
Abstract
The uniqueness of water originates from its three-dimensional hydrogen-bond network, but this hydrogen-bond network is suddenly truncated at the interface and non-hydrogen-bonded OH (free OH) appears. Although this free OH is the most characteristic feature of interfacial water, the molecular-level understanding of its dynamic property is still limited due to the technical difficulty. We study ultrafast vibrational relaxation dynamics of the free OH at the air/water interface using time-resolved heterodyne-detected vibrational sum frequency generation (TR-HD-VSFG) spectroscopy. With the use of singular value decomposition (SVD) analysis, the vibrational relaxation (T1) times of the free OH at the neat H2O and isotopically-diluted water interfaces are determined to be 0.87 ± 0.06 ps (neat H2O), 0.84 ± 0.09 ps (H2O/HOD/D2O = 1/2/1), and 0.88 ± 0.16 ps (H2O/HOD/D2O = 1/8/16). The absence of the isotope effect on the T1 time indicates that the main mechanism of the vibrational relaxation of the free OH is reorientation of the topmost water molecules. The determined sub-picosecond T1 time also suggests that the free OH reorients diffusively without the switching of the hydrogen-bond partner by the topmost water molecule.
Similar content being viewed by others

Introduction
Water is ubiquitous in nature and has unique physicochemical properties essential for realizing life, which attracts much attention in a broad range of scientific fields. In bulk, water molecules form a three-dimensional hydrogen-bond network that dynamically changes on a sub-picosecond time scale1,2,3,4,5,6. In contrast, at the air/water interface, such a three-dimensional hydrogen-bond network is truncated on the atomic scale, which is expected to cause distinct structures and dynamics of interfacial water. However, the molecular-level understanding of the properties of interfacial water is still limited owing to the technical difficulty of interface-selective measurements.
Vibrational sum-frequency generation (VSFG) is interface-selective spectroscopy based on the second-order nonlinear optical process, and it is a powerful method to investigate aqueous interfaces7,8,9,10,11,12,13,14. One of the most notable observations with VSFG is a sharp OH stretch band at ∼3700 cm−1 at the air/water interface, which appears with a broad hydrogen-bonded OH stretch band peaked at ∼3450 cm−115. Such a sharp high-frequency band is absent in either IR or Raman spectrum of liquid water, and it is characteristic of the air/water interface. In fact, this 3700-cm−1 band arises from the non-hydrogen-bonded OH (free OH) of topmost water molecules that have OH pointing out to the air. This free OH has been attracting much interest experimentally and theoretically16,17,18,19, and it has been found that the “free” OH exists not only at the air/water interface but also at other hydrophobic interfaces20. Indeed, the free OH is now considered a molecular-level indicator of the hydrophobicity that plays critical roles in various chemical/biological processes such as membrane functions, on-water catalytic reactions, etc21,22. Therefore, the elucidation of the dynamic property of the free OH is very important, as a cornerstone for understanding interfacial water.
The ultrafast vibrational dynamics of water at interfaces was initially studied by conventional homodyne-detected time-resolved (TR−) VSFG23,24,25, and the free OH dynamics at the air/water interface was investigated by Bonn and coworkers26,27,28. They examined the effect of isotopic dilution to discuss the mechanism of vibrational relaxation because the intramolecular energy transfer is expected to be decelerated in HOD species due to the very large mismatch between the OH and OD stretch frequencies29. Since the vibrational relaxation (T1) time of the excited free OH derived by their homodyne-detected TR-VSFG experiments looked significantly changed with isotope dilution, they concluded that intramolecular energy transfer is the major relaxation pathway of the vibrationally excited free OH28.
Although it appears that the previous studies using homodyne detection provide fair evidence for the major role of the energy transfer in the relaxation process of the free OH, it has recently been recognized that homodyne detection has crucial problems, in particular for TR-VSFG measurements13,30. Actually, they can only provide the temporal change of the square of the second-order susceptibility (|χ(2)|2). The time-resolved spectra are the difference between the spectra measured with and without pump pulse irradiation so that those measured with homodyne detection correspond to Δ|χ(2)(t)|2 (=χ(2)Δχ(2)* + χ(2)*Δχ(2) – |Δχ(2)|2). Obviously, it is very difficult to extract adequate spectral and dynamical information from the data obtained by homodyne TR-VSFG experiments. In contrast to homodyne VSFG, heterodyne-detected (HD−) VSFG provides the spectra that directly represent second-order susceptibility9,10. Therefore, time-resolved HD-VSFG (TR-HD-VSFG) allows us to obtain the pump-induced change of the imaginary part of χ(2) (ΔImχ(2)), which can directly be compared to TR-IR and TR-Raman spectra for the bulk13.
In the present study, the ultrafast vibrational relaxation dynamics of the free OH at the air/water interface is studied with TR-HD-VSFG for neat H2O and isotopically diluted water. Contrary to the previous homodyne TR-VSFG studies, we found that the relaxation T1 time of the free OH determined by TR-HD-VSFG does not change with isotopic dilution. The absence of the isotopic dilution effect as well as other observations indicates that the predominant vibrational relaxation mechanism of the excited free OH is not the intramolecular energy transfer but the reorientation of the topmost water molecule that has the free OH.
Results
Steady-state spectra of the air/water interfaces
Figure 1 shows the steady-state Imχ(2) spectra of the air/neat H2O interface and the air/isotopically diluted water interface (H2O/HOD/D2O = 1/2/1). We adopted this isotopic dilution ratio for direct comparison with the previous homodyne VSFG study28. Both spectra exhibit a broad negative OH stretch band around 3150–3550 cm−1 and a sharp positive OH stretch band at 3700 cm−1. The former low-frequency broad band is assigned to the hydrogen-bonded OH (HB OH) of interfacial water, and its frequency and bandwidth are very similar to those of the HB OH band in the bulk IR spectrum. The latter high-frequency band is due to the free OH31, which we address in this study. The Imχ(2) spectrum of the isotopically diluted water interface is similar to that of the H2O interface, except that the amplitude is about half due to the dilution. Time-resolved measurements are carried out by probing the whole OH stretch region with selective excitation of the free OH band.

Steady-state Imχ(2) spectra of the air/neat H2O interface (black) and the air/isotopically diluted water interface (red, H2O/HOD/D2O = 1/2/1).
Time-resolved spectra measured with the free OH excitation
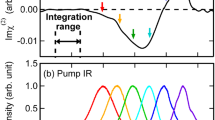
Figure 2a shows the time-resolved ΔImχ(2) spectra of the air/neat H2O interface obtained with the free OH excitation at the delay times from −3.0 to 6.0 ps. The bandwidth of the pump pulse is ~100 cm−1, which enables selective excitation of the free OH. At −3.0 ps without pump influence, no transient signal is observed. At 0.0 ps, a negative signal at ∼3700 cm−1, a positive signal at ∼3500 cm−1, and a small negative signal below ∼3400 cm−1 are observed. The negative signal at ∼3700 cm−1 is readily assignable to the bleach of the fundamental transition (v = 1 ← v = 0) of the free OH because we selectively excite the free OH. This bleach band is accompanied by a positive band due to the excited-state transition (v = 2 ← v = 1), i.e., the hot band, which is seen at ∼3500 cm−132,33,34. The small broad negative transient signal below 3400 cm−1 is attributable to the frequency shift of the HB OH band due to the anharmonic coupling between the HB OH and the free OH30,35,36. (This negative transient signal is more clearly seen in the expanded spectra shown in Supplementary Fig. 1). In other words, when the free OH of the topmost interfacial water molecule is excited, the intramolecular anharmonic coupling induces a redshift of the stretching frequency of another (HB−) OH in the same molecule. We note that this redshift of the negative HB OH band is expected to not only generate a negative feature in its low-frequency side but also give rise to a positive feature in its high-frequency side36. Therefore, the positive transient band around 3500 cm−1 should also have some contribution from this anharmonic frequency shift of the HB OH band. As the delay time increases, positive and negative signals appear at <3500 cm−1 and >3500 cm−1 in the HB OH region, respectively, reflecting a blue-shift of the HB OH band due to the temperature increase caused by the thermalization process30,37. This thermalized signal dominates the time-resolved spectra at later delay times.

Time-resolved ΔImχ(2) spectra from −3.0 to 6.0 ps obtained with selective excitation of the free OH, a neat H2O and b isotopically diluted water (H2O/HOD/D2O = 1/2/1) interfaces. The time shown along the vertical axis represents the delay time of each time-resolved ΔImχ(2) spectrum. Red and blue regions in the spectra are positive and negative changes from the steady-state Imχ(2) spectrum, respectively. The steady-state Imχ(2) spectrum and the spectrum of the pump pulse are also shown at the top, for comparison.
The time-resolved ΔImχ(2) spectra of the air/isotopically diluted water interface (H2O/HOD/D2O = 1/2/1) are shown in Fig. 2b. Basically, the temporal change of the ΔImχ(2) spectra of the air/isotopically diluted water interface is very similar to that observed for the air/neat H2O interface: the ground-state bleach and the hot band of the free OH are observed in the early delay times, whereas the thermalized spectrum gradually dominates the ΔImχ(2) spectra at the later delay times. However, the negative signal below ∼3400 cm−1 at 0.0 ps is not clearly recognized in the spectrum of the air/isotopically diluted water interface. (The difference from the ΔImχ(2) at the air/neat H2O interface is more clearly recognized in the expanded spectra shown in Supplementary Fig. 1). This confirms the assignment of the weak negative signal observed at the air/neat H2O interface to the frequency shift due to anharmonic coupling because only H2O molecules having two OH’s can have the anharmonic coupling between the HB OH and the free OH. Because isotopically diluted water contains only 25% of H2O compared to the neat H2O, the weak negative signal arising from the anharmonic redshift of the HB OH band of H2O becomes much weaker, so that it cannot be recognized clearly with the signal to noise ratio of the present measurements. Thus, the absence of the 3400-cm−1 band at the isotopically diluted water interface strongly supports the assignment of the broad negative band at the neat H2O interface to the frequency shift due to the anharmonic coupling and warrants the high reliability of the obtained ΔImχ(2) spectra as well.
T1 time of the free OH determined with SVD analysis
In order to extract quantitative information about the vibrational relaxation dynamics of the free OH by separating the thermalization signal, we carried out a singular value decomposition (SVD) analysis of the time-resolved ΔImχ(2) spectra obtained at the delay times from −0.2 to 6.0 ps. SVD is a mathematical technique to decompose a set of the time-resolved ΔImχ(2) spectra into independent spectral components and their temporal profiles (see Supplementary Note 2 for details)38,39,40. We stress that the SVD analysis is not applicable to time-resolved Δ|χ(2)|2 spectra measured with conventional homodyne detection and that it can be used for the time-resolved ΔImχ(2) spectra obtained with heterodyne detection. This is because the SVD analysis is only applicable to the spectra that are simple linear combinations of different spectral components. As shown in Fig. 3a, c, SVD successfully decomposes the ΔImχ(2) spectra into two spectral components. Component 1 represents the spectrum that is directly induced by the excitation of the free OH, whereas component 2 represents the spectrum after thermalization. The temporal profiles corresponding to these two components are shown in Fig. 3b, d. (Note that we assumed the exponential function for the temporal profiles of each component after SVD but the time constants themselves were determined by the analysis.) The most important result of this analysis is that the decay time constant of component 1, which corresponds to the T1 time of the free OH, is almost the same for the neat H2O (0.87 ± 0.06 ps) and the isotopically diluted water (0.84 ± 0.09 ps). This finding is very important for considering the relaxation mechanism of the free OH and is discussed in more detail later. The analysis also indicates a small difference between the T1 time of the free OH and the rise time of the thermalized spectrum. (1.13 ± 0.09 ps: neat H2O, 1.24 ± 0.15 ps: isotopically diluted water) This difference suggests the involvement of intermediate vibrational states (e.g., lower energy vibrational states) in the energy dissipation process of the excited free OH at the water interface.

a, c The spectra and b, d the temporal profiles of the two major components obtained from SVD analysis of the time-resolved ΔImχ(2) spectra from −0.2 ps to 6.0 ps: a, b neat H2O and c, d isotopically diluted water (H2O/HOD/D2O = 1/2/1). Red and blue regions in the spectra (a, c) correspond to the positive and negative changes from the steady-state Imχ(2) spectrum, respectively. The solid lines in b, d show the exponential fits for the decay and rise of the components, taking account of the instrumental response (200-fs Gaussian).
As already mentioned, we chose the isotopic dilution ratio of H2O/HOD/D2O = 1/2/1 for direct comparison with the results of the previous homodyne TR-VSFG study28. Nevertheless, to confirm no detectable isotopic effect on T1, we also carried out time-resolved HD-VSFG experiments using isotopically diluted water with a higher dilution radio (H2O/HOD/D2O = 1/8/16). Although the signal to noise ratio of the obtained ΔImχ(2) spectra was substantially lower, the SVD analysis showed that the T1 time of the free OH (0.88 ± 0.16 ps) is the same as T1’s obtained for neat H2O and isotopically diluted water of H2O/HOD/D2O = 1/2/1 within the error (see Supplementary Note 3 for details).
Vibrational relaxation mechanism of the free OH
There are three possible relaxation pathways for the vibrationally excited free OH28, which are depicted in Fig. 4a, i.e., (i) intramolecular energy transfer to the HB OH, (ii) intramolecular energy transfer to the bend overtone, and (iii) reorientation of the interfacial water molecules that change the free OH to the HB OH. In the case of (iii) the reorientation mechanism, while an excited free OH is relaxed with reorientation, an unexcited HB OH or an unexcited free OH that lies nearly horizontal becomes a new unexcited free OH, recovering the population of the free OH in the vibrationally ground state. Because the reorientation time of an OH group in HOD is considered essentially the same as that of an OH group in H2O, the relaxation time with this mechanism is not affected by the isotopic dilution. In the other two relaxation mechanisms, i.e., the intramolecular energy transfer from the free OH to HB OH stretch and that to the bend overtone, the relaxation is expected to be strongly decelerated in HOD generated with isotopic dilution, because of a large frequency mismatch between the OH and OD stretch vibrations and that between the OH stretch and the HOD bend overtone (Fig. 4b)41,42,43. The present experiment shows that the T1 time of the excited free OH does not change with the isotopic dilution. This absence of the isotope effect negates the primary role of the intramolecular energy transfer in the vibrational relaxation of the excited free OH. The present TR-HD-VSFG measurements strongly indicate that the reorientation of the topmost water molecules is the main relaxation mechanism of the free OH present at the air/water interface. In other words, the T1 time of the free OH is directly related to its reorientation time26.

a Three possible relaxation pathways for the vibrationally excited free OH. Intramolecular energy transfer from the excited free OH to (i) HB OH and (ii) bend overtone, and (iii) reorientation of the topmost water molecule. Arrows in (i), (ii), and (iii) depict the energy transfer, the bend vibration, and reorientational motion, respectively. Dotted lines represent the vibration in the excited state. b The vibrational energy levels of (i) H2O and (ii) HOD. The bending frequencies of H2O and HOD are the values of bulk water42,43.
HB OH region: comparison with homodyne detection
To justify the above conclusion derived from the isotope effect on the T1 time of the free OH, we also examine the rise of the transient signal in the HB OH region. Actually, in the previous homodyne-detected TR-VSFG studies27,28, the intensity change in the HB OH region was measured with excitation of the free OH, and it was argued that the rise of the transient signal was significantly delayed compared to the appearance of the transient in the free OH region. Then, this delayed rise was considered as evidence of the intramolecular energy transfer from the free OH to the HB OH. In our assignment, however, the transient signal in the HB OH region is due to the hot band of the free OH and the anharmonic coupling between the free OH and HB OH. Therefore, if our assignment is correct, the transient signal in the HB OH region is expected to appear simultaneously with the bleaching signal in the free OH region.
Figure 5a, c shows |χ(2) + Δχ(2)|2 – |χ(2)|2 calculated from the data obtained by the present TR-HD-VSFG measurements, which correspond to the signal obtained with the homodyne TR-VSFG measurement. (Time-resolved spectra of the real part of χ(2), ΔReχ(2), used for the calculation are shown in Supplementary Fig. 4). As clearly seen in Fig. 5a, the rise of the |χ(2) + Δχ(2)|2 – |χ(2)|2 signal in the HB OH region (3500 cm−1) is substantially delayed compared to that in the free OH region, which reproduces the observation in the previous homodyne-detected TR-VSFG study27,28. However, the temporal change of the time-resolved ΔImχ(2) signal in the HB OH and free OH regions is very similar to each other (Fig. 5b). This clearly demonstrates that the large delay reported for the rise of the transient signal in the HB OH region predominantly arises from the artifact due to the homodyne detection, not due to the intramolecular energy transfer. We also note that a significant isotope effect is seen for the decay of the |χ(2) + Δχ(2)|2 – |χ(2)|2 signal in the free OH region (Fig. 5c: 0.75 ± 0.04 ps (H2O) and 1.34 ± 0.55 ps (HOD)) as reported, whereas it is absent in the ΔImχ(2) signal (Fig. 5d: 0.74 ± 0.06 ps (H2O) and 0.76 ± 0.29 ps (HOD)). This demonstrates that the reported isotopic dilution effect on the decay of the transient signal in the free OH region is also due to the artifact originating from the homodyne detection. It is noted that the present analysis on |χ(2) + Δχ(2)|2 – |χ(2)|2 and the previous homodyne TR-VSFG study28 look to provide somewhat different decay time constants for the transient signal in the free OH region. The reason for this discrepancy is not clear at the moment, but it may be attributable to the difference in the excitation condition such as the pump pulse energy and the focus size. Indeed, the temporal change of |χ(2) + Δχ(2)|2 – |χ(2)|2 is changed with the relative magnitude between χ(2) and Δχ(2), and Δχ(2) strongly depends on the excitation condition.

a Homodyne TR-VSFG signals and b time-resolved ΔImχ(2) in the HB OH region (3500 cm−1, black) and the free OH region (3700 cm−1, red) at the air/neat H2O interface. Black and red dotted lines represent the peak of each temporal trace. c Homodyne TR-VSFG signals and d time-resolved ΔImχ(2) in the free OH region at the air/neat H2O interface (red) and isotopically diluted water interface (blue). Homodyne TR-VSFG signals in a and c are calculated from the data obtained in the present TR-HD-VSFG measurements as |χ(2) + Δχ(2)|2 – |χ(2)|2. Solid lines are the exponential fits convoluted by the instrumental response (200-fs Gaussian).
Although the large delay of the rise of the transient signal in the HB OH region is predominantly attributed to the artifact due to homodyne detection, there still exists a small (∼50 fs) difference between the rises of transient signals in the free OH and HB OH regions (Fig. 5b). This 50-fs difference observed in the time-resolved ΔImχ(2) signal is attributable to the effect of the perturbed free induction decay (PFID)44,45,46. It is known that when the pump pulse arrives before the complete dephasing of a probed vibration, the pump perturbs the free induction decay of the vibration and gives rise to transient signals in the negative delay time region. Indeed, the PFID is clearly recognized in the time-domain signals of the SFG electric field which are obtained with Fourier transformation. (See Supplementary Note 5 for the details.)
The time-resolved ΔImχ(2) data obtained in this study clearly show that the temporal evolution of the pump-induced changes in the HB OH region is essentially the same as that in the free OH region, except for the effect of PFID. This means that the transient signal in the HB OH region reflects the free OH dynamics and that it arises from the hot band of the free OH and the spectral shift due to the anharmonic coupling. Thus, the analysis of the transient signal in the HB OH region also supports that the excited free OH predominantly relaxes by the reorientation mechanism, not through intramolecular energy transfer. Actually, this conclusion is readily rationalized by the almost no overlap between the vibrational frequencies of the free OH and HB OH, which makes resonant energy transfer inefficient. The energetic isolation of the free OH also explains why the relaxation time of the excited free OH is substantially longer than the T1 time of the HB OH (0.29–0.35 ps in bulk6,47,48 and ∼0.4 ps at the neat H2O interface30).
Discussion
Lastly, we discuss the reorientation mechanism of the topmost water molecule based on the sub-picosecond relaxation time determined in this study. In bulk, the reorientation mechanism of water molecules has been discussed in the past decades, and it is now widely accepted that water molecules reorient with the jump mechanism in which hydrogen bonds are broken and reformed simultaneously in the reorientation process5,49. Polarization anisotropy decay measurements determined that this process proceeds with 2.5 ps50,51. At the interface, on the other hand, several theoretical studies suggest that the free OH reorients in a diffusive manner without switching the hydrogen-bond partner of the relevant topmost water molecule and that it proceeds faster than the reorientation by the jump mechanism52,53. The sub-picosecond relaxation time determined in this study accords with this theoretical prediction about the diffusive nature of the reorientation process of the topmost water molecules at the interface.
It may be worth noting that the picture of the reorientation-induced relaxation of the free OH obtained by the present TR-HD-VSFG study is in the same line with the conclusion of a recent homodyne-detected TR-VSFG study that reported the relaxation dynamics of the free OH at the water interface with the hydrophobic octadecylsilane self-assembled monolayer on fused silica54. Although experiments with heterodyne detection are desired also for such buried silica/monolayer interfaces, the similar conclusions for the two prototypical hydrophobic interfaces suggest the general importance of the reorientation relaxation pathway in the vibrational relaxation of the free OH at hydrophobic interfaces.
In summary, the ultrafast vibrational relaxation dynamics of the free OH at the air/water interface was studied using TR-HD-VSFG with neat H2O and isotopically diluted water. Using SVD analysis, we determined the T1 time of the excited free OH from the time-resolved ΔImχ(2) spectra, which are 0.87 ± 0.06 ps at the neat H2O interface, and 0.84 ± 0.09 ps (H2O/HOD/D2O = 1/2/1), and 0.88 ± 0.16 ps (H2O/HOD/D2O = 1/8/16) at the isotopically diluted water interfaces. The absence of the isotope effect on the T1 time strongly indicates that the vibrational relaxation of the excited free OH predominantly proceeds with the reorientation of interfacial water molecules, not through the intramolecular energy transfer. This means that the free OH is energetically isolated from the other vibrational modes. In addition, the sub-picosecond time constant of this reorientation-induced vibrational relaxation supports the diffusive nature of the free OH reorientation. Consequently, the picture of the vibrational relaxation process of the free OH obtained by this study is described as follows: When a free OH is vibrationally excited, the excited free OH is changed into an excited-state HB OH with diffusive reorientation. This process causes the loss of an excited free OH, which is compensated by the appearance of an unexcited free OH generated from an unexcited HB OH or an unexcited free OH lying almost horizontally, resulting in the recovery of the bleach of the unexcited free OH signal. The excited HB OH created by the reorientation of the free OH rapidly relaxes to the lower energy vibrational states as in the case of direct excitation of HB OH30. Finally, the pump energy absorbed by water converts to the thermal energy, which is observed as the appearance of the thermalized spectrum. A small difference between the T1 time of the free OH and the rise time of the thermalized spectrum suggests the involvement of other vibrational states (e.g., low-lying vibrational states) in this energy dissipation process at the water interface. The picture obtained in this study is sketched in Fig. 6. The air/water interface is the simplest and most fundamental hydrophobic interface. Therefore, the vibrational relaxation mechanism of the free OH at the air/water interface, which has been clarified in the present study, provides an indispensable basis for understanding the energy transport process at the hydrophobic interfaces.

When a free OH is vibrationally excited (process 1), the excited free OH diffusively reorients (process 2) to form an excited-state HB OH (process 3). The excited HB OH rapidly relaxes to the lower energy vibrational states (process 4). Finally, the pump energy absorbed by water converts to the thermal energy (process 5). Dotted OH bonds represent the vibration in the excited state.
Methods
Time-resolved heterodyne-detected vibrational sum-frequency generation (TR-HD-VSFG) spectroscopy
The optical setup for TR-HD-VSFG measurements was described in detail previously55. Briefly, a narrow-band visible ω1 pulse (center wavelength: 795 nm, bandwidth: 25 cm−1, pulse width: 0.5 ps, s-polarized) and a broadband infrared ω2 pulse (center frequency: 3450 cm−1, bandwidth: 300 cm−1, pulse width: 0.1 ps, p-polarized) were focused into a y-cut quartz crystal and then onto the air/water interface to generate sum frequency (ω1 + ω2, s-polarized). The former SFG was used as the local oscillator (LO) and passed through a glass plate (2 mm) in advance to be delayed by 3.2 ps with respect to the latter SFG from the sample. The spectral interferograms were recorded by using a polychromator with a charge-coupled device (CCD). The time-resolved measurements were carried out with pump ωpump pulses at ∼3700 cm−1 (bandwidth: 100 cm−1, pulse width: 0.2 ps, p-polarized, 30 μJ) for selective excitation of the free OH. The time-resolution of the measurements was evaluated by measuring the third-order nonlinear signal of ω1 + ω2 + ωpump that was generated with the z-cut quartz placed at the sample position while changing the ωpump delay. The FWHM of the temporal response was ∼200 fs. The signals obtained with a 20-second exposure were accumulated 380 times and 560 times for the neat H2O and isotopically diluted water (H2O/HOD/D2O = 1/2/1), respectively, at each delay time.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Woutersen, S., Emmerichs, U. & Bakker, H. J. Femtosecond mid-IR pump-probe spectroscopy of liquid water: evidence for a two-component structure. Science 278, 658–660 (1997).
Woutersen, S. & Bakker, H. J. Resonant intermolecular transfer of vibrational energy in liquid water. Nature 402, 507–509 (1999).
Fecko, C. J., Eaves, J. D., Loparo, J. J., Tokmakoff, A. & Geissler, P. L. Ultrafast hydrogen-bond dynamics in the infrared spectroscopy of water. Science 301, 1698–1702 (2003).
Cowan, M. L. et al. Ultrafast memory loss and energy redistribution in the hydrogen bond network of liquid H2O. Nature 434, 199–202 (2005).
Laage, D. & Hynes, J. T. A molecular jump mechanism of water reorientation. Science 311, 832–835 (2006).
Ramasesha, K., De Marco, L., Mandal, A. & Tokmakoff, A. Water vibrations have strongly mixed intra- and intermolecular character. Nat. Chem. 5, 935–940 (2013).
Shen, Y. R. & Ostroverkhov, V. Sum-frequency vibrational spectroscopy on water interfaces: polar orientation of water molecules at interfaces. Chem. Rev. 106, 1140–1154 (2006).
Gopalakrishnan, S., Liu, D., Allen, H. C., Kuo, M. & Shultz, M. J. Vibrational spectroscopic studies of aqueous interfaces: salts, acids, bases, and nanodrops. Chem. Rev. 106, 1155–1175 (2006).
Nihonyanagi, S., Mondal, J. A., Yamaguchi, S. & Tahara, T. Structure and dynamics of interfacial water studied by heterodyne-detected vibrational sum-frequency generation. Annu. Rev. Phys. Chem. 64, 579–603 (2013).
Shen, Y. R. Phase-sensitive sum-frequency spectroscopy. Annu. Rev. Phys. Chem. 64, 129–150 (2013).
Johnson, C. M. & Baldelli, S. Vibrational sum frequency spectroscopy studies of the influence of solutes and phospholipids at vapor/water interfaces relevant to biological and environmental systems. Chem. Rev. 114, 8416–8446 (2014).
Ishiyama, T., Imamura, T. & Morita, A. Theoretical studies of structures and vibrational sum frequency generation spectra at aqueous interfaces. Chem. Rev. 114, 8447–8470 (2014).
Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Ultrafast dynamics at water interfaces studied by vibrational sum frequency generation spectroscopy. Chem. Rev. 117, 10665–10693 (2017).
Inoue, K., Nihonyanagi, S. & Tahara, T. in Coherent Multidimensional Spectroscopy (ed. Cho, M.) 215–236 (Springer, Singapore, 2019).
Du, Q., Superfine, R., Freysz, E. & Shen, Y. R. Vibrational spectroscopy of water at the vapor/water interface. Phys. Rev. Lett. 70, 2313–2316 (1993).
Du, Q., Freysz, E. & Shen, Y. R. Surface vibrational spectroscopic studies of hydrogen bonding and hydrophobicity. Science 264, 826–828 (1994).
Wei, X. & Shen, Y. R. Motional effect in surface sum-frequency vibrational spectroscopy. Phys. Rev. Lett. 86, 4799–4802 (2001).
Gan, W., Wu, D., Zhang, Z., Feng, R.-R. & Wang, H.-F. Polarization and experimental configuration analyses of sum frequency generation vibrational spectra, structure, and orientational motion of the air/water interface. J. Chem. Phys. 124, 114705 (2006).
Sun, S. et al. Orientational distribution of free O-H groups of interfacial water is exponential. Phys. Rev. Lett. 121, 246101 (2018).
Scatena, L. F., Brown, M. G. & Richmond, G. L. Water at hydrophobic surfaces: weak hydrogen bonding and strong orientation effects. Science 292, 908–912 (2001).
Ball, P. Water as an active constituent in cell biology. Chem. Rev. 108, 74–108 (2008).
Jung, Y. & Marcus, R. A. On the theory of organic catalysis “on water”. J. Am. Chem. Soc. 129, 5492–5502 (2007).
McGuire, J. A. & Shen, Y. R. Ultrafast vibrational dynamics at water interfaces. Science 313, 1945–1948 (2006).
Ghosh, A., Smits, M., Bredenbeck, J. & Bonn, M. Membrane-bound water is energetically decoupled from nearby bulk water: an ultrafast surface-specific investigation. J. Am. Chem. Soc. 129, 9608–9609 (2007).
Eftekhari-Bafrooei, A. & Borguet, E. Effect of surface charge on the vibrational dynamics of interfacial water. J. Am. Chem. Soc. 131, 12034–12035 (2009).
Hsieh, C.-S. et al. Ultrafast reorientation of dangling OH groups at the air-water interface using femtosecond vibrational spectroscopy. Phys. Rev. Lett. 107, 116102 (2011).
Zhang, Z., Piatkowski, L., Bakker, H. J. & Bonn, M. Ultrafast vibrational energy transfer at the water/air interface revealed by two-dimensional surface vibrational spectroscopy. Nat. Chem. 3, 888–893 (2011).
Hsieh, C.-S. et al. Mechanism of vibrational energy dissipation of free OH groups at the air–water interface. Proc. Natl Acad. Sci. USA 110, 18780–18785 (2013).
Bakker, H. J. & Skinner, J. L. Vibrational spectroscopy as a probe of structure and dynamics in liquid water. Chem. Rev. 110, 1498–1517 (2010).
Inoue, K. et al. Efficient spectral diffusion at the air/water interface revealed by femtosecond time-resolved heterodyne-detected vibrational sum frequency generation spectroscopy. J. Phys. Chem. Lett. 7, 1811–1815 (2016).
Nihonyanagi, S. et al. Accurate determination of complex χ(2) spectrum of the air/water interface. J. Chem. Phys. 143, 124707 (2015).
Schofield, D. P. & Kjaergaard, H. G. Calculated OH-stretching and HOH-bending vibrational transitions in the water dimer. Phys. Chem. Chem. Phys. 5, 3100–3105 (2003).
Nizkorodov, S. A., Ziemkiewicz, M., Nesbitt, D. J. & Knight, A. E. W. Overtone spectroscopy of H2O clusters in the vOH = 2 manifold: Infrared-ultraviolet vibrationally mediated dissociation studies. J. Chem. Phys. 122, 194316 (2005).
Salmi, T. et al. Calculation of the O−H stretching vibrational overtone spectrum of the water dimer. J. Phys. Chem. A 112, 6305–6312 (2008).
Singh, P. C., Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Communication: ultrafast vibrational dynamics of hydrogen bond network terminated at the air/water interface: a two-dimensional heterodyne-detected vibrational sum frequency generation study. J. Chem. Phys. 139, 161101 (2013).
Ishiyama, T., Morita, A. & Tahara, T. Molecular dynamics study of two-dimensional sum frequency generation spectra at vapor/water interface. J. Chem. Phys. 142, 212407 (2015).
Nihonyanagi, S., Singh, P. C., Yamaguchi, S. & Tahara, T. Ultrafast vibrational dynamics of a charged aqueous interface by femtosecond time-resolved heterodyne-detected vibrational sum frequency generation. Bull. Chem. Soc. Jpn. 85, 758–760 (2012).
Yamaguchi, S. & Hamaguchi, H.-O. Femtosecond ultraviolet-visible absorption study of all-trans→13-cis⋅9-cis photoisomerization of retinal. J. Chem. Phys. 109, 1397–1408 (1998).
Matsuzaki, K. et al. Partially hydrated electrons at the air/water interface observed by UV-excited time-resolved heterodyne-detected vibrational sum frequency generation spectroscopy. J. Am. Chem. Soc. 138, 7551–7557 (2016).
Ahmed, M., Inoue, K., Nihonyanagi, S. & Tahara, T. Hidden isolated OH at the charged hydrophobic interface revealed by two-dimensional heterodyne-detected VSFG spectroscopy. Angew. Chem. Int. Ed. 59, 9498–9505 (2020).
Xu, X., Shen, Y. R. & Tian, C. Phase-sensitive sum frequency vibrational spectroscopic study of air/water interfaces: H2O, D2O, and diluted isotopic mixtures. J. Chem. Phys. 150, 144701 (2019).
Kundu, A. et al. Bend vibration of surface water investigated by heterodyne-detected sum frequency generation and theoretical study: dominant role of quadrupole. J. Phys. Chem. Lett. 7, 2597–2601 (2016).
Bodis, P., Larsen, O. F. A. & Woutersen, S. Vibrational relaxation of the bending mode of HDO in liquid D2O. J. Phys. Chem. A 109, 5303–5306 (2005).
Hamm, P. Coherent effects in femtosecond infrared spectroscopy. Chem. Phys. 200, 415–429 (1995).
Inoue, K., Watanabe, K. & Matsumoto, Y. Instantaneous vibrational frequencies of diffusing and desorbing adsorbates: CO/Pt(111). J. Chem. Phys. 137, 024704–024706 (2012).
Inoue, K., Watanabe, K., Sugimoto, T., Matsumoto, Y. & Yasuike, T. Disentangling multidimensional nonequilibrium dynamics of adsorbates: CO desorption from Cu(100). Phys. Rev. Lett. 117, 186101 (2016).
Lock, A. J. & Bakker, H. J. Temperature dependence of vibrational relaxation in liquid H2O. J. Chem. Phys. 117, 1708–1713 (2002).
van der Post, S. T. et al. Strong frequency dependence of vibrational relaxation in bulk and surface water reveals sub-picosecond structural heterogeneity. Nat. Commun. 6, 8384 (2015).
Laage, D., Stirnemann, G., Sterpone, F., Rey, R. & Hynes, J. T. Reorientation and allied dynamics in water and aqueous solutions. Annu. Rev. Phys. Chem. 62, 395–416 (2011).
Fecko, C. J., Loparo, J. J., Roberts, S. T. & Tokmakoff, A. Local hydrogen bonding dynamics and collective reorganization in water: ultrafast infrared spectroscopy of HOD/D2O. J. Chem. Phys. 122, 054506 (2005).
Rezus, Y. L. & Bakker, H. J. On the orientational relaxation of HDO in liquid water. J. Chem. Phys. 123, 114502 (2005).
Vila Verde, A., Bolhuis, P. G. & Campen, R. K. Statics and dynamics of free and hydrogen-bonded OH groups at the air/water interface. J. Phys. Chem. B 116, 9467–9481 (2012).
Ni, Y., Gruenbaum, S. M. & Skinner, J. L. Slow hydrogen-bond switching dynamics at the water surface revealed by theoretical two-dimensional sum-frequency spectroscopy. Proc. Natl Acad. Sci. USA 110, 1992–1998 (2013).
Xiao, S., Figge, F., Stirnemann, G., Laage, D. & McGuire, J. A. Orientational dynamics of water at an extended hydrophobic interface. J. Am. Chem. Soc. 138, 5551–5560 (2016).
Inoue, K., Nihonyanagi, S., Singh, P. C., Yamaguchi, S. & Tahara, T. 2D heterodyne-detected sum frequency generation study on the ultrafast vibrational dynamics of H2O and HOD water at charged interfaces. J. Chem. Phys. 142, 212431 (2015).
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 18H05265. K.I. acknowledges the Special Postdoctoral Researchers (SPDR) program of RIKEN.
Author information
Authors and Affiliations
Contributions
K.I., S.N., and T.T. designed the research. K.I. and M.A. performed the experiments and analyzed the data. All authors discussed the results and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Inoue, Ki., Ahmed, M., Nihonyanagi, S. et al. Reorientation-induced relaxation of free OH at the air/water interface revealed by ultrafast heterodyne-detected nonlinear spectroscopy. Nat Commun 11, 5344 (2020). https://doi.org/10.1038/s41467-020-19143-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19143-8
This article is cited by
-
Unified picture of vibrational relaxation of OH stretch at the air/water interface
Nature Communications (2024)
-
Barrier-free molecular reorientations in polyhedral water clusters
Structural Chemistry (2023)
-
Can electric fields drive chemistry for an aqueous microdroplet?
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
