in the catalytic enantioselective 1,6-conjugate allylation of α,β,γ,δ-unsaturated diesters with B2Pin2 and allenes30,31. In 2011, the Feng group achieved an asymmetric conjugate allylation of activated unsaturated lactones with a bimetallic catalytic system32. In 2019, the same group reported a formal catalytic asymmetric 1,4-allylation of β,γ-unsaturated α-ketoesters4. In fact, the formal conjugate allylation was enabled by the allylation of the ketone group and the following oxy-Cope rearrangement. Unfortunately, alkyl was not well tolerated at the γ-position as only moderate enantioselectivity was observed. Moreover, Shibasaki and Kumagai uncovered a catalytic asymmetric conjugate allylation of α,β-unsaturated thioamides with allyl cyanide under proton-transfer conditions33. In view of the above achievements, we are interested in developing a catalytic asymmetric conjugate 1,6-allylation with more general substrate structure and broader substrate scope.
Copper(I)-catalyzed asymmetric 1,6-addition with alkyl metal reagents (such as organozinc reagent and Grignard reagent) has been reported as a powerful tool to regio-selectively construct carbon-carbon bonds34,35,36,37,38,39,40,41,42,43,44,45,46. Herein, we disclose an asymmetric 1,6-conjugate allylation of 2,2-dimethyl-6-alkenyl-4H-1,3-dioxin-4-one with a copper(I)-NHC catalyst (Fig. 1c). The 2,2-dimethyl-4H-1,3-dioxin-4-one moiety is an equivalent of the synthetically versatile β-keto-ester group and the product containing both an allyl group and a dioxinone group allows further structure elaboration. Furthermore, in view of the bulky steric hindrance around the carbonyl group and the relative stability of the lactone moiety, it is envisioned that the highly nucleophilic allylcopper(I) species would not touch the carbonyl group in the dioxinone and thus would attack the less hindered conjugate carbon-carbon double bond to give the desired 1,6-allylation.
Results
Conditions optimization
First of all, the catalytic asymmetric conjugate allylation of (E)-2,2-dimethyl-6-(4-phenylbut-1-en-1-yl)-4H-1,3-dioxin-4-one (1a) with bench-stable allylboronate 2 was studied in the presence of 5 mol % Cu(CH3CN)4PF6, 6 mol % (R)-BINAP, and 1 equiv LiOtBu (Table 1, entry 1). The conjugate allylation proceeded smoothly to afford product 3a in 25% yield with 9% ee. Then, screening of commercially available bisphosphine ligands was performed and proved to be fruitless (entries 2-6). Especially, (R,R)-Ph-BPE, the previously reported best ligand for the copper(I)-catalyzed allylation of ketones9,11,13, only led to 38% ee (entry 3). Moreover, ferrocene-embedded bisphosphine ligands, such as (R,RP)-TANIAPHOS and (R)-(S)-JOSIPHOS, were not effective either (entries 5-6). Obviously, copper(I)-bisphosphine catalyst did not suit this conjugate 1,6-allylation.
Then, we turned our attention to NHC ligands (Table 1). Five NHC ligands were synthesized according to literature methods47,48,49. NHC-L1 was completely ineffective to get asymmetric induction in this 1,6-conjugate allylation (entry 7). A small but promising 8% ee was obtained in the reaction with NHC-L2 (entry 8). Since phenol-containing NHCs (including NHC-L3-L5) were identified as powerful ligands in some copper(I)-catalyzed enantioselective reactions by the Sawamura Group49,50,51,52, NHC-L3 was tried in our reaction, which provided 3a in 10% yield with 64% ee (entry 9). To our joy, 90% ee was observed with NHC-L4 (entry 10). However, decreased enantioselectivity (82% ee) was obtained in the reaction with bulkier NHC-L5 (entry 11). The yield was increased to 50% by using 10 mol % copper(I) salt and 12 mol % NHC-L4 (entry 12). By increasing the amount of LiOtBu from 1 equiv to 2 equiv, the yield was further increased to 85% with 89% ee (entry 13). Performing the reaction at −20 °C resulted in increased enantioselectivity (95% ee) but with decreased yield (53%) (entry 14). The yield was enhanced to 84% yield with 94% ee at −20 °C by using 3 equiv allylboronate and 3 equiv LiOtBu (entry 15).
Substrate scope
With the optimized reaction conditions in hand, the substrate scope of (E)-2,2-dimethyl-6-alkyl-4H-1,3-dioxin-4-ones was studied (Fig. 2). Linear alkyls, including ethyl (3b), npropyl (3c), and nheptyl (3d), were well tolerated and the corresponding products were isolated in good yields with high enantioselectivity. β-Branched alkyl (ibutyl) (3e) was also accepted at the δ-position. The substrates bearing a α-branched alkyl with bigger steric hindrance (3f and 3g), afforded the allylated products in moderate yields and slightly decreased enantioselectivity. Then, substrates with an alkyl containing a functional group, such as benzyl (3a), terminal alkene (3h), internal alkyne (3i), alkyl chloride (3j), ester (3k), TBS-ether (3l), and N-Boc (3n) were examined. To our joy, the products were obtained in moderate to high yields and high enantioselectivity. Notably, alkyl chloride and ester group were not touched by the nucleophilic allylcopper-NHC species, demonstrating that allylcopper-NHC species was less nucleophilic than allylcopper-bisphosphine species. Unfortunately, the substrate containing a free alcohol (3m) was not tolerated. A substrate with a preexisting chiral center (3o) was also studied. The allylated product was generated in 72% yield with 91% de, indicating that the asymmetric induction was mainly controlled by the copper(I) catalyst. It should be noted that in some cases, the reaction temperature was increased to get good yields.

Various aliphatic substituents at δ-position are studied.
The reaction conditions were applied to the catalytic asymmetric allylation of (E)-2,2-dimethyl-6-aryl-4H-1,3-dioxin-4-ones with 4 equiv allylboronate (2) as 3 equiv 2 generally resulted in inferior yields (Fig. 3). The reaction was not very sensitive to the position of a substituent on the phenyl ring. As the allylated products containing a para-substituent, ortho-substituent, or meta-substituent were isolated in moderate to high yields with uniformly high enantioselectivity (5a–5o). It was noted that substrates with electron-withdrawing groups led to lower yields but with maintained enantioselectivity (5d–5f, 5 m, and 5o). Moreover, substrates with electron-donating groups served as competent substrates as the corresponding products were furnished in good yields with high enantioselectivity (5b–5c, 5h–5i, and 5k–5l).

Various aromatic substituents at δ-position are studied.
The phenyl group at δ-position was successfully changed to 2-naphthyl group without affecting both yield and enantioselectivity significantly (5p). Moreover, several heteroaryl groups, including 3-pyridyl (5q), 2-furanyl (5r), 2-benzofuranyl (5s), 3-benzothienyl (5t), and 3-N-Boc-indolyl (5u), were successfully tolerated at the δ-position. The corresponding products were furnished in moderate yields with uniformly high enantioselectivity. It should be pointed out that the reaction temperature varied in order to get good yields. The absolute configuration of 5a was determined to be S by its transformation to a known compound (for the details, see SI). The absolute configurations of other products (3 and 5) were deduced by analogy.
Then, the 1,6-conjugate allylation with 2-substituted allylboronates (6–8) was investigated as shown in Fig. 4. Several aryl groups, including phenyl, 2-F-phenyl, and 3-methyl-phenyl, were well tolerated at the δ-position in the reaction with 6. The corresponding products (9a–9c) were obtained in 57%-63% yield with 93%-97% ee. An alkyl group, such as 2-phenyl-ethyl, was also acceptable at the δ-position (9d). 2-Methyl group in allylboronate 6 was successfully extended to 2-benzyl and nhexyl without eroding both yields and enantioselectivity (10–11). Moreover, the reactions of both 3-methyl-(E)-allylboronate (12a) and 3-methyl-(Z)-allylboronate (12b) were studied as shown in Fig. 5. The diastereoselective allylation of 4j and 12a proceeded smoothly to afford 13 in moderate yield with moderate diastereoselectivity and excellent enantioselectivity. Surprisingly, the reaction with 12b also furnished 13 as the product in decreased yield and slightly decreased enantioselectivity. At this stage, it is difficult to understand such experimental results. However, it is speculated that the addition of the (Z)-allylcopper(I) is kinetically unfavored and the isomerization of (Z)-allylcopper(I) species to (E)-allylcopper(I) might occur through 1,3-translocation in the present reaction conditions53. The absolute configurations of 9, 10, and 11 were deduced analogically based on the stereochemical structure of 5a. Moreover, the absolute configurations of the two stereogenic carbon centers in 13 were determined by its transformation (For the details, see SI). In addition, the present catalytic system was extended to the asymmetric additions with PhMgBr and EtMgBr. However, only racemic products were obtained54.

Both aromatic and aliphatic substituents at δ-position are tried.

a The 1.6-conjugate allylation with (E)-allylBPin. b The 1,6-conjugate allylation with (Z)-allylBPin.
Demonstration of the importance of the phenol group in NHC ligands
Several ligand variants of NHC-L4 were prepared to investigate the importance of the naphthol group (Fig. 6). The allylation with NHC-L6 containing a protected naphthol group did not afford the product 3a at all. Moreover, the reaction using NHC-L7 without the naphthol moiety was fruitless. Interestingly, NHC-L8 bearing a naphthol group and a protected naphthol group was found as a good ligand as product 3a was generated in 32% yield with 85% ee. These control experiments demonstrate that a free naphthol moiety is indispensable for this reaction to proceed. Furthermore, the steric hindrances on the both aryls are responsible for the asymmetric induction. Our finding of the essentiality of the free phenol or naphthol in this type of NHC ligands in asymmetric catalysis with copper(I) is in accordance with Sawamura’s original findings49,50,51,52.

The importance of the presence of a phenol group in NHC-ligands is demonstrated.
Transformation
At last, transformations of 5d were performed as described in Fig. 7. An Ir-catalyzed hydroborylation of terminal olefin moiety in 5d afforded boronate 14 in 62% yield55. The olefin-metathesis between 5d and 4-methylstyrene with 10 mol % Hoveyda-Grubbs catalyst 2nd generation produced (E)-olefin 15 in 71% yield at 40 °C (>20/1 (E) form/(Z) form ratio). Removal of the propylidene group in 5d was accomplished in MeOH to give β-keto-ester 16 in 75% yield. Then, a synthetic sequence, including the reduction of ketone unit, the formation of a mesylate, and the subsequent elimination furnished α,β-unsaturated ester 17 in 60% total yield. It should be noted that 16 serves as a formal 1,4-conjugate allylation product of α,β-unsaturated ketone and 17 serves as a formal 1,6-conjugate allylation product of α,β,γ,δ-unsaturated ester, which are difficult to access by known methods. Moreover, the preparation of pyrazole 18 was achieved by means of the β-keto-ester motif through a reported procedure56.

a Ir-catalyzed hydroboration. [Ir(COD)Cl]2 (5 mol %), dppm (Ph2PCH2PPh2) (10 mol %), HBPin (2 equiv), CH2Cl2, rt. b Olefin Metathesis. Hoveyda-Grubbs II catalyst (10 mol %), 4-methylstyrene (2 equiv), CH2Cl2, 40 °C. c Transformation to β-keto-ester. MeOH, K2CO3, rt. d Transformation to α,β-unsaturated ester. (1) NaBH4, MeOH, 0 °C; (2) MsCl, Et3N, CH2Cl2, 0 °C to rt. e Transformation to pyrazole. NH2NH2•HCl (2 equiv), MeOH, reflux.
Discussion
In summary, a catalytic asymmetric 1,6-conjugate allylation was achieved in moderate to high yields with high enantioselectivity. NHC ligands containing a phenolic hydroxyl group were found to be indispensable to enable this reaction. Both 2,2-dimethyl-6-alkenyl-4H-1,3-dioxin-4-one and allylboronate enjoyed broad substrate scopes. Several functional groups, especially alkyl halide and ester, were well tolerated in this reaction. The allyl group in the product allowed facile both hydroboration and olefin metathesis to give synthetically useful products. Moreover, the versatile dioxinone group was easily transformed to β-keto-ester moiety and α,β-unsaturated ester moiety, which generated a formal 1,4-conjugate allylation product of α,β-unsaturated ketone and a formal 1,6-conjugate allylation product of α,β,γ,δ-unsaturated ester. Detailed investigations of the mechanism are currently undertaken in our laboratory.
Methods
A general procedure for the catalytic asymmetric 1,6-conjugate allylation
A dried 25 ml Schlenk tube equipped with a magnetic stirring bar was charged with CuPF6(CH3CN)4 (3.7 mg, 0.01 mmol, 0.1 equiv), NHC-L4 (6.2 mg, 0.012 mmol, 0.12 equiv) and LiOtBu (24.0 mg, 0.3 mmol, 3 equiv) in a glove box under Ar atmosphere. Anhydrous THF (1 ml, 0.1 M) was added to the tube via a syringe. The resulting mixture was stirred under room temperature for 7 min. Then 1 (0.1 mmol, 1 equiv) was added to the reaction mixture. It was cooled down to the stated temperature before adding 2 (50.4 mg, 0.3 mmol, 3 equiv) by a syringe. This mixture was stirred for 12–36 h at that temperature. Then the reaction was quenched by adding silica gel and the mixture was purified by flash silica gel column chromatography to give product 3.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information file. Any further relevant data are available from the authors on request.
References
Alexakis, A., Bäckvall, J. E., Krause, N., Pàmies, O. & Diéguez, M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev. 108, 2796–2823 (2008).
Jerphagnon, T., Pizzuti, M. G., Minnaard, A. J. & Feringa, B. L. Recent advances in enantioselective copper-catalyzed 1,4-addition. Chem. Soc. Rev. 38, 1039–1075 (2009).
Thaler, T. & Knochel, P. Copper-catalyzed asymmetric Michael addition of magnesium, zinc, and aluminum organometallic reagents: efficient synthesis of chiral molecules. Angew. Chem. Int. Ed. 48, 645–648 (2009).
Tang, Q. et al. Asymmetric catalytic formal 1,4-allylation of β,γ-unsaturated α-ketoesters: allylboration/oxy-Cope rearrangement. Angew. Chem. Int. Ed. 58, 11846–11851 (2019).
Wada, R., Oisaki, K., Kanai, M. & Shibasaki, M. Catalytic enantioselective allylboration of ketones. J. Am. Chem. Soc. 126, 8910–8911 (2004).
Wada, R. et al. Catalytic enantioselective allylation of ketoimines. J. Am. Chem. Soc. 128, 7687–7691 (2006).
Shi, S.-L., Xu, L.-W., Oisaki, K., Kanai, M. & Shibasaki, M. Identification of modular chiral bisphosphines effective for Cu(I)-catalyzed asymmetric allylation and propargylation of ketones. J. Am. Chem. Soc. 132, 6638–6639 (2010).
Vieira, E. M., Snapper, M. L. & Hoveyda, A. H. Enantioselective synthesis of homoallylic amines through reactions of (pinacolato)allylborons with aryl-, heteroaryl-, alkyl-, or alkene-substituted aldimines catalyzed by chiral C1-symmetric NHC−Cu complexes. J. Am. Chem. Soc. 133, 3332–3335 (2011).
Wei, X.-F., Xie, X.-W., Shimizu, Y. & Kanai, M. Copper(I)-catalyzed enantioselective addition of enynes to ketones. J. Am. Chem. Soc. 139, 4647–4650 (2017).
Yue, W.-J., Zhang, C.-Y. & Yin, L. Asymmetric vinylogous aldol-type reactions of aldehydes with allyl phosphonate and sulfone. iScience 14, 88–99 (2019).
Kawai, J., Chikkade, P. K., Shimizu, Y. & Kanai, M. In situ catalytic generation of allylcopper species for asymmetric allylation: toward 1H-isochromene skeletons. Angew. Chem. Int. Ed. 52, 7177–7180 (2013).
Meng, F., Jang, H., Jung, B. & Hoveyda, A. H. Cu-catalyzed chemoselective preparation of 2-(pinacolato)boron-substituted allylcopper complexes and their in situ site-, diastereo-, and enantioselective additions to aldehydes and ketones. Angew. Chem. Int. Ed. 52, 5046–5051 (2013).
Chikkade, P. K., Shimizu, Y. & Kanai, M. Catalytic enantioselective synthesis of 2-(2-hydroxyethyl)indole scaffolds via consecutive intramolecular amido-cupration of allenes and asymmetric addition of carbonyl compounds. Chem. Sci. 5, 1585–1590 (2014).
Jiang, L. et al. Highly diastereo- and enantioselective Cu-catalyzed borylative coupling of 1,3-dienes and aldimines. Angew. Chem. Int. Ed. 55, 13854–13858 (2016).
Tsai, E. Y., Liu, R. Y., Yang, Y. & Buchwald, S. L. A regio- and enantioselective CuH-catalyzed ketone allylation with terminal allenes. J. Am. Chem. Soc. 140, 2007–2011 (2018).
Liu, R. Y., Zhou, Y., Yang, Y. & Buchwald, S. L. Enantioselective allylation using allene, a petroleum cracking byproduct. J. Am. Chem. Soc. 141, 2251–2256 (2019).
Cui, Y., Yamashita, Y. & Kobayashi, S. Facile preparation of allylzinc species from allylboronates and zinc amide via a boron-to-zinc exchange process and their reactions with carbonyl compounds, imines and hydrazones. Chem. Commun. 48, 10319–10321 (2012).
Cui, Y., Li, W., Sato, T., Yamashita, Y. & Kobayashi, S. Catalytic use of zinc amide for transmetalation with allylboronates: general and efficient catalytic allylation of carbonyl compounds, imines, and hydrazones. Adv. Synth. Catal. 355, 1193–1205 (2013).
Wadamoto, M., Ozasa, N., Yanagisawa, A. & Yamamoto, H. BINAP/AgOTf/KF/18-Crown-6 as new bifunctional catalysts for asymmetric Sakurai−Hosomi allylation and Mukaiyama aldol reaction. J. Org. Chem. 68, 5593–5601 (2003).
Wadamoto, M. & Yamamoto, H. Silver-catalyzed asymmetric Sakurai−Hosomi allylation of ketones. J. Am. Chem. Soc. 127, 14556–14557 (2005).
Naodovic, M., Wadamoto, M. & Yamamoto, H. Enantioselective Ag-catalyzed allylation of aldimines. Eur. J. Org. Chem. 2009, 5129–5131 (2009).
Wadamoto, M., Naodovic, M. & Yamamoto, H. Stereochemical studies of Ag-catalyzed Hosomi–Sakurai reaction using chiral silanes. Eur. J. Org. Chem. 2009, 5132–5134 (2009).
Chakrabarti, A., Konishi, H., Yamaguchi, M., Schneider, U. & Kobayashi, S. Indium(I)-catalyzed asymmetric allylation, crotylation, and α-chloroallylation of hydrazones with rare constitutional and high configurational selectivities. Angew. Chem. Int. Ed. 49, 1838–1841 (2010).
Huang, Y.-Y., Chakrabarti, A., Morita, N., Schneider, U. & Kobayashi, S. A catalytic asymmetric borono variant of Hosomi-Sakurai reactions with N,O-aminals. Angew. Chem. Int. Ed. 50, 11121–11124 (2011).
Shizuka, M. & Snapper, M. L. Catalytic enantioselective Hosomi-Sakurai conjugate allylation of cyclic unsaturated ketoesters. Angew. Chem. Int. Ed. 47, 5049–5051 (2008).
Sieber, J. D., Liu, S. & Morken, J. P. Catalytic conjugate addition of allyl groups to styryl-activated enones. J. Am. Chem. Soc. 129, 2214–2215 (2007).
Sieber, J. D. & Morken, J. P. Asymmetric Ni-catalyzed conjugate allylation of activated enones. J. Am. Chem. Soc. 130, 4978–4983 (2008).
Brozek, L. A., Sieber, J. D. & Morken, J. P. Catalytic enantioselective conjugate allylation of unsaturated methylidene ketones. Org. Lett. 13, 995–997 (2011).
Li, X., Meng, F., Torker, S., Shi, Y. & Hoveyda, A. H. Catalytic enantioselective conjugate additions of (pin)B-Substituted allylcopper compounds generated in situ from butadiene or isoprene. Angew. Chem. Int. Ed. 55, 9997–10002 (2016).
Meng, F. et al. Catalytic erantioselective 1,6-conjugate additions of propargyl and allyl groups. Nature 537, 387–393 (2016).
Huang, Y., Torker, S., Li, X., del Pozo, J. & Hoveyda, A. H. Racemic vinylallenes in catalytic enantioselective multicomponent processes: rapid generation of complexity through 1,6-conjugate additions. Angew. Chem. Int. Ed. 58, 2685–2691 (2019).
Kuang, Y. et al. Catalytic asymmetric conjugate allylation of coumarins. Org. Lett. 13, 3814–3817 (2011).
Yanagida, Y., Yazaki, R., Kumagai, N. & Shibasaki, M. Asymmetric synthesis of isothiazoles through Cu catalysis: direct catalytic asymmetric conjugate addition of allyl cyanide to α,β-unsaturated thioamides. Angew. Chem. Int. Ed. 50, 7910–7914 (2011).
Silva, E. M. P. & Silva, A. M. S. 1,6-conjugate addition of nucleophiles to α,β,γ,δ-diunsaturated systems. Synthesis 44, 3109–3128 (2012).
Mauduit, M., Baslé, O., Clavier, H., Crévisy, C. & Denicourt-Nowicki, A. Metal Catalyzed Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes in Comprehensive Organic Synthesis II, Vol. 4 (eds Molander, G. A. & Knochel, P.) 189 (Elsevier, Amsterdam, 2014).
den Hartog, T., Harutyunyan, S. R., Font, D., Minnaard, A. J. & Feringa, B. L. Catalytic enantioselective 1,6-conjugate addition of Grignard reagents to linear dienoates. Angew. Chem. Int. Ed. 47, 398–401 (2008).
Hénon, H., Mauduit, M. & Alexakis, A. Regiodivergent 1,4 versus 1,6 asymmetric copper-catalyzed conjugate addition. Angew. Chem. Int. Ed. 47, 9122–9124 (2008).
Wencel-Delord, J., Alexakis, A., Crévisy, C. & Mauduit, M. Enantioselective 1,6-conjugate addition to cyclic dienones catalyzed by the Cu−DiPPAM complex. Org. Lett. 12, 4335–4337 (2010).
Magrez, M., Wencel-Delord, J., Alexakis, A., Crévisy, C. & Mauduit, M. Significant asymmetric amplification in enantioselective Cu/DiPPAM-catalyzed 1,6- and 1,4-conjugate additions of diethylzinc to (di)enones. Org. Lett. 14, 3576–3579 (2012).
Tissot, M. & Alexakis, A. Enantio- and regioselective conjugate addition of organometallic reagents to linear polyconjugated nitroolefins. Chem. Eur. J. 19, 11352–11363 (2013).
Magrez-Chiquet, M. et al. Enantioselective 1,6-conjugate addition of dialkylzinc reagents to acyclic dienones catalyzed by Cu-DiPPAM complex-extension to asymmetric sequential 1,6/1,4-conjugate addition. Chem. Eur. J. 19, 13663–13667 (2013).
Jahier-Diallo, C. et al. Multicomponent synthesis of chiral bidentate unsymmetrical unsaturated n-heterocyclic carbenes: copper-catalyzed asymmetric C-C bond formation. Chem. Eur. J. 21, 993–997 (2015).
den Hartog, T. et al. On the mechanism of Cu-catalyzed enantioselective extended conjugate additions: a structure-based approach. ACS Catal. 5, 560–574 (2015).
Blons, C. et al. Asymmetric sequential Cu-catalyzed 1,6/1,4-conjugate additions of hard nucleophiles to cyclic dienones: determination of absolute configurations and origins of enantioselectivity. Chem. Eur. J. 23, 7515–7525 (2017).
Guo, Y., Kootstra, J. & Harutyunyan, S. R. Catalytic regio- and enantioselective alkylation of conjugated dienyl amides. Angew. Chem. Int. Ed. 57, 13547–13550 (2018).
Halbert, S. et al. Catalytically active species in copper/DiPPAM-catalyzed 1,6-asymmetric conjugate addition of dialkylzinc to dienones: a computational overview. ChemCatChem 11, 4108–4115 (2019).
Clavier, H., Coutable, L., Guillemin, J.-C. & Mauduit, M. New bidentate alkoxy-NHC ligands for enantioselective copper-catalysed conjugate addition. Tetrahedron 16, 921–924 (2005).
Park, J. K. et al. A chiral 6-membered N-heterocyclic carbene copper(i) complex that induces high stereoselectivity. Org. Lett. 12, 5008–5011 (2010).
Harada, A., Makida, Y., Sato, T., Harada, A. & Sawamura, M. Copper-catalyzed enantioselective allylic alkylation of terminal alkyne pronucleophiles. J. Am. Chem. Soc. 136, 13932–13939 (2014).
Ohmiya, H., Zhang, H., Shibata, S., Harada, A. & Sawamura, M. Construction of quaternary stereogenic carbon centers through copper-catalyzed enantioselective allylic alkylation of azoles. Angew. Chem. Int. Ed. 55, 4777–4780 (2016).
Yasuda, Y., Ohmiya, H. & Sawamura, M. Copper-catalyzed enantioselective allyl–allyl coupling between allylic boronates and phosphates with a phenol/N-heterocyclic carbene chiral ligand. Angew. Chem. Int. Ed. 55, 10816–10820 (2016).
Hojoh, K., Ohmiya, H. & Sawamura, M. Synthesis of α-quaternary formimides and aldehydes through umpolung asymmetric copper catalysis with isocyanides. J. Am. Chem. Soc. 139, 2184–2187 (2017).
Lipshutz, B. H. & Hackmann, C. Conjugate addition reactions of allylic copper species derived from Grignard reagents: synthetic and spectroscopic aspects. J. Org. Chem. 59, 7437–7444 (1994).
Wipf, P. & Grenon, M. Toward the total synthesis of lophotoxin—New methodologies and synthetic strategies. Can. J. Chem. 84, 1226–1241 (2006).
Yamamoto, Y., Fujikawa, R., Umemoto, T. & Miyaura, N. Iridium-catalyzed hydroboration of alkenes with pinacolborane. Tetrahedron 60, 10695–10700 (2004).
Guillou, S., Bonhomme, F. J. & Janin, Y. L. An improved preparation of 3-alkoxypyrazoles. Synthesis 2008, 3504–3508 (2008).
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21672235, No. 21871287, and No. 21922114), the Program of Shanghai Academic/Technology Research Leader (20XD1403600), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB20000000), CAS Key Laboratory of Synthetic Chemistry of Natural Substances and Shanghai Institute of Organic Chemistry.
Author information
Authors and Affiliations
Contributions
L.Y. and P.T. conceived and designed the study. C.Y.S. and Z.Z.P. performed the synthetic experiments and analyzed data for all new compounds. L.Y. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Senmiao Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shi, CY., Pan, ZZ., Tian, P. et al. Copper(I)-catalyzed asymmetric 1,6-conjugate allylation. Nat Commun 11, 5480 (2020). https://doi.org/10.1038/s41467-020-19293-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19293-9
This article is cited by
-
Copper-catalysed asymmetric reductive cross-coupling of prochiral alkenes
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
