
Abstract
The Human Proteome Organization (HUPO) launched the Human Proteome Project (HPP) in 2010, creating an international framework for global collaboration, data sharing, quality assurance and enhancing accurate annotation of the genome-encoded proteome. During the subsequent decade, the HPP established collaborations, developed guidelines and metrics, and undertook reanalysis of previously deposited community data, continuously increasing the coverage of the human proteome. On the occasion of the HPP’s tenth anniversary, we here report a 90.4% complete high-stringency human proteome blueprint. This knowledge is essential for discerning molecular processes in health and disease, as we demonstrate by highlighting potential roles the human proteome plays in our understanding, diagnosis and treatment of cancers, cardiovascular and infectious diseases.
Similar content being viewed by others


Introduction
A decade after the release of the draft Human Genome Project (HGP), the Human Proteome Organization (HUPO) leveraged this genomic encyclopedia to launch a visionary international scientific collaboration called the Human Proteome Project (HPP)1,2,3,4. Utilizing substantial community data, the HPP connects scientists, clinicians, industry, institutions and knowledgebase (KB) partners to create a framework for collaboration, data sharing and quality assurance—all targeted at discovering credible evidence for the entire complement of human genome-coded proteins (Box 1).
Here we report and discuss HUPO’s first high-stringency HPP blueprint (https://www.nextprot.org/about/statistics, data release 17-01-2020). This blueprint was assembled over 10 years by the HPP and covers >90% of the human proteome, paralleling progress made by the HGP5. This effort relied heavily upon community efforts that enabled HPP data inspection and re-analysis, culminating in the creation of a high-stringency human proteome KB. To illustrate the many historical innovations driving growth in proteomics, HUPO has created a historical timeline that will be released coincidentally with this publication (https://hupo.org/Proteomics-Timeline).
HPP mission and strategic aims
The HPP mission is to assemble and analyse community data, bringing increased granularity to our molecular understanding of the dynamic nature of the proteome, its modifications and relationships to human biology and disease. This aligns closely with HUPO’s aim of ‘translating the code of life’, providing crucial biochemical and cell biological information that genomics per se cannot deliver, while laying better foundations for diagnostic, prognostic, therapeutic and precision medicine applications.
From its inception, the HPP stated two strategic objectives as follows:
-
1.
To credibly catalogue the human proteome parts list and discover its complexity (including posttranslational modifications (PTMs), splice variants, interactions and functions) by:
-
a.
Establishing agreed, stringent, reliable standards,
-
b.
Identifying >1 protein product from each protein-coding gene and
-
c.
Detecting expression of the remaining missing proteins (see below).
-
a.
-
2.
To make proteomics an integrated component of multi-omics studies to advance life sciences, biomedical sciences and precision medicine.
Comparisons with the HGP are numerous. Both global projects are ambitious cooperative community efforts seeking to identify how genes (HGP) or proteins (HPP) help define the molecular mechanisms underlying health and disease. Both groups have implemented exhaustive data sharing and stringent quality control efforts. However, we now know that sequencing human genomes is necessary but not sufficient to understand the complexity of human biology or pathology. Knowledge of expressed proteins (including concentration, spatio-temporal localization, activities, protease-processed forms, transport, interactions, splice isoforms, PTMs and the many proteoforms derived from the proteome) cannot be predicted by genome sequencing alone.
HPP structure and achievements
HUPO launched the HPP in 2010 at the 9th HUPO Annual World Congress in Sydney, with Gil Omenn as the inaugural chair. The HPP started without long-term funding. Financial support over the decade came through individual principal investigator projects, institutional infrastructure/core facilities and philanthropy, without large-scale, long-term, integrated multi-government strategic funding.
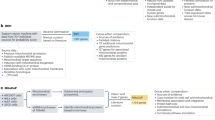
The HPP grew from several archetypal HUPO projects (plasma, liver, brain, cardiovascular, kidney/urine proteomes). At present, the HPP comprises two strategic initiatives—chromosome-centric (C-HPP; 25 teams) and biology/disease-centric (B/D-HPP; 19 teams)—organized in a strategic matrix underpinned by four Resource Pillars: antibodies (AB), mass spectrometry (MS), KB and Pathology (Fig. 1a). Before explaining these elements in more detail, we will describe HPP’s criteria underlying the establishment of the high-stringency human proteome.

a The HPP matrix formed by creating two major initiatives (C-HPP and B/D-HPP). The initiatives and their teams are underpinned by 4 Resource Pillars (AB, MS, KB and pathology). b The HPP KB pipeline demonstrates how MS, AB and other biological data are collected, processed, re-analysed and presented annually for FAIR (see below) use by the scientific community. MS datasets are deposited, tagged with a PXD identifier, and stored by PX repositories (PRIDE, PeptideAtlas, MassIVE, Panorama, iProX, JPOST). Data selection, extraction and re-analysis by PeptideAtlas and MassIVE results in processed data that is transmitted to neXtProt. Subsequently, neXtProt annotates and curates other biological data (like Sanger sequencing, protein : protein interaction and other structural/crystallographic data) that is aggregated, integrated and then disseminated to the community. The HUPO HPP KB uses reverse date versions (e.g., the latest 2020 neXtProt HPP reference release 17-01-2020).
Defining the human proteome at high-stringency
The HPP relies upon a coordinated system developed by neXtProt and UniProtKB, which attributes five levels of supporting evidence for protein existence (PE)6. Evidence at PE1 indicates clear experimental evidence for the existence of at least one proteoform, based on credible identification by MS, Edman sequencing, X-ray, nuclear magnetic resonance (NMR) structure of purified natural protein, reliable protein–protein interaction and/or antibody data. PE2 indicates evidence limited to the corresponding transcript (cDNA, reverse-transcriptase PCR, northern blotting). PE3 indicates the existence of orthologs in closely related species. PE4 refers to entries based on gene models without evidence at protein, transcript or homology levels. PE5 classifications indicate that coding evidence is doubtful and/or probably corresponds to an incorrect in silico translation of a non-coding element. As PE5 entries are largely non-coding, the HPP preferentially tracks only PE1,2,3,4 protein-coding entries. Proteins classified as PE2,3,4 are colloquially referred to as missing proteins7,8. Since 2013, neXtProt and the HPP (Fig. 1b) have issued annual tallies of PE1,2,3,4,5 status (www.nextprot.org/about/protein-existence), which have been reported in annual collaborative metrics manuscripts (ref. 9 and references therein).
The latest neXtProt HPP reference release (https://www.nextprot.org/about/statistics, data release: 17-01-2020) designates credible PE1 evidence for 90.4% of the human proteome (17,874 PE1s from the 19,773 PE1,2,3,4 protein entries excluding the dubious 577 PE5 entries). This leaves 1899 (9.6%) PE2,3,4 missing human proteome entries to be identified at high-stringency.
Here, the term high-stringency refers to rigorous HPP standards for post-acquisition processing and any protein inference made from raw MS peptide spectral data10. This term avoids confusion with the pre-existing term ‘high-accuracy’ frequently used in MS parlance, which relates to the generation of high (mass)-accuracy spectra from modern instruments.
The use of high-stringency influences the quality of all protein inferences derived from any raw MS data. The HPP routinely applies high-stringency protein inference analytics10, generated through the Trans-Proteomic Pipeline11. Claims for detection of new PE1s and/or detection of coding elements not previously in neXtProt, should meet minimum evidence thresholds. The current HPP guidelines10 require at least two uniquely mapping peptides (using neXtProt’s Peptide Uniqueness Checker Tool12), which are at least nine amino acid residues long. Peptides must be non-nested (i.e., one not fully contained within another) but may overlap partially so coverage exceeds >18 residues. The HPP also requires full declaration of false discovery rate (FDR) calculation procedures at peptide and protein levels, with a maximum allowable protein-level FDR of 1%. Within HPP missing protein publications, further validation is required through synthetic peptide spectra matching13. Going forward, the HPP also requests association of Universal Spectrum Identifiers with every MS spectrum10.
We emphasize that MS-based data from high-accuracy14,15 MS instruments combined with subsequent high-stringency protein inference analysis provide definitive best-practice confirmation of protein identification and abundance. To ensure high-quality analyses, increasingly stringent criteria have been applied10,16 and the latest HPP MS Guidelines v3.010 are designed to make spectral data Findable, Accessible, Interoperable and Reusable (FAIR)17.
Many previous studies use high (mass)-accuracy instruments with subsequent protein inference identifications undertaken at lower default settings, such as accepting single peptides or those only seven amino acids in length and/or not conforming to more rigid neXtProt proteotypic analysis18,19. These analyses can result in spurious identification of many more false positives20, with lower-quality single non-proteotypic spectra data colloquially referred to as one-hit wonders, better explained by sequence variation in other highly observed proteins21.
Chromosome-centric (C)-HPP
The C-HPP (https://www.hupo.org/C-HPP) aims to annotate all genome-encoded proteins7,8 in an unbiased and high-stringency manner. It explores proteins that have not previously been confidently observed by MS or other analytical methods7,8,22. International C-HPP teams are organized according to chromosomes (Chrs), namely Chrs 1–22, Chr X, Chr Y and mitochondrial (Mt) genome teams.
From 2017, the C-HPP expanded its mission to include functional characterization of the 1899 PE2,3,4 proteins that have not been confidently observed and 1254 PE1s that have no neXtProt curated function (uncharacterized PE1s or uPE1s), cumulatively referred to as the dark proteome23,24.
Biology/disease-centric (B/D)-HPP
The B/D-HPP (https://www.hupo.org/B/D-HPP) measures and interprets human proteome data under a range of physiological and pathological conditions. It focuses on the following: (i) elucidating the hallmark protein drivers of biology/disease and (ii) promoting development of new proteomics analytical tools such as Ab-based approaches and targeted selected/multiple/parallel reaction monitoring (SRM/MRM/PRM) assays.
As an example, the initial HUPO liver proteome project grew into a B/D-HPP team focussing on liver expression profiles, PTMs, tissue expression, subcellular localization, interactions, physiology and pathologies25. The Chinese CN-HPP have characterized four liver cell types, emphasizing benefits of acquiring cell-type specific maps to understand underlying biology/pathology26. In addition, they mapped landscapes of early hepatocellular and lung carcinoma, generating cancer subtype alterations where proteomic signatures identified poor prognosis patients and/or those benefiting from targeted therapy27,28. In other studies, they analysed microdissected cell types with gross anatomical resolution using MS26,29, revealing circadian cycles and spatio-temporal proteome expression in the liver, brain, heart and stomach30, providing resources to better understand organ biochemistry, physiology and pathology.
Significant discoveries continue to be made from all B/D-HPP teams across personalized cancer immunotherapy and therapeutic modalities (e.g., lymphoma31, ovarian32, liver27 and lung28 cancers), with PTMs orchestrating many outcomes including response to therapy33,34,35.
B/D-HPP resources include the Human SRMAtlas36, a unique compendium of high-resolution spectra and multiplexed SRM/MRM/PRM assays developed from 166,174 synthetic proteotypic peptides. This assay library enables targeted identification and quantification of a theoretical maximum of 99.7% of the human proteome36, provided proteins are expressed spatiotemporally at concentrations amenable to MS detection. For example, SRMAtlas supported the C-HPP Chr X team’s confident identification of missing proteins37.
HPP resource pillars
The B/D-HPP and C-HPP are supported by four HPP Resource Pillars that ensure effective data generation, integration and implementation, including the establishment of metrics and guidelines, enhancement of technology platforms, reagent development and optimal use of existing and emerging data streams (Fig. 1a).
The HPP MS Resource Pillar informs the community about MS technology/workflow advances, appropriate high-stringency standards and liaises with industry regarding instrument development, all leading to improved depth and accuracy of proteome identification, quantification and modification. These include methods like matrix-assisted laser desorption ionization time of flight (MALDI-TOF)-MS, electrospray-MS, bottom-up (shotgun) MS, data-dependent acquisition MS, data-independent acquisition (DIA) MS, targeted SRM/MRM/PRM, top-down MS, cross-linking MS, PTM analysis, N- and C-termini measurement, MS data computational analysis and interactomics.
The MS Resource Pillar previously undertook a SWATH/DIA-MS reproducibility study38 and are currently coordinating a phosphopeptide challenge involving >20 participating labs with partners SynPeptide Shanghai and Resyn Biosciences South Africa, who have provided a human phosphopeptide standard set with unphosphorylated counterparts, as peptide mixtures spiked into a yeast tryptic digest background. This will result in a better understanding of phosphopeptide enrichment, MS data analytics and informatics tools.
The HPP Ab Resource Pillar, ostensibly led by the Human Protein Atlas (HPA; www.proteinatlas.org), was initiated in 2003 and uses Ab-based strategies to analyse spatio-temporal aspects of the proteome39. Linking the identification of proteins with ‘real-time’ localization at tissue, cell and subcellular levels supports a more comprehensive understanding of biology, health and disease. This requires information at resolution not currently available by MS (see single-cell section below). Approaches for spatio-temporal proteomics include single-cell in situ MS, fractionated cell lysates, proximity labelling or imaging-based proteomics40,41. Imaging-based proteomics has a clear advantage, namely analysing proteins in their native location at single-cell resolution. To this end, the HPA has developed industrial scale epitope-directed Abs for community use.
HPA also integrates multi-omics data. It contains extensive transcriptome data and neXtProt PE assignments, and contributes to the open-access catalogue Antibodypedia, containing >4 M Abs (www.antibodypedia.org)42 against >19,000 targets that assist the community to select application-appropriate Abs.
At HPP launch, the HPA had detected >50% of the protein-coding genome43. Currently, ~87% of the proteome is targeted by >1 HPA Ab, detected though an encyclopaedia of >10 M annotated high-resolution digital images, partitioned into a number of sub-atlases that are interconnected44,45,46,47,48. These currently comprise the; Tissue Atlas (protein distribution across all major tissues), Cell Atlas (subcellular localization and heterogeneity in single cells), Pathology Atlas (correlations between gene expression and patient survival in major human cancer types), Blood Atlas (protein profiles across major immune cells and blood levels), Brain Atlas (protein distribution in the brain), and the Metabolic Atlas (various tissue metabolic enzymes localizations). Over the decade, HPA’s open-access database44,45,46,47,48 has become one of the world’s most visited biological resources (>3.6 M visits in toto annually).
The HPA also plays an emerging role in establishing guidelines around the appropriate use of Abs and ensuring immunoassay validation49,50,51. It recently spent considerable effort validating the selectivity of their Abs, including championing efforts of the International Working Group for Ab Validation proposing many new approaches implemented across >10,000 Abs42. In addition, HPA’s massive collection of images has supported a multitude of publications and become a citizen science resource for developing AI classification learning models52,53. The HPA is sustained through community contribution to ELIXIR54, that allows scientists from academia and industry to explore spatio-temporal aspects of the human proteome55,56,57.
Since 2018, the HPP Pathology Resource Pillar has coordinated identification of areas of unmet clinical need, develops fit-for-purpose clinical assay guidelines/standards, promotes best-practice awareness, coordinates access to quality clinical samples/metadata and liaises with pathology organizations, diagnostic companies and regulatory agencies to promote professional application of proteomics in pathology.
The HPP KB Resource Pillar captures, collects, collates, analyses and re-distributes all human proteome data. As cohesive knowledge transfer plays such a crucial role in big data science, including the HPP, the KB pillar’s activities are addressed in the expanded section that follows.
The human proteome in the neXtProt HPP reference KB
Assembly and curation of neXtProt
Prior to the HPP, HUPO established a Protein Standards Initiative (PSI; www.hupo.org/Proteomics-Standards-Initiative), emphasizing from the outset their priority for defining high-quality community standards, minimal requirements for experimental information58 and high-stringency data metrics. PSI continues to work cooperatively with the HPP KB to inform HPP initiatives, pillars and teams.
In 2013, neXtProt59,60 was officially designated as the HPP reference KB61. Annually, a neXtProt release is designated as the ‘HPP release’ and this serves as the basis for subsequent HPP high-stringency analyses, planning and reporting progress10. It receives and curates data from UniProtKB/SwissProt62, adding MS evidence from PeptideAtlas63 and since 2019 from MassIVE64. neXtProt also curates Ab-based, genome, transcriptome and other biological data to create an assembled snapshot of the human proteome6,65.
PeptideAtlas (http://www.peptideatlas.org) uses sequence search engines Comet66, X!Tandem67 and SpectraST68 to reprocess publicly uploaded MS/MS data deposited through ProteomeXchange (PX). Data are aggregated using rigorous criteria including peptide spectral matching with FDR ~ 0.0009% in the latest PeptideAtlas build to achieve ≤1% FDR at the protein level. MassIVE searches public datasets using the MS-GF+ search engine69, also with strict criteria70 to enforce global <1% FDR at the protein level for single-peptide identifications and stricter <0.01% FDR for proteins identified by >2 peptides. PeptideAtlas and MassIVE peptide lists are integrated into current neXtProt builds. neXtProt then cross-references all peptides to protein entries and validates PE levels, requiring at least two MS-identified uniquely mapping 9-mer non-nested peptides coming from either PeptideAtlas or MassIVE.
neXtProt builds on UniProtKB/SwissProt PE1 entries that include MS, partial or complete Edman sequencing, X-ray or NMR structure, reliable protein–protein interaction data or Ab detection by considering additional PeptideAtlas/MassIVE data that meets minimum peptide uniqueness, number, length, nestedness and other requirements to upgrade entries to PE1. Noticeably, neXtProt is becoming more reliant on MS than non-MS data (e.g., 1860 non-MS data PE1s in 2016 down to 950 in 2020).
To ensure only high-quality Ab data are used, in 2018 neXtProt/HPP Ab pillar revised criteria to upgrade entries to PE1, including specificity and other rigorous criteria suggested by the International Working Group for Ab Validation50. As an example, neXtProt recently analysed 41 Ab-based publications and upgraded three PE2,3,4 entries to PE1. Discussions regarding data provenance delivered by non-MS data for PE1 assignment actively continues in the HPP71.
Expansion of the high-stringency proteome
The baseline number of protein-coding genes in the reference proteome given by neXtProt is managed by UniProtKB/SwissProt. Every protein-coding gene is assigned a protein entry (inclusive of all proteoforms), with chromosome location and other data organized under these entries. The number of protein-coding genes originally estimated by the HGP dropped from >100,00072 to a relatively stable ~19,700 at HGP draft release, a number that is close to the current neXtProt’s 2020 release of 19,773 protein-coding genes (Supplementary Fig. 1)60.
Confident detection of the human proteome has consistently risen from 69.8% neXtProt PE1 entries in 2011 to 90.4% in 2020 (Fig. 2a). However, progress has recently slowed (Fig. 2a), suggesting that it may be difficult to confidently identify all 1899 missing proteins. Parenthetically, the HGP celebrated a provisional 90% completion ten years after its launch5 and annotation of the human genome still remains incomplete or uncertain, especially in regions of repeat sequences and Z-DNA inserts.

a Annual neXtProt HPP evidence of protein existence (PE1,2,3,4,5) metrics from 2010 to 2020. This data demonstrates a strong and progressive increase in PE1 identifications across the decade (13,588 in 2011 to 17,874 in 2020), correlative equivalent decrease in PE2 (5,696 to 1596), a post-2015 rise in PE3 coincident with revised guideline implementation (239 to 253) and decrease in PE4 identifications (90 down to 50). PMS Pantone colours employed match in the figure match for all past annual neXtProt HPP KB reference PE1,2,3,4,5 data release colours, namely PE1: light green, PE2: teal, PE3: yellow, PE4: orange, and PE5: red). b Decadal Sankey diagram of changes in PE1,2,3,4,5 status of neXtProt entries between 2011 and 2020, where arrow widths are proportional to the number of decadal PE entries that change category. This Sankey diagram displays fluidity in PE status of neXtProt entries. PMS Pantone colours match those used for all past annual neXtProt HPP KB reference PE1,2,3,4,5 data releases https://www.nextprot.org/about/protein-existence (i.e., PE1: light green, PE2: teal, PE3: yellow, PE4: orange and PE5: cerise). All neXtProt protein entries that were deleted or newly introduced during the decade are represented in black, noting that 432 neXtProt entries were deleted and 676 introduced. Sankey analysis demonstrates that 2011 PE2 entries were the most significant (but not exclusively) the source for the majority of additional 2020 PE1s. Year-by-year transition data can be found in metrics publications associated with annual (2013–2019) HPP special issues9 and refs therein, guided by high-stringency HPP Guidelines10.
In each of the seven annual HPP metrics papers published to date (9 and refs therein), PE1,2,3,4,5 entries have been added, deleted, renamed, merged and/or de-merged, indicating ongoing fine-tuning of the HPP reference proteome. The Sankey flow diagram73 (Fig. 2b) illustrates that significant fluidity has occurred across all PE classifications since 2011. The largest shifts occur with increases in PE1s (13,603 up to 17,874), followed by decreases in PE2s (5,587 down to 1,596), despite increasingly stringent HPP MS data guidelines adopted in 2015 (v2.1) and 2019 (v3.0). Linear regression of PE1 increases against PE2 decreases results in a strong (R2 = 97%) inverse correlation, suggesting new PE1s come from PE2s where mRNA expression has been previously observed. Extrapolation of this PE1 discovery curve suggests that 95% PE1s may be reached sometime between 2024 and 2027. Minor conversions occurred between other PE categories, with downgrading of PE1 or PE2s and upgrading of PE4s, and even a few PE5s, to PE1s. Although a plethora of studies show low linear correlations (40–60%) between mRNA level and protein abundance74,75, our binary data (Fig. 2) supports the contention that once a neXtProt entry has mRNA expression verified, those PE2s are amenable to upgrade to PE1, whereas PE3s, PE4s and particularly PE5s are more resistant to PE1 upgrade.
Missing protein analyses
neXtProt protein descriptors and associated data can be used to analyse protein groups/families according to their Chr location or PE classification. Missing protein (PE2,3,4) analysis indicates that some groups/families have been upgraded to PE1 more successfully than others (Fig. 3a). For example, between 2011 and 2020, 372 zinc (Zn) finger proteins, 171 transmembrane proteins, 93 carbohydrate metabolism proteins, 90 testes-, sperm-, prostate-associated proteins, 78 coiled-coil domain-containing proteins and 58 homeobox proteins have been upgraded from PE2,3,4 to PE1. These represent the six most prominent protein groups upgraded to PE1 over the decade (Fig. 3a, green). In contrast, two G protein-coupled receptor (GPCR) chemosensory families prove particularly resistant to PE1 upgrade: many olfactory receptors (ORs) are still missing in 202076 (417 of the 2011 PE2,3,4 entries, including putative and uncharacterized ones) and 17 of 20 taste receptors remain PE2,3,4 s (Fig. 3a, magenta). In addition, some groups with a large number of PE1-upgraded protein entries still contain many PE2,3,4 entries (e.g., 85 non-GPCR transmembrane, 69 Zn finger and 33 keratin-associated proteins remain PE2,3,4 s (Fig. 3a).

a The top 15 neXtProt protein descriptor groups/families with the highest number of 2020 PE2,3,4 missing protein members (i.e., lacking high-stringency PE1 evidence of protein existence data; magenta, left) and the top 15 protein descriptor groups that have been upgraded to PE1 since 2011 (green, right). The data illustrates that the OR family has the highest number of 2020 missing PE2,3,4 proteins (magenta bars) and the Zn finger protein family has the highest number of discovered PE2,3,4 entries upgraded to PE1 since 2011 b Human chromosomal distribution of the OR and Zn finger families neXtProt protein descriptor groups/families. This example data clearly illustrates that the positioning of multiple ORs (magenta vertical bars) or Zn finger protein-coding genes (green vertical bars) on certain chromsomes explains why Chr 11 appears more resistant and Chr 19 more susceptible to PE1 discovery over the decade.
When Chr distribution of the most resistant (ORs) or most discovered (Zn-finger proteins) groups were plotted (Fig. 3b), difficult-to-find ORs mapped mostly to Chr 11 (~55%) and Zn-finger proteins mapped mostly to Chr 19 (~55%). Our data demonstrate that 46% of all Chr 19 proteins elevated to PE1 were Zn-finger family members, illustrating this protein family has been highly amenable to confident MS detection over the decade. Therefore, it was not surprising that most Zn-finger protein members were coded on Chr 19 (i.e., 255/698 total Zn-finger genes), which has the highest Chr PE1 reclassification rate over the decade (Fig. 3b).
These data suggest that gene family duplication on particular Chrs explains why some families are resistant or sensitive to PE1 reclassification. In agreement, PE1 discovery has occurred productively, but not uniformly, across all chromosomes (Fig. 4). Missing protein decadal upgrade statistics range from 16% for Chr Y up to 29% for Chr 1, with raw data representing 425 protein entries for Chr 1 down to only 5 for Chr Y. A higher percentage of Chr 19 proteins (29%) ascended to PE1 compared to those on Chrs 11, 14, 21 or Y (<17%). The HPA chromosome viewer (www.proteinatlas.org/humanproteome/proteinevidence) illustrates many recently-evolved protein family members are present on Chr Y, many ORs on Chr 11 and many keratin-associated proteins on Chr 21. Proteins on these three Chrs have proven relatively resistant to PE1 reassignment. Notably, there was just a single Mt missing protein in 2011 and over the decade all Mt protein-coding genes have now been identified.

a The percentage of missing proteins (PE2,3,4) relative to all protein-coding genes (PE1,2,3,4) plotted annually according to human Chrs 1–22, X and Y location from the first neXtProt release (23-08-2011) to the latest HPP reference release (17-01-2020). b The relative percentage (magenta bars) and absolute number (green dots) of all neXtProt PE2,3,4 missing protein entries specifically upgraded to PE1 since 2011 across Chrs 1–22, X and Y.
PE1 upgrade success by the 25 C-HPP teams could be pre-determined by the presence of resistant or more easily-identified missing protein families on particular Chrs. However, the current HPP KB processing pipeline utilizing PeptideAtlas, MassIVE and neXtProt (Fig. 1b) makes it impossible to isolate decadal PE1 contributions from any particular C-HPP team as opposed to the overall community. Although the HPP explores capturing full data provenance (i.e., from quantification/identification back to original data source) for FAIR data practices17, we can only historically estimate PE1s emanating from community deposition.
In silico analysis reveals only 22 human proteins cannot produce the characteristic >2 high-stringency proteotypic peptides of the required length after tryptic digestion36. However, many missing proteins may be present at levels below detection limits or in under-studied cell types/tissues, expressed under particular conditions (e.g., stress/infection) or only found in developmental stages (e.g., embryo/fetus)77. Equally, difficult-to-solubilize, hydrophobic multi-transmembrane domain membrane proteins may only generate short tryptic peptides that do not meet high-stringency guidelines or are indistinguishable from other family member sequences76. Furthermore, transmembrane protein regions cut at single sites are unlikely to release embedded hydrophobic membrane-anchored protein strands76.
We anticipate that finding the 9.6% remaining PE2,3,4 missing proteins will require exceptional future effort, including careful sampling of rare cells/tissues78 combined with better sample fractionation and improved detection limits. Low abundance proteins might be enriched using Abs prior to MS. To this end, the HPA has developed Abs against proteotypic sequences in many missing proteins79. Several other labs are working on improved protocols for insoluble keratin-associated cross-linked missing proteins, non-tryptic or chemical digestion strategies to increase proteotypic peptide productivity78,80, higher efficiency search engines81, and compendia of missing protein biological evidence (e.g., MissingProteinPedia)71. Future HPP projects anticipate a shift to detection of biologically functional proteoforms82, noting their numbers are far larger and more difficult to measure83 because of heterogeneous nuclear RNA splicing, many PTMs and detection of peptides with single amino acid variants (SAAVs). Considerable PTM and splicing isoform data are already available through neXtProt, including 190,938 PTM sites and 9,193,365 SAAVs84.
Community impact on the human proteome
One barometer of community engagement in the HPP is the magnitude of investigator-submitted raw MS data that have been re-analysed. Many journals require raw data submission and HPP action was a significant factor in journals adopting requirements aligned to HPP data deposition guidelines. Raw data deposition occurs through PX, which registers and standardizes capture/dissemination of public MS data from partner repositories, including founders PRIDE62 and PeptideAtlas and recent members MassIVE64, jPOST85, iProX86 and Panorama Public87. As of 2020, a total of 4634 human MS datasets have been received. Each PX dataset is branded with a unique PXD identifier with depositors, publications and voluntary metadata noted88,89. Illustrating the magnitude of this community data, ~470 TBs of data have come from 5658 human datasets (~47% of 1 petabyte PRIDE volume), with only 358 (6.3%) of these specifically tagged by depositors as from the HPP. The HPP encourages raw human MS data/metadata submission (including association to HPP) through PX, and that journals request that PXD identifiers be published in accordance with FAIR principles17 as discussed above.
To provide an additional measure of global scientific impact, HUPO commissioned the website construction of the Human Proteome Reference Library (HPRL; https://hupo.org/HPP-HPRL/), where all HPP-associated PubMed searches are hyperlinked and can be accessed and re-run routinely by the community. These hyperlinked searches automatically produce the latest PubMed outputs in a manner where all PubMed filtering, ranking and timeline tools can be applied subsequently by a user as required. As an example of the fascinating data unearthed, a ‘human proteome project’ search showed that the structural biologists Montelione and Anderson first suggested the possibility of building a HPP in their 1999 Nature Structural Biology publication90—well before HUPO or the HPP began.
The HPRL can also be used to measure impact (Fig. 5), where PubMed identifiers (PMIDs) from 2010 to 2019 references can be captured using web APIs or software, e.g., NCBI E-utilities like https://www.ncbi.nlm.nih.gov/books/NBK25501 or the easyPubMed R interface (www.rdrr.io/cran/easyPubMed) that enable extraction and aggregation of PubMed bibliometric records. Paralleling the low percentage of PX datasets tagged as emanating from the HPP, HPRL data show only ~1000 PubMed title and abstract searches that specifically and cumulatively mention either the terms HPP, C-HPP and/or B/D-HPP (or their unabbreviated counterparts) out of a total of more than 50,000 ‘human AND proteomics’ title/abstract search outcomes over the decade. In contrast to this and more reassuringly, HPRL searches of the community’s biology/ or disease studies covered by nine of the B/D-HPP teams generated >5000 publications each since 2010 (Fig. 5b), with ‘cancer proteomics’ topping rankings at around 20,000 publications. Although this may seem surprising, a PubMed search of ‘human genome project’ produced a similarly modest number of ~1900 hits during the HGP’s first decade. To visualize outputs from various HPP teams and the proteomics community in general, we have employed VOSviewer91 to construct and visualize bibliometric networks from collective PubMed searches (example shown in Fig. 5c). This particular analysis revealed a remarkable number of highly-interconnected relationships that have evolved between HPP teams, pillars and initiatives—providing evidence for the impressive level of established international collaboration developed over the decade.

Data show cumulative PubMed search references emanating since HPP launch in 2010 up until 2019. a PubMed search for the terms HPP, C-HPP and B/D-HPP (including unabbreviated version). b PubMed community-at-large bibliometric impacts that parallel the research disciplines (e.g., ‘human’ AND ‘cancer proteomics’) addressed and undertaken by key B/D-HPP teams. All B/D-HPP PubMed bibliometric searches are listed as full searches and as hyperlinked current PubMed searches on the HUPO website at https://hupo.org/HPP-HPRL/. All NCBI PubMed filters and tools are fully accessible to users and searches can be selected and modified in a user-friendly manner, allowing decadal (from 2010 to 2019) and other bibliometric analyses to be undertaken routinely. c VOSviewer HPP collaborations analysis. All co-author geographical affiliations for PubMed publications emanating from Fig. 5a were transposed onto a world map.
Translating proteomics to precision medicine
A key aspect of biomedical research lies in translating discovery into clinical use. Protein assays remain a cornerstone of diagnostics. Although individual proteins can be measured diagnostically with high precision (i.e., sensitivity and specificity), some assays suffer low specificity due to cross-reactivity with interfering substances including autoantibodies (e.g., thyroglobulin immunoassays). Modern SRM/MRM/PRM assays allow multiple proteins to be measured simultaneously, accurately, sensitively and with high specificity. In addition, the use of liquid chromatography MS with immunocapture assays has been reported to eliminate interferences92. Moreover, as most diseases are heterogeneous and multigenic, it is likely that multiplexed proteomic or multi-omics panels will achieve higher accuracy (e.g., optimized biomarkers for ovarian malignancy with adnexal masses93). The HPP assists in the development of proteomic educational programmes with pathology societies to train the pathology community on the potential impact of proteomic technologies.
Below, in recognition of the impact human proteomics can and is having in precision medicine, we highlight examples demonstrating the role of the HPP and proteomics in tackling contemporary medical grand challenges.
Cancer precision medicine
As mentioned above, PubMed extracts ~20,000 published human cancer proteome studies since 2011 (Fig. 5b). Although genomics can routinely determine high-risk, predisposition and aspects related to tumour burden and recurrence, effective targeted cancer treatment is still not available for all cancers. For example, systematic genome-wide studies like the Pan-Cancer Analysis integrated analyses of >2600 whole genomes from 38 tumour types with matching normal tissues, uncovered many cancer-associated genes94, chromosome rearrangements, some unknown drivers but few new therapeutic targets. This is mainly because mutations do not automatically cause predicted changes in the proteome, making it difficult to establish which changes are crucial biochemical drivers from those that are not.
Integrating genomic and proteomic data (i.e., proteogenomics) has the potential to provide insights into causes and mechanisms underlying diseases, including the hallmarks of cancer biology95. This can facilitate the implementation of effective therapeutic intervention. The value of a proteogenomic analysis of functional consequences of cancer somatic mutations has assisted in narrowing down candidate driver genes within large deletions and amplified regions96. Reviewing the underlying causes of breast cancer also demonstrates that coupling genomic/transcriptomic data with proteomic/phosphoproteomic analysis was more insightful than any individual approach. Of note, melanoma tumour genomic BRAF driver mutations match corresponding protein sequences97, illustrating that proteomic landscapes add value to genomic data, when considered with patient tumour histopathology and clinical metadata98.
Proteomics substantially benefits a comprehensive understanding of precision medicine. To illustrate this, NCI’s Clinical Proteomic Tumor Analysis Consortium (CPTAC; proteomics.cancer.gov) in collaboration with the B/D-HPP cancer team, established guidelines, data sharing, and standards in analytical and computational workflows to ensure rigour in designing and performing research99,100,101,102,103. CPTAC applied these standards and workflows to tumours previously genomically characterized by The Cancer Genome Atlas. In doing so, CPTAC pioneered the integration of proteomics with genomics (i.e., proteogenomics) to produce a more unified and comprehensive understanding of cancer biology and implemented its transition into cancer clinical research studies96,104,105. Largely due to these efforts, NCI hosts open-access repositories of unified proteogenomics datasets, assays and reagents, including the Proteomic Data Commons (pdc.cancer.gov), a fit-for-purpose targeted assay site (assays.cancer.gov)102,106 and an Ab portal (antibodies.cancer.gov)107. These activities empowered the recent creation of the International Proteogenome Consortium (ICPC; icpc.cancer.gov)108. Collectively, CPTAC and ICPC collaborators have comprehensively characterized 13 cancer types at the proteogenomics level, with all datasets publicly accessible109,110,111,112,113,114,115,116,117.
Cardiovascular diseases
Cardiovascular disease (CVD) research is challenged by daunting structural heterogeneity and molecular complexity. For example, cardiac circuitry function/dysfunction cannot be reduced to differentially expressed single genes. On the other hand, proteogenomics allows assessment of interactions, pathways and networks and informs diagnosis and therapy of multifactorial CVDs. Over the decade, CVD proteomics has broadened from identifying single canonical proteins to mapping proteoforms derived from combinations of alternative splicing, cleavage, and PTMs33,83. Now, identification of proteoforms (e.g., genetic variations, alternatively spliced products, phosphorylation118, glycosylation119, oxidative120 and other PTMs121) allow CVD sub-classification. In parallel, the heart is uniquely sensitive to alternative splicing (frequently altered in congenital heart diseases), explaining the B/D-HPP’s interest in developing assays for splice isoform-specific changes in cardiomyocyte development and maturation122,123.
Many technological developments have been prominent, including phospho-PTM analysis to identify PDE5A targets in heart failure therapy118 and proximity labelling to assess protein–protein interactions involved in β-adrenergic signalling of contractility in cardiac fight-or-flight responses124 and evaluation of regenerative stem cell therapy efficacy in post-infarct hearts125. Proteomic studies have also addressed the kilometres of vascular beds and extracellular matrix responsible for transporting blood, giving valuable insights into the molecular anatomy of aneurysms and atherosclerosis126,127. Targeted MS methods have been developed, again especially where interferences obfuscate immunoassays128 or where additional biological context is required129. Likewise, volumetric absorptive micro-sampling VAMS blood collection devices (e.g., Mitra devices or dried blood spots) allow patients to mail samples from home to analytical labs to undertake SRM/MRM/PRM assays quantifying CVD risk-associated apolipoproteins130 or other markers131. In summary, consumer-based CVD proteomics-based precision medicine testing services are now coming of age.
Microorganism detection
Proteomics and the HPP have made fundamental contributions to understanding pathogenic infection, providing diagnostics and developing therapies132. The B/D-HPP Infectious Diseases team promotes international proteomics collaborations investigating viral, bacterial, fungal and parasitic diseases.
MALDI–time-of-flight (TOF)–MS, once considered revolutionary, is now established as a routine tool in clinical microbiology133. Classical phenotypic tests identify unknown and potentially pathogenic microorganisms, but may require incubation for several days, with misidentification resulting in adverse treatment consequences. MALDI–TOF–MS provides significantly shortened analyses (now minutes) with improved accuracy on single colony or bacterial pellets for difficult-to-detect microorganisms, using automated spectra acquisition and extensive reference spectra databases134. Minor spectral differences enable typing below species levels135, allowing subspecies identification through epidemiological analyses. Bacteria and yeasts (most clinical identifications), mycobacteria136 and moulds137 can now be identified accurately and rapidly. Further MS clinical diagnostic applications are being investigated (e.g., antibiotic resistance (ART) and susceptibility testing (AST) based on hydrolytic β-lactamase activity138), with kits under development commercially through STAR-Carba, STAR-Cepha and Bruker Daltonics.
SARS-CoV-2 virology
The recent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak that causes COVID-19 disease represents a major threat to human health and our economies139,140,141. The pandemic underscores our need to understand virus pathobiology, identify host-pathogen interactions that support replication, find biomarkers correlative with clinical outcome and expand surveillance.
Many omics studies followed the 2003 SARS-CoV-1 and related MERS and IBV coronavirus outbreaks142,143,144,145,146. The cell surface receptor for the CoV-1 and CoV-2 surface spike protein has been identified by affinity-MS to be angiotensin-converting enzyme 2 (ACE2)147, which in a recent large-scale study based on antibody-based proteomics was shown to be mainly localized to the digestive system, kidney, heart, testis, placenta, eye and upper respiratory epithelia148. Virus binding leads to proteolysis by the transmembrane serine protease TMPRSS2 expressed in airway epithelia149, thus a clinically approved TMPRSS2 inhibitor (camostat mesylate) is being investigated to block infections150,151. Furthermore, proteomics has characterized the infectious CoV-1 viral particle143, temporal changes in host cells during infection142 and virus-induced endoplasmic reticulum membrane remodelling into double-membrane vesicles152,153 that house viral replication compartments146. Proximity labelling revealed >500 host and 14 viral protein associations with the viral replicase NPS2146, highlighting vesicular trafficking, autophagy and splicing proteins in coronavirus replication, which if shown to also be the case for CoV-2, indicate potential drug targets.
Building on this knowledge of coronavirus infection, recent proteomics studies have focused on SARS-CoV-2, uncovering additional potential therapeutic targets154,155. MS and array-based proteomics serology has screened for potential biomarkers and Abs against infection156,157. Clinical isolate infection models have been developed using Caco-2 cells154 with temporal proteome changes identified during infection using multiplexed MS by combining metabolic labelling with tandem mass tagging methods. Consistently, host vesicular trafficking, translation, RNA splicing, nucleotide synthesis and glycolysis pathway proteins were upregulated following infection143,144 and targeting these processes with inhibitors revealed potential therapeutic targets158,159. Additionally, affinity-MS interactome studies examined 26 of 29 total SARS-CoV-2 proteins expressed within HEK293T human cells155, suggesting 69 existing drugs merit further investigation. Moreover, a recent phosphoproteome analysis pointed to the regulation of viral proteins through PTMs160.
The SARS-CoV-2 pandemic highlights the need for applying proteomic approaches to the development of serologic testing and preclinical and computational model systems to evaluate patient responses to infection156. Serological biomarkers of asymptomatic/symptomatic infection, disease severity, risk of re-infection and/or vaccine efficacy are being characterized157,161. In addition to this accumulating omics knowledge, many aspects of SARS-CoV-2 pathobiology await further exploration including development of additional methods for clinical virus detection, identification of infection stage, and an in-depth understanding of functional spatio-temporal virus–host protein interactions and organelle remodelling162,163,164. For example, recent studies that utilize targeted MS for SARS-CoV-2 protein detection and proteomic characterization of serological immune responses161,165,166,167 from patient samples may bolster PCR screening for the assessment of disease severity168. Other proteomics approaches can also be deployed to further expand the understanding of SARS-CoV-2 biology. Among these is the application of TAILS N-terminomics that promises to identify many SARS-CoV-2 protease substrates and those cellular pathways inactivated by viral proteolysis, as reported for other viruses169.
In summary, proteomics plays increasingly important roles in understanding viral outbreak biology, accurate diagnosis and effective treatment and is positioned to continue to co-ordinate and drive international collaborations towards these goals.
Conclusions and future directions
Western and Eastern cultures urge us to know thyself and thy enemy. These axioms resonate with precision medicine where future benefits arise from a detailed omics understanding of the hallmarks of health and disease. Here, we reviewed the construction of a community-endorsed, high-stringency blueprint of the human proteome. The decadal neXtProt HPP PE metrics shown here demonstrate the community’s progressive success in PE1 identification from 13,588 in 2011 to 17,874 PE1s in 2020, marking the completion of >90% of the human proteome parts list (see strategic objective 1 above). We also present specific examples demonstrating proteomics will be an integrated component (with genomics and other omics) in future biomedical science discovery and precision medicine.
HUPO recommits to its original HPP strategic aims as well as the FAIR data principles17, while anticipating the following future priorities:
-
1.
Unearth credible proteomics data for the majority of current PE2 proteins: Since most PE1 identifications come from former PE2s, our future strategy is to find credible data for 95–99% of current PE2s, allowing reclassification of these to PE1. PE3 also remain promising, as homologous proteins are detected in related species.
-
2.
Unravel currently unknown proteome functionalities: Fill functional annotation gaps for all protein-coding elements, with a priority on credibly identified proteins170 and develop, expand and apply function prediction tools24,171,172.
-
3.
Expand the HPP KB: Maintain a sustainable knowledge-transfer HPP KB infrastructure with funding that captures/displays high-stringency partner omics data streams and publication data (HPRL) to researchers and the public in an accessible and compelling manner.
-
4.
Develop community-approved multi-omics technology guidelines: Explore DIA-MS, Ab and aptamer-based multiplexed assays, top-down MS and other not yet invented multi-omics technologies.
-
5.
Champion collaborative multi-omics health/disease approaches: In addition to extensions in HPP KB partnerships, the HPP will collaborate with international and regional initiatives (including Human Variome, Human Cell Atlas, hPOP/iPOP, MoTrPAC, HuBMAP, Cancer Moonshot, HTAN, EDRN, CPTAC, ICPC and upcoming international initiatives) around multi-omics approaches to human disease processes, biomarker discovery and therapeutic development.
-
6.
Apply and improve single-cell proteomic technology: Develop technologies that allow detection and quantification of proteomes in single cells to further understand cellular/tissue heterogeneity, differentiation, diseases and the intrinsic biological noise in health and disease173,174. Many single-cell proteomics advances will be explored to analyse cellular heterogeneity175,176,177,178. Sensitivity increases (analogous to PCR) and trade-offs maximizing coverage per cell with throughput/accuracy will be studied179.
-
7.
Champion dual Ab-capture with MS identification: Enhance the accuracy of Ab-based epitope/antigen detection by confirmation using high-accuracy, high-stringency MS identifications across real-life spatio-temporal biological settings. Collectively, there is recognition within the HPP that future stringent co-registration of MS with Ab data are required to achieve the ultimate spatio-temporal human proteome expression atlas180.
-
8.
Exploit massively parallel MS174 to increase throughput: For rapid, sensitive and higher-throughput MS analysis.
-
9.
Capitalize on human disease biobanks: The HPP will work with biobanking consortia to improve access for proteomics researchers to highly curated and accurately annotated clinical samples collected in a standardized manner.
-
10.
Encourage higher levels of community engagement: The HPP will continue to reach out to the community, supporting findable, accessible, interoperable and reusable (FAIR) data principles17, while encouraging and appropriately recognizing all contributions to the re-analysis of proteomics data.
The post SARS-CoV-2 pandemic world will be different. It is likely that new paradigms to accelerate precision medicine will emerge. These will undoubtedly involve global collaboration (even between competing entities) using multidisciplinary approaches that enable the fast-tracking of novel diagnostic tests and precision therapeutics. Almost certainly these outcomes will require knowledge involving the human proteome—celebrated here in the inaugural HPP High-Stringency Blueprint.
References
Humphery-Smith, I. A human proteome project with a beginning and an end. Proteomics 4, 2519–2521 (2004).
Baker, M. S. Building the ‘practical’ human proteome project—the next big thing in basic and clinical proteomics. Curr. Opin. Mol. Ther. 11, 600–602 (2009).
Rabilloud, T., Hochstrasser, D. & Simpson, R. J. Is a gene-centric human proteome project the best way for proteomics to serve biology? Proteomics 10, 3067–3072 (2010).
Legrain, P. et al. The human proteome project: current state and future direction. Mol. Cell. Proteomics. 10, M111.009993 (2011). First HPP publication after launch, outlining systematic global effort to map the human proteome (abundance, distribution, temporal-spatial and subcellular localisation, interactions and function) and decribing reource pillars (MS, Ab and KB), with a biologically-driven and a chromosome-based mapping initiatives to deliver a protein parts list, reagents and tools. HUPO urged funding agencies and the community to participate in identifying pathways.
Pennisi, E. Human genome. Finally, the book of life and instructions for navigating it. Science 288, 2304–2307 (2000).
Lane, L. et al. Metrics for the human proteome project 2013-2014 and strategies for finding missing proteins. J. Proteome Res. 13, 15–20 (2014).
Paik, Y. K. et al. Standard guidelines for the chromosome-centric human proteome project. J. Proteome Res. 11, 2005–2013 (2012).
Marko-Varga, G., Omenn, G. S., Paik, Y. K. & Hancock, W. S. A first step toward completion of a genome-wide characterization of the human proteome. J. Proteome Res. 12, 1–5 (2013).
Omenn, G. S. et al. Progress on identifying and characterizing the human proteome: 2019 Metrics from the HUPO human proteome project. J. Proteome Res. 18, 4098–4107 (2019). 2019 annual HPP metrics publication (NeXtProt 2019-01-11) reporting progress in credibly identifying and characterising the human proteome.
Deutsch, E. W. et al. Human proteome project mass spectrometry data interpretation guidelines 3.0. J. Proteome Res. 18, 4108–4116 (2019). Details current high-stringency MS guidelines criteria used by the HPP for PE1 status (evidence for protein existence), where only protein entries with two or more neXtProt uniquely-mapping, non-nested peptides with length 9 amino acids or greater are deemed to have sufficient evidence to be labelled as confidently detected PE1 protein entries.
Deutsch, E. W. et al. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteom. Clin. Appl. 9, 745–754 (2015).
Schaeffer, M. et al. The neXtProt peptide uniqueness checker: a tool for the proteomics community. Bioinformatics 33, 3471–3472 (2017).
Vandenbrouck, Y. et al. Looking for missing proteins in the proteome of human spermatozoa: an update. J. Proteome Res. 15, 3998–4019 (2016).
Zubarev, R. & Mann, M. On the proper use of mass accuracy in proteomics. Mol. Cell. Proteomics 6, 377–381 (2007).
Mann, M. & Kelleher, N. L. Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl Acad. Sci. USA 105, 18132–18138 (2008).
Deutsch, E. W. et al. Human proteome project mass spectrometry data interpretation guidelines 2.1. J. Proteome Res. 15, 3961–3970 (2016).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Kim, M. S. et al. A draft map of the human proteome. Nature 509, 575–581 (2014).
Wilhelm, M. et al. Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 (2014).
Ezkurdia, I., Vazquez, J., Valencia, A. & Tress, M. Analyzing the first drafts of the human proteome. J. Proteome Res. 13, 3854–3855 (2014).
Mendoza, L. et al. Flexible and fast mapping of peptides to a proteome with ProteoMapper. J. Proteome Res. 17, 4337–4344 (2018).
Paik, Y. K. et al. The chromosome-centric human proteome project for cataloging proteins encoded in the genome. Nat. Biotechnol. 30, 221–223 (2012).
Paik, Y. K., Omenn, G. S., Hancock, W. S., Lane, L. & Overall, C. M. Advances in the chromosome-centric human proteome project: looking to the future. Expert Rev. Proteomics 14, 1059–1071 (2017).
Paik, Y. K. et al. Launching the C-HPP neXt-CP50 pilot project for functional characterization of identified proteins with no known function. J. Proteome Res. 17, 4042–4050 (2018).
He, F. Human liver proteome project: plan, progress, and perspectives. Mol. Cell. Proteomics 4, 1841–1848 (2005).
Ding, C. et al. A cell-type-resolved liver proteome. Mol. Cell. Proteomics 15, 3190–3202 (2016).
Jiang, Y. et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 (2019).
Xu, J. Y. et al. Integrative proteomic characterization of human lung adenocarcinoma. Cell 182, 245–261.e17 (2020). Integrated multiomics example (proteome, phosphoproteome, transcriptome, and whole-exome sequencing) analyses leading to clinical outcomes, performed on 103 lung adenocarcinomas, revealing many cancer-associated features (e.g., tumour-associated variants, patient clinical outcome correlation with EGFR or TP53 mutational status, proteomic stratification of lung cancer subtypes and potential drug targets.
Ding, C. et al. Proteome-wide profiling of activated transcription factors with a concatenated tandem array of transcription factor response elements. Proc. Natl Acad. Sci. USA 110, 6771–6776 (2013).
Ni, X. et al. A region-resolved mucosa proteome of the human stomach. Nat. Commun. 10, 39 (2019).
Khodadoust, M. S. et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 543, 723–727 (2017).
Schuster, H. et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl Acad. Sci. USA 114, E9942–E9951 (2017).
Fert-Bober, J., Murray, C. I., Parker, S. J. & Van Eyk, J. E. Precision profiling of the cardiovascular post-translationally modified proteome: where there is a will, there is a way. Circ. Res. 122, 1221–1237 (2018).
Ren, L. et al. In vivo phosphoproteome analysis reveals kinome reprogramming in hepatocellular carcinoma. Mol. Cell. Proteomics 17, 1067–1083 (2018).
Wang, S. B. et al. Protein s-nitrosylation controls glycogen synthase kinase 3β function independent of its phosphorylation state. Circ. Res. 122, 1517–1531 (2018).
Kusebauch, U. et al. Human SRMAtlas: a resource of targeted assays to quantify the complete human proteome. Cell 166, 766–778 (2016). Generation and verification of a compendium of highly specific SRM assays that enable quantification of >95% of all annotated human proteins. Provides definitive MS data on 166,174 proteotypic peptides that facilitate the design of multiple, independent assays to quantify most human proteins, numerous splice-variants, non-synonymous mutations, and post-translational modifications.
Elguoshy, A. et al. Identification and validation of human missing proteins and peptides in public proteome databases: data mining strategy. J. Proteome Res. 16, 4403–4414 (2017).
Collins, B. C. et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 8, 291 (2017).
Uhlen, M. et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 4, 1920–1932 (2005).
Lundberg, E. & Borner, G. H. H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302 (2019).
Gingras, A. C., Abe, K. T. & Raught, B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 48, 44–54 (2019).
Bjorling, E. & Uhlen, M. Antibodypedia, a portal for sharing antibody and antigen validation data. Mol. Cell. Proteomics 7, 2028–2037 (2008).
Uhlen, M. et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 28, 1248–1250 (2010).
Uhlen, M. et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 366, eaax9198 (2019).
Sjöstedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947 (2020).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017).
Robinson, J. L. et al. An atlas of human metabolism. Sci. Signal 13, eaaz1482 (2020).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). First of many encyclopedic HPA atlases that map the spatio-temporal expression human proteome based on integrated multi-omics data involving quantitative transcriptomics and Ab microarray-based tissue immunohistochemistry and delivering spatial localisation of human proteins down to the single-cell level.
Uhlen, M. et al. A proposal for validation of antibodies. Nat. Methods 13, 823–827 (2016).
Edfors, F. et al. Enhanced validation of antibodies for research applications. Nat. Commun. 9, 4130 (2018).
Sullivan, D. P. et al. Deep learning is combined with massive-scale citizen science to improve large-scale image classification. Nat. Biotechnol. 36, 820–828 (2018).
Ouyang, W. et al. Analysis of the Human Protein Atlas Image Classification competition. Nat. Methods 16, 1254–1261 (2019).
Drysdale, R. et al. The ELIXIR Core Data Resources: fundamental infrastructure for the life sciences. Bioinformatics 36, 2636–2642 (2020).
Sjostedt, E. et al. Integration of transcriptomics and antibody-based proteomics for exploration of proteins expressed in specialized tissues. J. Proteome Res. 17, 4127–4137 (2018).
Pineau, C. et al. Cell type-specific expression of testis elevated genes based on transcriptomics and antibody-based proteomics. J. Proteome Res. 18, 4215–4230 (2019).
Regev, A. et al. The human cell atlas. Elife 6, e27041 (2017).
Taylor, C. F. et al. The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 25, 887–893 (2007).
Lane, L. et al. neXtProt: a knowledge platform for human proteins. Nucleic Acids Res. 40, D76–D83 (2012).
Zahn-Zabal, M. et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res. 48, D328–D334 (2020).
Gaudet, P. et al. neXtProt: organizing protein knowledge in the context of human proteome projects. J. Proteome Res. 12, 293–298 (2013).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Deutsch, E. W. et al. State of the human proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J. Proteome Res. 14, 3461–3473 (2015).
Pullman, B. S., Wertz, J., Carver, J. & Bandeira, N. ProteinExplorer: a repository-scale resource for exploration of protein detection in public mass spectrometry data sets. J. Proteome Res. 17, 4227–4234 (2018).
Gaudet, P. et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 45, D177–D182 (2017).
Eng, J. K., Jahan, T. A. & Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013).
Bjornson, R. D. et al. X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. J. Proteome Res. 7, 293–299 (2008).
Lam, H. et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 (2007).
Kim, S. & Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277 (2014).
Wang, M. et al. Assembling the community-scale discoverable human proteome. Cell Syst. 7, 412–421.e5 (2018).
Baker, M. S. et al. Accelerating the search for the missing proteins in the human proteome. Nat. Commun. 8, 14271 (2017). Community encouragement to identify biological data that complement high-stringency MS strategies to accelerate discovery and understanding of human proteome PE2,3,4 missing proteins. Database allows unpublished, preliminary or proprietary data (e.g., antibody, MS, cell biology and genetic studies) to be shared with collaborators via a protected interface.
Pertea, M. et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 19, 208 (2018).
Icay, K., Liu, C. & Hautaniemi, S. Dynamic visualization of multi-level molecular data: the Director package in R. Comput. Methods Prog. Biomed. 153, 129–136 (2018).
Fortelny, N., Overall, C. M., Pavlidis, P. & Freue, G. V. C. Can we predict protein from mRNA levels? Nature 547, E19–E20 (2017).
Bludau, I. & Aebersold, R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol. Cell Biol. 21, 327–340 (2020).
Adhikari, S., Sharma, S., Ahn, S. B. & Baker, M. S. In silico peptide repertoire of human olfactory receptor proteomes on high-stringency mass spectrometry. J. Proteome Res. 18, 4117–4123 (2019).
Paik, Y. K. et al. Toward completion of the human proteome parts list: progress uncovering proteins that are missing or have unknown function and developing analytical methods. J. Proteome Res. 17, 4023–4030 (2018).
Bell, P. A., Solis, N., Kizhakkedathu, J. N., Matthew, I. & Overall, C. M. Proteomic and N-terminomic TAILS analyses of human alveolar bone proteins: improved protein extraction methodology and LysargiNase digestion strategies increase proteome coverage and missing protein identification. J. Proteome Res. 18, 4167–4179 (2019).
Thul, P. J. & Lindskog, C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 27, 233–244 (2018).
Sun, J. et al. Multiproteases combined with high-pH reverse-phase separation strategy verified fourteen missing proteins in human testis tissue. J. Proteome Res. 17, 4171–4177 (2018).
Sun, J. et al. Open-pFind enhances the identification of missing proteins from human testis tissue. J. Proteome Res. 18, 4189–4196 (2019).
Smith, L. M. & Kelleher, N. L. Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 (2013).
Aebersold, R. et al. How many human proteoforms are there? Nat. Chem. Biol. 14, 206–214 (2018).
Zahn-Zabal, M. & Lane, L. What will neXtProt help us achieve in 2020 and beyond?. Expert Rev. Proteomics 17, 95–98 (2020).
Moriya, Y. et al. The jPOST environment: an integrated proteomics data repository and database. Nucleic Acids Res. 47, D1218–D1224 (2019).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Sharma, V. et al. Panorama public: a public repository for quantitative data sets processed in Skyline. Mol. Cell. Proteomics 17, 1239–1244 (2018).
Vizcaino, J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014).
Deutsch, E. W. et al. The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 48, D1145–D1152 (2020).
Montelione, G. T. & Anderson, S. Structural genomics: keystone for a Human Proteome Project. Nat. Struct. Biol. 6, 11–12 (1999).
van Eck, N. J. & Waltman, L. Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics 84, 523–538 (2010).
Kushnir, M. M. et al. Measurement of thyroglobulin by liquid chromatography-tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies. Clin. Chem. 59, 982–990 (2013).
Zhang, Z. & Chan, D. W. The road from discovery to clinical diagnostics: lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiol. Biomark. Prev. 19, 2995–2999 (2010).
Campbell, P. J. et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). Details the hallmarks of cancer acquired during multistep development of human tumors (i.e., sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion/metastasis, genome instability, inflammation, reprogramming of energy metabolism and evading immune destruction). Creates a roadmap for the understanding of multi-omics based cancer data.
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
Betancourt, L. H. et al. The hidden story of heterogeneous B-raf V600E mutation quantitative protein expression in metastatic melanoma-association with clinical outcome and tumor phenotypes. Cancers (Basel) 11, 1981 (2019).
Gil, J. et al. Clinical protein science in translational medicine targeting malignant melanoma. Cell Biol. Toxicol. 35, 293–332 (2019).
Tabb, D. L. et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 9, 761–776 (2010).
Addona, T. A. et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 (2009).
Kennedy, J. J. et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat. Methods 11, 149–155 (2014).
Carr, S. A. et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 (2014).
Regnier, F. E. et al. Protein-based multiplex assays: mock presubmissions to the US Food and Drug Administration. Clin. Chem. 56, 165–171 (2010).
Zhang, B. et al. Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 (2014).
Zhang, H. et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 (2016).
Whiteaker, J. R. et al. CPTAC assay portal: a repository of targeted proteomic assays. Nat. Methods 11, 703–704 (2014).
Withers, N. Antibody characterisation—an essential researchers’ resource. Drug Target Rev. 7, 4 (2019).
Rodriguez, H. & Pennington, S. R. Revolutionizing precision oncology through collaborative proteogenomics and data sharing. Cell 173, 535–539 (2018).
Clark, D. J. et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964–983.e31 (2019).
Dou, Y. et al. Proteogenomic characterization of endometrial carcinoma. Cell 180, 729–748.e26 (2020).
Gillette, M. A. et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182, 200–225.e35 (2020).
Vasaikar, S. et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049.e19 (2019). Example of a CPTAC/ICPC proteogenomics study highlighting new therapeutic insights into colorectal cancer. Comparative proteomic and phosphoproteomic analysis of a prospectively-collected paired colon cancer tumour and adjacent normal tissue catalogue of colon cancer-associated proteins and phosphorylation sites, including known and putative new biomarkers, drug targets, and cancer/testis antigens.
McDermott, J. E. et al. Proteogenomic characterization of ovarian HGSC implicates mitotic kinases, replication stress in observed chromosomal instability. Cell Rep. Med. 1, 100004 (2020).
Chen, T. W. et al. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat. Commun. 8, 465 (2017).
Mun, D. G. et al. Proteogenomic characterization of human early-onset gastric cancer. Cancer Cell 35, 111–124.e10 (2019).
Gao, Q. et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell 179, 561–577.e22 (2019).
Chen, Y. J. et al. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell 182, 226–244.e17 (2020).
Lee, D. I. et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519, 472–476 (2015). Demonstrates the potential of proteomics and PTM analyses to increase our understanding of hypertrophic heart disease. Transcription factor activation and phosphoproteome analyses of myocytes revealed PDE9A regulates cGMP signalling independent of the NO-pathway in stress-induced heart disease, suggesting potential as a therapeutic target.
Ashwood, C., Waas, M., Weerasekera, R. & Gundry, R. L. Reference glycan structure libraries of primary human cardiomyocytes and pluripotent stem cell-derived cardiomyocytes reveal cell-type and culture stage-specific glycan phenotypes. J. Mol. Cell. Cardiol. 139, 33–46 (2020).
Wang, J. et al. Integrated dissection of cysteine oxidative post-translational modification proteome during cardiac hypertrophy. J. Proteome Res. 17, 4243–4257 (2018).
Janssens, J. V. et al. Cardiac troponins may be irreversibly modified by glycation: novel potential mechanisms of cardiac performance modulation. Sci. Rep. 8, 16084 (2018).
Lau, E. et al. Splice-junction-based mapping of alternative isoforms in the human proteome. Cell Rep. 29, 3751–3765.e5 (2019).
Cai, W. et al. An unbiased proteomics method to assess the maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 125, 936–953 (2019).
Liu, G. et al. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577, 695–700 (2020).
Arrell, D. K., Rosenow, C. S., Yamada, S., Behfar, A. & Terzic, A. Cardiopoietic stem cell therapy restores infarction-altered cardiac proteome. NPJ Regen. Med. 5, 5 (2020).
Yin, X. et al. Glycoproteomic analysis of the aortic extracellular matrix in Marfan patients. Arterioscler. Thromb. Vasc. Biol. 39, 1859–1873 (2019).
Langley, S. R. et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Invest. 127, 1546–1560 (2017).
Camparini, L. et al. Targeted aproach to distinguish and determine absolute levels of GDF8 and GDF11 in mouse serum. Proteomics 20, e1900104 (2020).
Huth, C. et al. Protein markers and risk of type 2 diabetes and prediabetes: a targeted proteomics approach in the KORA F4/FF4 study. Eur. J. Epidemiol. 34, 409–422 (2019).
van den Broek, I. et al. Application of volumetric absorptive microsampling for robust, high-throughput mass spectrometric quantification of circulating protein biomarkers. Clin. Mass Spectrom. 4–5, 25–33 (2017).
Eshghi, A. et al. Concentration determination of >200 proteins in dried blood spots for biomarker discovery and validation. Mol. Cell. Proteom. 19, 540–553 (2020).
Greco, T. M., Diner, B. A. & Cristea, I. M. The impact of mass spectrometry-based proteomics on fundamental discoveries in virology. Annu. Rev. Virol. 1, 581–604 (2014).
Seng, P. et al. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis. 49, 543–551 (2009).
Kostrzewa, M., Nagy, E., Schrottner, P. & Pranada, A. B. How MALDI-TOF mass spectrometry can aid the diagnosis of hard-to-identify pathogenic bacteria—the rare and the unknown. Expert Rev. Mol. Diagn. 19, 667–682 (2019).
Sauget, M., Valot, B., Bertrand, X. & Hocquet, D. Can MALDI-TOF mass spectrometry reasonably type bacteria? Trends Microbiol. 25, 447–455 (2017).
Alcaide, F. et al. How to identify non-tuberculous Mycobacterium species using MALDI-TOF mass spectrometry. Clin. Microbiol. Infect. 24, 599–603 (2018).
Sanguinetti, M. & Posteraro, B. Identification of molds by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 55, 369–379 (2017).
Burckhardt, I. & Zimmermann, S. Susceptibility testing of bacteria using MALDI-TOF mass spectrometry. Front. Microbiol. 9, 1744 (2018).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Dong, E., Du, H. & Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 20, 533–534 (2020).
Anderson, R. M., Heesterbeek, H., Klinkenberg, D. & Hollingsworth, T. D. How will country-based mitigation measures influence the course of the COVID-19 epidemic? Lancet 395, 931–934 (2020).
Zhang, L., Zhang, Z. P., Zhang, X. E., Lin, F. S. & Ge, F. Quantitative proteomics analysis reveals BAG3 as a potential target to suppress severe acute respiratory syndrome coronavirus replication. J. Virol. 84, 6050–6059 (2010).
Neuman, B. W. et al. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 82, 5279–5294 (2008).
Emmott, E. et al. The cellular interactome of the coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J. Virol. 87, 9486–9500 (2013).
Menachery, V. D. et al. MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc. Natl Acad. Sci. USA 115, E1012–E1021 (2018).
V’kovski, P. et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. Elife 8, e42037 (2019).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003).
Hikmet, F. et al. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 16, e9610 (2020).
Matsuyama, S. et al. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 84, 12658–12664 (2010).
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and Is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). Illustrates the rapidity at which modern proteomics can contribute to the understanding of a recent pandemic, revealing insights into viral transmission and therapeutic targets. Indicates SARS-CoV-2 uses the receptor ACE2 for human cell entry and serine protease TMPRSS2 for SARS-CoV-2 spike protein priming. A clinically approved TMPRSS2 inhibitor blocked virus entry and sera from convalescent SARS-CoV-2 patients cross-neutralized viral entry, suggestive of potential therapeutic approaches.
Sasvari, Z. & Nagy, P. D. Making of viral replication organelles by remodeling interior membranes. Viruses 2, 2436–2442 (2010).
Lum, K. K. & Cristea, I. M. Proteomic approaches to uncovering virus-host protein interactions during the progression of viral infection. Expert Rev. Proteomics 13, 325–340 (2016).
Bojkova, D. et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583, 469–472 (2020).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Jiang, H. W. et al. SARS-CoV-2 proteome microarray for global profiling of COVID-19 specific IgG and IgM responses. Nat. Commun. 11, 3581 (2020).
Shen, B. et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72.e15 (2020).
Saijo, M. et al. Inhibitory effect of mizoribine and ribavirin on the replication of severe acute respiratory syndrome (SARS)-associated coronavirus. Antivir. Res. 66, 159–163 (2005).
Pruijssers, A. J. & Denison, M. R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 35, 57–62 (2019).
Davidson, A. D. et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 12, 68 (2020).
Messner, C. B. et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 11, 11–24.e14 (2020).
Liu, J., Qian, C. & Cao, X. Post-translational modification control of innate immunity. Immunity 45, 15–30 (2016).
Murray, L. A., Sheng, X. & Cristea, I. M. Orchestration of protein acetylation as a toggle for cellular defense and virus replication. Nat. Commun. 9, 4967 (2018).
Hashimoto, Y., Sheng, X., Murray-Nerger, L. A. & Cristea, I. M. Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat. Commun. 11, 806 (2020).
Ihling, C. et al. Mass spectrometric identification of SARS-CoV-2 proteins from gargle solution samples of COVID-19 patients. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.0c00280 (2020).
Gouveia, D. et al. Proteotyping SARS-CoV-2 virus from nasopharyngeal swabs: a proof-of-concept focused on a 3 min mass spectrometry window. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.0c00535 (2020).
Jean Beltran, P. M., Mathias, R. A. & Cristea, I. M. A portrait of the human organelle proteome in space and time during cytomegalovirus infection. Cell Syst. 3, 361–373.e6 (2016).
Wee, S. et al. Multiplex targeted mass spectrometry assay for one-shot flavivirus diagnosis. Proc. Natl Acad. Sci. USA 116, 6754–6759 (2019).
Jagdeo, J. M. et al. N-Terminomics TAILS identifies host cell substrates of poliovirus and coxsackievirus B3 3C proteinases that modulate virus infection. J. Virol. 92, e02211–e02217 (2018).
Kim, C. Y. et al. FusionPro, a versatile proteogenomic tool for identification of novel fusion transcripts and their potential translation products in cancer cells. Mol. Cell. Proteomics 18, 1651–1668 (2019).
Zhang, C., Wei, X., Omenn, G. S. & Zhang, Y. Structure and protein interaction-based gene ontology annotations reveal likely functions of uncharacterized proteins on human chromosome 17. J. Proteome Res. 17, 4186–4196 (2018).
Zhang, C., Lane, L., Omenn, G. S. & Zhang, Y. Blinded testing of function annotation for uPE1 proteins by I-TASSER/COFACTOR pipeline using the 2018-2019 additions to neXtProt and the CAFA3 challenge. J. Proteome Res. 18, 4154–4166 (2019).
Heath, J. R., Ribas, A. & Mischel, P. S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 15, 204–216 (2016).
Kagan, J. et al. National Cancer Institute think-tank meeting report on proteomic cartography and biomarkers at the single-cell Level:interrogation of premalignant lesions. J. Proteome Res. 19, 1900–1912 (2020).
Minakshi, P. et al. in Single-Cell Omics (eds Barh, D. & Azevedo, V.) (Academic Press, 2019).
Specht, H. et al. Single-cell mass-spectrometry quantifies the emergence of macrophage heterogeneity. Preprint at https://doi.org/10.1101/665307 (2019).
Magness, A. J. et al. Multiplexed single cell protein expression analysis in solid tumours using a miniaturised microfluidic assay. Convergent Sci. Phys. Oncol. 3, 024003 (2017).
Palii, C. G. et al. Single-cell proteomics reveal that quantitative changes in co-expressed lineage-specific transcription factors determine cell fate. Cell Stem Cell 24, 812–820.e5 (2019).
Specht, H. & Slavov, N. Transformative opportunities for single-cell proteomics. J. Proteome Res. 17, 2565–2571 (2018).
Boström, T., Johansson, H. J., Lehtiö, J., Uhlén, M. & Hober, S. Investigating the applicability of antibodies generated within the human protein atlas as capture agents in immunoenrichment coupled to mass spectrometry. J. Proteome Res. 13, 4424–4435 (2014).
Acknowledgements
HUPO acknowledges collaborators, proteomic scientists, independent partners, industry vendors and members of the scientific community who have contributed to the HPP. A full alphabetical listing of the Human Proteome Project members appears in the Supplementary Information. In recognition of the many accomplishments, HUPO has produced a publicly available HPP timeline available through https://hupo.org/Proteomics-Timeline to be released with this HPP Blueprint. Parts of this work were supported by grants to ProteoRed PRB3-ISCIII, PT17/0019/0001 Comunidad de Madrid Grant B2017/BMD-3817 (F.J.C.); Korean Ministry of Health and Welfare HI13C2098 and HI16C0257 (Y.K.P.); NIH grants P30ES017885 and U24CA210967 (G.S.O.), 5U01HL-13104204, PADOM-SPO11347 and PARYB-SPO112285 (M.P.S.); NCI CPTAC U24CA210985 and NCI EDRN U24CA115102 (D.W.C.); NIH National Institute of General Medical Sciences R01GM087221 (E.W.D./R.L.M.) and R24GM127667 (E.W.D.); NIH National Institute on Aging U19AG023122 (R.L.M.); NSF DBI-1933311 (E.W.D.); CIHR COVID-19 Rapid Research Funding (F20-01013), CIHR Foundation Grant FDN:14840 and Canada Research Chair (C.M.O.); Investissement d’Avenir Infrastructures Nationales en Biologie et Santé ANR-10-INBS-08 (Proteomics French Infrastructure ProFI (Y.V.); Wellcome Trust WT101477MA and 208391/Z/17/Z (J.A.V.); Knut and Alice Wallenberg Foundation (M.U., C.L., J.M.S., E.L.); Brazilian CAPES 88887.130697, CNPq 440613/2016-7, FAPERJ E-26/210.173/2018 (G.B.D.) and FAPERJ E-26/202.650/2018 (F.C.S.N.), Australian Commonwealth NCRIS (M.S.B.); NHMRC 1010303 (M.S.B., E.C.N.); Cancer Council NSW RG19-04 (M.S.B., S.B.A., E.C.N.); Cancer Institute NSW Fellowship 15/ECF/1-38 (S.B.A.), Sydney Vital CINSW Translational Cancer Research Centre grant (M.S.B., S.B.A., S.A.), ‘Fight on the Beaches’ (M.S.B., S.B.A., E.C.N., S.A.) funding and an International Macquarie Research Excellence Scholarship (S.A.). M.S.B. thanks the Faculty of Medicine, Stanford University for a sabbatical visiting professorship.
Author information
Authors and Affiliations
Contributions
M.S.B. was corresponding author, conceptualized HPP decadal blueprint, HUPO proteomics timeline and HPRL, and he designed/integrated all aspects of the manuscript. E.C.N. assisted in assembly of most blueprint sections. S.A., E.C.N., E.W.D., L.L., G.S.O., S.R.P., Y.K.P., C.M.O., F.J.C., J.E.V.E., M.U., C.L., D.W.C., A.B., J.L.J., J.L., H.R., F.H., M.K., P.P., R.L.G., P.S., Sanjeeva Srivastava, Sudhir Srivastava, F.C.S.N., G.B.D., Y.V., M.P.Y.L., S.W., M.W., J.M.S., E.L., G.MV., C.P., U.K., R.L.M., S.B.A., M.P.S., R.A. and M.S.B. made academic co-contributions to manuscript section drafting, editing, review and/or proof-reading. S.A., L.L., A.B., E.W.D., J.A.V., E.C.N., N.B., G.S.O. and M.S.B. captured and analysed communal neXtProt HPP data. S.A. and M.S.B. prepared Figs. 1–5a, b and Supplementary Fig. 1. M.P. performed VOSviewer analyses for Fig. 5c. S.B.A. and M.S.B. managed referencing. M.L., S.W. and M.S.B. constructed HPRL, while J.W., S.P. and M.S.B. constructed the HUPO proteomics timeline.
Corresponding author
Ethics declarations
Competing interests
S.R.P. is founder and chief scientific officer of Atturos, a clinical diagnostics company. M.K. is an employee of Bruker Daltonics, a manufacturer of MS systems. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adhikari, S., Nice, E.C., Deutsch, E.W. et al. A high-stringency blueprint of the human proteome. Nat Commun 11, 5301 (2020). https://doi.org/10.1038/s41467-020-19045-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-19045-9
This article is cited by
-
A palmitoyl transferase chemical–genetic system to map ZDHHC-specific S-acylation
Nature Biotechnology (2024)
-
Protein prognostic biomarkers in stage II colorectal cancer: implications for post-operative management
BJC Reports (2024)
-
Global detection of human variants and isoforms by deep proteome sequencing
Nature Biotechnology (2023)
-
KATP channel dependent heart multiome atlas
Scientific Reports (2022)
-
Understudied proteins: opportunities and challenges for functional proteomics
Nature Methods (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
