
Abstract
The equilibrium and non-equilibrium optical properties of single-layer transition metal dichalcogenides (TMDs) are determined by strongly bound excitons. Exciton relaxation dynamics in TMDs have been extensively studied by time-domain optical spectroscopies. However, the formation dynamics of excitons following non-resonant photoexcitation of free electron-hole pairs have been challenging to directly probe because of their inherently fast timescales. Here, we use extremely short optical pulses to non-resonantly excite an electron-hole plasma and show the formation of two-dimensional excitons in single-layer MoS2 on the timescale of 30 fs via the induced changes to photo-absorption. These formation dynamics are significantly faster than in conventional 2D quantum wells and are attributed to the intense Coulombic interactions present in 2D TMDs. A theoretical model of a coherent polarization that dephases and relaxes to an incoherent exciton population reproduces the experimental dynamics on the sub-100-fs timescale and sheds light into the underlying mechanism of how the lowest-energy excitons, which are the most important for optoelectronic applications, form from higher-energy excitations. Importantly, a phonon-mediated exciton cascade from higher energy states to the ground excitonic state is found to be the rate-limiting process. These results set an ultimate timescale of the exciton formation in TMDs and elucidate the exceptionally fast physical mechanism behind this process.
Similar content being viewed by others


Introduction
Single-layer (1L) TMDs are attracting growing interest because of their peculiar properties that make them highly suitable for optoelectronics applications1. The reduced dielectric screening that is caused by the strong spatial confinement results in an optical response that is dominated even at room temperature by strongly bound excitons with large (hundreds of meV) binding energies2,3. The enhanced Coulomb interaction gives rise to additional effects such as the occurrence of a Rydberg series of excitonic states4 and many-body complexes like trions5 and biexcitons6 whose physical properties can be tuned by changing the dielectric environment7 or by applying external stimula such as light8, strain9, electric10, and magnetic fields11. Ultrashort laser pulses offer an additional way to change and potentially control the properties of excitons on a fast timescale. The non-equilibrium optical response of TMDs has been extensively explored both experimentally and theoretically12,13. The dynamical response of TMDs and, in general, all semiconductors can be divided into coherent and incoherent regimes14,15. While the incoherent exciton dynamics of TMDs, including processes, such as exciton thermalization16, radiative/non radiative recombination17, intra and intervalley scattering18,19,20,21,22, exciton dissociation23, and exciton–exciton annihilation24, have been extensively studied, the coherent exciton response and the corresponding early stage dynamics of the exciton formation process are still almost unexplored largely because the limited available temporal resolution (i.e., >100 fs for pump-probe optical spectroscopy and >1 ps for time-resolved photoluminescence) has prevented the study of the primary early stage dynamics of exciton photo-generation processes17,25,26.
Here, we push the temporal resolution of ultrafast differential reflectivity (ΔR/R) spectroscopy to the regime of less than 30 fs in order to investigate the dynamics of exciton formation in 1L-MoS227. Experimentally, we employ a pump-probe technique that uses ΔR/R to monitor the evolution of the excited state population. Particularly, we elucidate how long it takes for an initial population of high-energy photoexcited electrons and holes to relax to the lowest-energy exciton states (i.e., the 1s states of the A and B excitons), and how this formation time depends upon the energy of the initial state (c.f. Fig. 1). The measured dynamics are found to be remarkably fast, suggesting that lowest-energy excitons form from these high-energy populations with a characteristic timescale as fast as 10 fs. Upon increasing the energy of the initial state of photoinjected carriers (i.e., by increasing the pump photon energy), we find that (1) the formation time of the excitons increases monotonically with increasing energy; (2) an initial, sub-100 fs fast decay component of the excitons vanishes; and (3) the dynamics of a slower decay process (on the timescale of picoseconds) associated with the relaxation of a thermal population of excitons does not substantially change. Simulations based on the TMD Bloch equations attribute the excitation energy dependence of the formation dynamics to a phonon-induced cascade-like relaxation process of high-energy incoherent excitons down to the excitonic ground state28. This process, although extremely fast, is predicted to have a characteristic time constant between 20 and 30 fs, corroborating our experimental observations. The early sub-100 fs relaxation dynamics are also well captured by the simulations, which reveal that they arise from the decay of the pump-induced coherent optical polarization into ultimately thermal exciton populations (i.e., incoherent excitons). These results are of great relevance for optoelectronic applications of TMDs as they directly define a timescale for an efficient extraction of hot carriers in 1L-TMDs and TMD-based heterostructures before the exciton formation process.

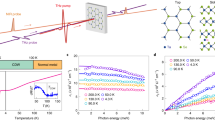
a Cartoon of the exciton formation process after photo-injection of free electron-hole pairs. b Schematic illustration of the single particle band structure of 1L-MoS2 at the K/K' points. The two arrows represent A/B excitonic transitions, split due to the strong spin-orbit interaction at the K/K' points of the Brillouin zone. c Sketch of the pump-probe experiment. A few-optical-cycle laser pulse injects free electron/hole pairs at increasing energies above the exciton continuum (EG). These quasiparticles lose their initial kinetic energy and scatter down via a cascade process mediated by phonons to lower-lying discrete excitonic states until they reach the 1s excitonic state. The timescale τ(E) of this relaxation process is determined by measuring the absorption change of a probe beam, tuned on resonance with the 1s state, due to the Pauli blocking effect.
Results
Ultrafast exciton dynamics
Figure 1 schematically summarizes the exciton formation process (Fig. 1a) and the key optical excitonic transitions (Fig. 1b) of 1L-MoS2. The 1L-MoS2 used here is grown by chemical vapor deposition on a SiO2/Si substrate. All measurements were made on as-grown samples. From the steady-state reflectance measurements that are reported in Supplementary Fig. 1, the lowest energy A and B excitonic resonances that arise from optical transitions between the two-highest energy valence bands and the lowest-energy conduction bands at the K and K’ points in the Brillouin zone are centered at the energies of 1.88 eV and 2.03 eV, respectively (see sketch in Fig. 1b). As illustrated in Fig. 1c, both the A and B optical transitions form two manifolds of Rydberg-like series of bound states that merge into a continuum of unbound electron-hole states3,29. These resonances correspond to the lowest-energy (or ground state) excitons in the A and B manifolds (i.e., the A1s and B1s states, respectively), possess the largest oscillator strengths, and dominate the optical response over the corresponding higher-energy unbound states. Based on previous experimental studies3,29 and refined theoretical models18, we estimate that the exciton binding energy (i.e., the energetic difference between the 1s exciton and the lowest energy state in the exciton continuum) is of the order of 350 meV. In our measurements the pump photon energies span from 2.29 eV to 2.75 eV (with bandwidths spanning from 70 meV to 140 meV) and thus, at all energies, photoexcitation predominantly creates an initial population of excitons at or well-into the exciton continuum for the A excitons (as well as for the B excitons at energies above ~2.4 eV). To monitor the formation dynamics and quantify the formation time (i.e., τ(E) in Fig. 1c), our probe energies monitor the rise and decay dynamics of the ΔR/R signals for the 1s states of the A and B excitons.
We use tunable pump pulses to photo-inject electron-hole pairs with increasing excess energy with respect to EG. The time-delayed broadband probe pulses measure the build-up and early decay dynamics of both A1s and B1s excitons. The experimental temporal resolution of the setup is sub-30-fs (see Methods and Supplementary Fig. 5). Figure 2a and b reports ΔR/R(Eprobe,τ) maps as a function of the pump-probe delay τ and the probe photon energy Eprobe for two different pump photon energies: 2.29 eV and 2.75 eV, i.e., respectively, across and well above EG. Due to the presence of the reflecting Si substrate, ΔR/R is equivalent to a double-pass differential transmission (ΔT/T). Both measurements show comparable qualitative responses: positive strong features associated with the A1s (centered at Eprobe = 1.88 eV) and B1s (centered at Eprobe = 2.03 eV) exciton states. The ΔR/R spectrum displays a symmetric profile around each excitonic resonance with no shift of the peak maximum, within the considered temporal window (i.e., from −100 to 200 fs). Weak and broad negative features are detected at higher and lower probe energies as better shown in Supplementary Fig. 4. These features are strongly quenched with respect to previously published pump-probe measurements on 1L-MoS2 performed in transmission geometry12. We attribute the different intensities of these signals away from the excitonic transitions to a photonic effect caused by the interference of multiple reflections of the incoming beam in the thin SiO2 substrate.

ΔR/R maps measured on 1L-MoS2 photoexcited a at 2.29 eV and b at 2.75 eV. The measurements are performed at room temperature. The horizontal dashed lines pass through the maximum of the ΔR/R spectrum at the energies of the A1s/B1s exciton transitions, while the vertical lines mark the temporal range from 10 to 90% of the build-up signal. The excitation fluence is 5 μJcm−2. Pump and probe beams have parallel and linear polarizations. The time zero is defined, for each measurement, as the maximum of the cross-correlation signal between the pump and the probe pulses as explained in the Methods. c Temporal cuts of the ΔR/R maps measured across the A1s and B1s excitonic resonances for increasing pump photon energy. d Pump photon energy dependence of τrise. Horizontal error bars are determined by the bandwidth of the pump pulses; vertical error bars are obtained from the fits of the time traces.
In the temporal window explored in the experiment (i.e., ~200 fs), the ΔR/R spectrum displays a symmetric profile around each excitonic resonance with no shift of the peak maximum, showing that the signal is dominated by Pauli blocking (see Supplementary Fig. 2).
A closer inspection of the maps reveals that the formation time τrise of the ΔR/R signals of the A1s and B1s excitons is longer for higher pump photon energies. When the excitation energy is close to EG, the signal displays a quasi-instantaneous (i.e., pulse-width limited) build-up, while for increased pump photon energy, τrise significantly increases. We stress that this result can be observed only thanks to the high temporal resolution of the setup. To better quantify this effect, Fig. 2c reports the temporal cuts of the maps taken at the A1s and B1s exciton peak for increasing pump photon energies. The build-up times are estimated by fitting the temporal traces in the temporal window between −100 and 200 fs, to the function:
where H is the Heaviside function centered at τ = 0; τ1 and τ2 are decay constants and A1 and A2 are the amplitudes of each decay component. The fitting function is convoluted with a Gaussian profile corresponding to the instrumental response function, which is the experimentally measured cross-correlation profile between the pump and the probe (see Methods and Supplementary Note 5 for the temporal characterization of the pulses). We stress that only after including a finite exponential rise time in the fitting function, we can satisfactorily reproduce the experimental formation time (see Supplementary Fig. 6). Figure 2d unambiguously shows that τrise monotonically increases with the initial excess energy of the photoinjected carriers. Interestingly, for a higher pump energy excitation (i.e., 3.75 eV), we observe that τrise sensitively deviates from the increasing energy trend reported in Fig. 2d (see Supplementary Note 3). We can tentatively explain this flattening of the formation dynamics as a result of different phonon-mediated scattering process involving electronic states far away from the K/K’ points. The weak negative dip observed for negative times in the B exciton signal for 2.29 eV excitation photon energy is attributed to the so-called pump-perturbed free-induction decay (PPFID)30. In this process that occurs when the probe pulse precedes the pump, the free-induction decay field emitted by the sample excited by the probe pulse is perturbed by the interaction with the pump pulse, giving rise to oscillating signals at negative delays. The PPFID effect can be safely disregarded from the measured build-up timescale of the sample because its amplitude is more than one order of magnitude lower than the bleaching of the excitonic peak. Another interesting effect is the extremely fast decay observed at lower pump photon energy, which occurs on a timescale comparable to that of the build-up and fades away as the pump is tuned to higher photon energies. This effect is particularly clear on the A exciton ΔR/R trace (see Fig. 2c).
Theoretical model
To identify the essential mechanisms that underlie the early-time non-equilibrium optical response of 1L-MoS2, we performed simulations based on the TMD Bloch equations31. Our model describes the temporal dynamics of the excitons after the photoexcitation of free electron-hole pairs above or close to EG. The ΔR/R at the A/B exciton transitions due to the photoexcitation process is also calculated and compared with experimental results. Excitonic excitations are theoretically described by solving the Wannier equation (see Supplementary Note 8)31,32. The exciton kinetics are determined by a set of coupled differential equations (TMD Bloch equations) describing the temporal evolution of the polarization (in our theory an excitonic scattering state) and the excitonic population (incoherent excitons) where the phonon-mediated relaxation from energetically higher densities is described with effective rates Γν+1→ν determined by independent density functional theory calculations (see Methods section and the sketch in Fig. 3a). Solving the set of equations of motion for the coherent polarization as well as incoherent exciton population gives access to the temporal dynamics of the differential reflectance at the 1s exciton resonance frequencies.
The results of the simulations, reported in Fig. 3b, agree remarkably well with the experimental results. For all pump photon energies, the calculated ΔR/R exhibits sub-100 fs build-up followed by a decay time on the picosecond timescale. At higher pump energies, the peak of the signal is progressively delayed as a result of a slower rise time. The timescale and the pump photon energy dependence of the build-up dynamics are in remarkable quantitative agreement with the experimental results. The slowing of τrise with increasing pump photon energy is the result of a high-energy exciton cascade scattering process down to the excitonic ground state. A sketch of the scattering processes involving photoexcited excitons is reported in Fig. 3a. The nonresonant pump pulse induces an almost instantaneous coherent interband exciton polarization which oscillates with the same frequency of the driving pulse. This polarization rapidly dephases by exciton–phonon scattering and leads to a delayed formation of an incoherent exciton population involving electronic states above EG. These high-energy weakly bound excitons quickly lose their energy and scatter into lower-lying continuum and discrete excitonic states. The scattering process continues until most of the excitonic population reaches the lowest energy 1s exciton state. With increasing pump photon energy, the number of intermediate scattering events, required to complete the cascade process, increases. This increase in needed scattering events results in the experimentally observed delayed formation of the bleaching signal measured at the A1s/B1s optical transitions.

a Schematic illustration of the relaxation model in the exciton picture where Q denotes the exciton center-of-mass wave vector. After optical excitation of continuum states with the pump pulse, the sample is probed at the A1s (left) and B1s (right) exciton resonance energy. The measured signal exhibits contributions originating from instantaneous coherent polarizations as well as incoherent exciton densities, which are formed in the continuum and relax down to the energetically lowest 1s states (relaxation rate Γν+1→ν). Finally, the exciton density associated with the lowest 1s states decays slowly with a rate Γdecay. b Calculated ΔR/R signal at the A1s (left) and B1s (right) exciton resonance energy for varying pump photon energies. All the calculated traces are normalized to the maximum value. The insets report the pump photon energy dependence of τrise of the calculated ΔR/R traces. The calculated τrise values are estimated by fitting the traces with a rising exponential convoluted with a Gaussian profile accounting for the finite temporal duration of the pump and probe pulses used in the calculation (i.e., respectively 20 fs and 15 fs). The B exciton traces exhibit, as expected, a slightly shorter rise time. This difference between the timescales is well below the temporal resolution of the pump-probe experiment and cannot be resolved by the measurements. Similar experimental τrise for the A1s and B1s excitonic resonances might also be due to the mixing of excitonic states due to intra and intervalley exchange coupling mechanism22.
The simulations also capture the occurrence of a sub-100 fs decay component, which is particularly evident in the ΔR/R temporal trace for the A1s state and progressively vanishes at higher pump photon energies. We attribute this effect to an interplay between a coherent exciton polarization and an incoherent exciton density. The coherent contribution adiabatically follows the pump pulse and gives rise to an instantaneous coherent build-up, while the incoherent signal is characterized by a delayed formation time involving additional exciton–phonon scattering processes. Figure 4 compares the full transient signal incorporating both contributions (solid line) as well as the underlying coherent (shaded area) and incoherent (dashed line) contributions to the differential signal obtained by artificially turning off the coherent contribution for low and high pump photon energies. For low pump photon energies, the full signal and the incoherent part of the signal display different dynamics in the first tens of femtoseconds and almost overlap after ~100 fs. While the build-up dynamics are dominated by the coherent excitons, the energetically lowest incoherent exciton densities play a dominant role after the polarization to population transfer is complete. We stress that, in this excitation regime, coherent exciton contribution could be more clearly disentangled from the incoherent exciton dynamics by performing pump-probe measurements that utilized extremely short, linearly, and circularly polarized pulses and polarization resolved detection schemes. However, the use of broadband polarization optics makes it difficult to preserve the extremely high temporal resolution needed to observe such a process.

a Calculated ΔR/R dynamics at the energy of the A1s resonance considering only the coherent exciton contribution (shaded areas), only the incoherent exciton contribution (dashed lines) and both the contributions (continuous lines) for low (orange traces) and high (blue traces) pump photon energy, i.e., 2.29 and 2.75 eV, respectively. b The same for the dynamics at the energy of the B1s resonance.
For high pump photon energies, the difference between full and incoherent curves diminishes since the coherent part inversely depends on the detuning between pump and probe pulses. This difference is related to the polarization, which rapidly decays before a significant 1s exciton density builds up that can be detected by the probe pulse. Thus, the transient signal in this energy regime is strongly dominated by the dynamics of incoherent exciton densities. In this excitation regime, the extracted rise time (~30 fs) of the ΔR/R traces is a direct estimation of the timescale of the incoherent exciton formation process.
Discussion
Our combined experimental and theoretical work defines a different timescale for the exciton formation process in 1L-TMDs, which is much faster than the one previously estimated by intraexcitonic mid-IR33,34 and interband visible optical spectroscopy25. We also stress that this exciton formation process is faster than the ~1 ps trion formation time in TMDs5 and, remarkably, orders of magnitude faster than the formation time of excitons in quantum wells (i.e., ~1 ns) measured by time-resolved photoluminescence35 and transient terahertz spectroscopy36. This result points to a correlation between the formation time and the binding energy of the excitons. The reduced Coulomb screening in TMDs makes this process more rapid and effective than for weakly bound excitons in quantum wells or trions. Two different mechanisms have been proposed to describe the exciton formation process in semiconductors: geminate and bimolecular37. In the geminate mechanism excitons are directly created upon photoexcitation by simultaneous emission of phonons, while in the bimolecular process, excitons are created from thermalized electron-hole pairs. The observed sub-100 fs build-up dynamics suggests that the geminate mechanism mediated by strong exciton–phonon scattering is the dominant process responsible for the formation of excitons. This conclusion is further supported by the pump fluence dependent measurements (see Supplementary Fig. 8), where no change of τrise is observed for different densities of photoexcited excitons, contrary to what expected for a bimolecular formation process.
In summary, we have studied the exciton formation process in 1L-MoS2 by measuring its transient optical response upon excitation with energy tunable sub-30-fs laser pulses. We resolve extremely fast and pump photon energy-dependent build-up dynamics of the ΔR/R signal around the A1s and B1s excitonic transitions. Microscopic calculations based on the TMD Bloch equations quantitatively reproduce the experimental results and explain the delayed formation of the transient signal as a result of a phonon-induced cascade-like relaxation process of high-energy incoherent excitons down to the excitonic ground state. These results shed light on the poorly explored mechanism of the exciton formation in 2D semiconductors, redefining the timescale of this process and are extremely important in view of optoelectronic applications of these materials.
Methods
Sample preparation
The large area 1L-MoS2 sample was grown by chemical vapor deposition on a SiO2/Si substrate. The SiO2 layer thickness is 300 nm. The growth procedure was carried on in a dual-zone tube furnace filled by sulfur and MoO3 precursors. Further details on the growth process are reported in ref. 3. Contrast reflectivity and photoluminescence measurements have been carried out to characterize the static optical response of the sample (see Supplementary Fig. 1).
Pump-probe setup
The ultrafast pump-probe experiments were carried out using a regeneratively amplified Ti:Sapphire system (Coherent Libra II), emitting 100 fs pulses centered at 1.55 eV at 2 kHz repetition rate with 4W average power. The laser drives two home-made Non-collinear Optical Parametric Amplifiers (NOPAs), which serve, respectively, as the pump and the probe. The first NOPA (probe beam) is pumped at 3.1 eV by the second harmonic of the laser and is seeded by a white-light continuum (WLC), generated in a 1-mm thick sapphire plate. The seed is amplified in a 1-mm thick beta-barium borate (BBO) crystal and compressed to nearly transform-limited duration (i.e., 7 fs) by a pair of custom-designed chirped mirrors. The probe spectrum extends from 1.75 to 2.4 eV, covering the A/B excitonic resonances of 1L-MoS2. The second NOPA, used to produce the pump beam, is also pumped at 3.1 eV and uses a BBO crystal. It can be configured to amplify either the visible or the near-infrared parts of the WLC. In the former case one obtains pulses tunable from 2 to 2.5 eV, compressed to sub-20-fs duration by chirped mirrors. In the latter case one obtains pulses tunable between 1.1 and 1.4 eV, which are compressed to a nearly transform-limited sub-15-fs duration by a pair of fused silica prisms. These pulses are then frequency-doubled in a 50μm-thick BBO crystal to obtain pulses tunable between 2.29 and 2.75 eV. The overall temporal resolution of the setup is characterized via Cross-Frequency Resolved Optical Gating (X-FROG), as extensively explained in Supplementary Note 5. Pump and probe pulses are temporally synchronized by a motorized translation stage, and non-collinearly focused on the sample by a spherical mirror, resulting in spot-size diameters of ~100 μm and ~70 μm, respectively. After the interaction with the pump-excited sample, the probe pulse is spectrally dispersed on a Silicon CCD camera with 532 pixels working at the same repetition rate as the laser. The pump beam is modulated at 1 kHz by a mechanical chopper. The detection sensitivity is on the order of 104–105, with an integration time of 2s. The fluence is ~5 μJcm−2 for different pump photon energies (i.e., well below the fluence threshold for the Mott transition measured for 1L-TMDs38,39). No pump fluence dependence of the build-up and the relaxation dynamics was observed in the pump fluence range between 1 and 20 μJcm−2 (see Supplementary Fig. 8). In all the experiments, pump and probe beams have parallel linear polarizations. No pump-probe polarization dependence of the build-up dynamics was detected (see Supplementary Fig. 7).
Theory
To get insight into the static optical properties properties of 1L-MoS2 on SiO2 substrate, we first solve the Wannier equation, obtaining a set of bound and continuum exciton wavefunctions φλ,q with energies ϵλ, cf. Supplementary Note 8. This allows to work in a convenient excitonic basis including coherent exciton polarizations Pλ, biexcitons and exciton–exciton scattering states Bη, as well as incoherent exciton populations \({N}_{{\lambda }_{1},{\lambda }_{2},q}\). The optical response is determined by the TMD Bloch equations for the exciton amplitude Pλ where λ as a compound index includes the exciton state with bound and continuum excitonic states and the valley and spins of involved electrons and holes31,40:
The left-hand side of Eq. (2) describes the oscillation of the exciton transition damped by a dephasing rate \({\gamma }_{{\lambda }_{1}}\)41,42,43. The first term on the right-hand side represents the optical pump generating excitons with zero center-of-mass motion at the corners of the hexagonal Brillouin zone with σ+ (ξ = K) or σ− (ξ = K\(^{\prime}\)) circularly polarized light. Here, \({d}_{{\lambda }_{1}}\) is the optical transition matrix element and \({{\mathcal{E}}}_{{\sigma }_{\pm }}(t)\) the light field at the position of the monolayer. The latter is obtained by solving Maxwell’s wave equation, which determines the reflected light44,45. The next contribution characterizes Pauli blocking by coherent excitons (~∣P∣2) and incoherent densities (~N). The third term on the right-hand side of Eq. (2) characterizes nonlinear exciton–exciton interactions on a Hartree–Fock level (~P∣P∣2), which couples bound and continuum excitonic states. Finally, the last line of Eq. (2) describes the coupling of excitons to biexcitons Bη=b, and two-exciton scattering continua Bη≠b46. Here, the index η serves as a compound index and includes the high-symmetry points and spins of the two electrons and two holes. All matrix elements are defined in ref. 18.
In order to decrease the complexity of the problem for the description of the experiment, the excitonic occupations (N) are treated with effective occupation numbers λ2, which average over the details of the complex momentum distribution of incoherent exciton occupations: \({\sum }_{{\lambda }_{2},{\lambda }_{3},q}{\hat{d}}_{{\lambda }_{1},{\lambda }_{2},{\lambda }_{3},q}\,{N}_{{\lambda }_{2},{\lambda }_{3},q}\approx {\sum }_{{\lambda }_{2}}{\tilde{d}}_{{\lambda }_{1},{\lambda }_{2}}\,{N}_{{\lambda }_{2}}\) with \({\tilde{d}}_{{\lambda }_{1},{\lambda }_{2}}={\hat{d}}_{{\lambda }_{1},{\lambda }_{2},{\lambda }_{2},0}/2\). However, the bleaching cross sections are explicitly evaluated as a function of the exciton state number λ1/2. These cross sections exhibit a drastic decrease with increasing state number. The equations of motion of the exciton densities are given by:
Equations (3) and (4) describe the dynamics of the incoherent exciton densities associated with the energetically lowest A1s/B1s states \({N}_{{{\rm{A}}}_{{\rm{1s}}}/{{\rm{B}}}_{{\rm{1s}}}}\) and higher states Nλ, respectively. Γdecay = 1 meV/ℏ characterizes the relaxation of the incoherent exciton densities into the ground state. Its value is adjusted to the experimental results. The first contributions to the right-hand sides of Eqs. (3) and (4) represent the formation of incoherent exciton densities out of optically excited coherent excitations by exciton–phonon scattering47. The terms proportional to Γλ+1→λ characterize the phonon-mediated relaxation from energetically higher densities with effective rates \({\Gamma }_{\lambda +1\to \lambda }=\frac{1\ {\rm{eV}}}{50\ {\rm{fs}}}\frac{1}{{\epsilon }_{\lambda +1}-{\epsilon }_{\lambda }}\) adapted to density functional theory calculations48,49. The solution of the Schrdinger equation for two electrons and two holes accesses biexcitons as well as exciton–exciton scattering continua18,50,51. The Heisenberg equation of motion for bound biexcitons as well as continuous exciton–exciton scattering states Bη characterize damped (\({\gamma }_{{\lambda }_{1}}+{\gamma }_{{\lambda }_{2}}\)) oscillations (energy ϵxx,η), which are driven by two excitons \({P}_{{\lambda }_{1}}{P}_{{\lambda }_{2}}\) mediated by Coulomb interactions (\({\hat{W}}_{\eta ,{\lambda }_{1},{\lambda }_{2}}\)):
Solving the set of equations of motion, Eqs. (2)–(5), together with Maxwell’s wave equation44,45 gives access to the differential reflection signal. Our simulations include coherent exciton binding energy renormalizations and other many-body effects related to the transient variation of the Coulomb screening due to optically excited A1s and B1s excitons. For the incoherent contributions we restricted our model to the Pauli blocking effect of reduced complexity whereas incoherent Coulomb renormalizations in the TMD Bloch equations, originating from incoherent exciton populations, which are formed on a similar timescale were neglected. The agreement between the calculated and measured dynamics suggests that the out-of-equilibrium optical response of 1L-MoS2 on a sub-100 fs timescale is well captured by the incoherent dynamics to phase-space filling.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References
Mueller, T. & Malic., E. Exciton physics and device application of two-dimensional transition metal dichalcogenide semiconductors. npj 2D Mater. Appl. 2, 29 (2018).
Qiu, D. Y., da Jornada, F. H. & Louie, S. G. Optical spectrum of MoS2: many-body effects and diversity of exciton states. Phys. Rev. Lett. 111, 216805 (2013).
Yao, K. et al. Optically discriminating carrier-induced quasiparticle band gap and exciton energy renormalization in monolayer MoS2. Phys. Rev. Lett. 119, 087401 (2017).
Chernikov, A. et al. Exciton binding energy and nonhydrogenic Rydberg series in monolayer WS2. Phys. Rev. Lett. 113, 076802 (2014).
Singh, A. et al. Trion formation dynamics in monolayer transition metal dichalcogenides. Phys. Rev. B 93, 041401(R) (2016).
Steinhoff, A. et al. Biexciton fine structure in monolayer transition metal dichalcogenides. Nat. Phys. 14, 1199–1204 (2018).
Raja, A. et al. Dielectric disorder in two-dimensional materials. Nat. Nanotechnol. 14, 832–837 (2019).
Kim, J., Hong, X., Jin, C., Shi, S.-F. & Chang, C.-Y. S. et al. Ultrafast generation of pseudo-magnetic field for valley excitons in WSe2 monolayers. Science 346, 1205–1208 (2014).
Mennel, L. et al. Optical imaging of strain in two-dimensional crystals. Nat. Commun. 9, 516 (2018).
Roch, J. G. et al. Quantum-confined Stark effect in a MoS2 monolayer van der Waals heterostructure. Nano Lett. 18, 1070–1074 (2018).
Stier, A. V., McCreary, K. M., Jonker, B. T., Kono, J. & Crooker, S. A. Exciton diamagnetic shifts and valley Zeeman effects in monolayer WS2 and MoS2 to 65 Tesla. Nat. Commun. 7, 10643 (2016).
Pogna, E. A. A. et al. Photo-induced bandgap renormalization governs the ultrafast response of single-layer MoS2. ACS Nano 10(1), 1182–1188 (2016).
Moody, G., Schaibley, J. & Xu, X. Exciton dynamics in monolayer transition metal dichalcogenides. J. Opt. Soc. Am. B 33, C39–C49 (2016).
Thränhardt, A., Kuckenburg, S., Knorr, A., Thomas, P. & Koch, S. W. Interplay between coherent and incoherent scattering in quantum well secondary emission. Phys. Rev. B 62, 16802 (2000).
Kira, M. & Koch, S.W. Semiconductor Quantum Optics 1st edn. (Cambridge University Press, 2012).
Selig, M., Berghäuser, G., Richter, M., Bratschitsch, R. & Knorr, A. et al. Dark and bright exciton formation, thermalization, and photoluminescence in monolayer transition metal dichalcogenides. 2D Mater. 5, 035017 (2018).
Robert, C. et al. Exciton radiative lifetime in transition metal dichalcogenide monolayers. Phys. Rev. B 93, 205423 (2016).
Katsch, F., Selig, M. & Knorr, A. Theory of coherent pump-probe spectroscopy in monolayer transition metal dichalcogenides. 2D Mater. 7, 015021 (2020).
Glazov, M. M. et al. Exciton fine structure and spin decoherence in monolayers of transition metal dichalcogenides. Phys. Rev. B 89, 201302(R) (2014).
Schmidt, R. et al. Ultrafast Coulomb-induced intervalley coupling in atomically thin WS2. Nano Lett. 16, 2945–2950 (2016).
Dal Conte, S. et al. Ultrafast valley relaxation dynamics in monolayer MoS2 probed by nonequilibrium optical techniques. Phys. Rev. B 92, 235425 (2015).
Selig, M. et al. Ultrafast dynamics in monolayer transition metal dichalcogenides: interplay of dark excitons, phonons, and intervalley exchange. Phys. Rev. Research 1, 022007(R) (2019).
Massicotte, M. et al. Dissociation of two-dimensional excitons in monolayer WSe2. Nat. Commun. 9, 1633 (2018).
Sun, D. et al. Observation of rapid exciton-exciton annihilation in monolayer molybdenum disulfide. Nano Lett. 14, 5625–5629 (2014).
Ceballos, F., Cui, Q., Bellus, M. Z. & Zhao, H. Exciton formation in monolayer transition metal dichalcogenides. Nanoscale 8, 11681–11688 (2016).
Cunningham, P. D., Hanbicki, A. T., McCreary, K. M. & Jonker, B. T. Photoinduced bandgap renormalization and exciton binding energy reduction in WS2. ACS Nano 11, 12601–12608 (2017).
Trovatello, C. et al. The ultrafast onset of exciton formation in 2D semiconductors. Preprint at https://arxiv.org/abs/2002.07108 (2020).
Brem, S., Selig, M., Berghaeuser, G. & Malic, E. Exciton relaxation cascade in two-dimensional transition metal dichalcogenides. Sci. Rep. 8, 8238 (2018).
Borys, N. J. et al. Anomalous above-gap photoexcitations and optical signatures of localized charge puddles in monolayer molybdenum disulfide. ACS Nano 11, 2115–2123 (2017).
Brito-Cruz, C. H., Gordon, J. P., Becker, P. C., Fork, R. L. & Shank, C. V. Dynamics of spectral hole burning. IEEE J. Quantum Electron. 24, 261–269 (1988).
Katsch, F. et al. Theory of exciton-exciton interactions in monolayer transition metal dichalcogenides. Phys. Stat. Solidi b 255, 1800185 (2018).
Kira, M. & Koch, S. W. Many-body correlations and excitonic effects in semiconductor spectroscopy. Prog. Quantum Electron. 30, 155–296 (2006).
Steinleitner, P. et al. Direct observation of ultrafast exciton formation in a monolayer of WSe2. Nano Lett. 17, 1455–1460 (2017).
Cha, S. et al. 1s-intraexcitonic dynamics in monolayer MoS2 probed by ultrafast mid-infrared spectroscopy. Nat. Commun. 7, 10768 (2016).
Amand, T. et al. Exciton formation and hole-spin relaxation in intrinsic quantum wells. Phys. Rev. B 50, 11624 (1994).
Kaindl, R. A., Carnahan, M. A., Hägele, D., Lövenich, R. & Chemla, D. S. Ultrafast terahertz probes of transient conducting and insulating phases in an electron-hole gas. Nature 423, 734–738 (2003).
Piermarocchi, C., Tassone, F., Savona, V., Quattropani, A. & Schwendimann, P. Exciton formation rates in GaAs/Alx Ga1−x As quantum wells. Phys. Rev. B 55, 1333 (1997).
Steinhoff, A., Florian, M., Rösner, M., Schönhoff, G., Wehling, T. O. & Jahnke, F. Exciton fission in monolayer transition metal dichalcogenide semiconductors. Nat. Commun. 8, 1166 (2017).
Chernikov, A., Ruppert, C., Hill, H. M., Rigosi, A. F. & Heinz, T. F. Population inversion and giant bandgap renormalization in atomically thin WS2 layers. Nat. Photonics 9, 466–470 (2015).
Lindberg, M. & Koch, S. W. Effective Bloch equations for semiconductors. Phys. Rev. B 38, 3342 (1988).
Selig, M. et al. Excitonic linewidth and coherence lifetime in monolayer transition metal dichalcogenides. Nat. Commun. 7, 13279 (2016).
Christiansen, D. et al. Phonon sidebands in monolayer transition metal dichalcogenides. Phys. Rev. Lett. 119, 187402 (2017).
Lengers, F., Kuhn, T. & Reiter, D. E. Theory of the absorption line shape in monolayers of transition metal dichalcogenides. Phys. Rev. B 101, 155304 (2020).
Knorr, A., Hughes, S., Stroucken, T. & Koch, S. W. Theory of ultrafast spatio-temporal dynamics in semiconductor heterostructures. Chem. Phys. 210, 27–47 (1996).
Stroucken, T., Knorr, A., Thomas, P. & Koch, S. W. Coherent dynamics of radiatively coupled quantum-well excitons. Phys. Rev. B 53, 2026 (1996).
Katsch, F., Selig, M. & Knorr, A. Exciton-scattering-induced dephasing in two-dimensional semiconductors. Phys. Rev. Lett. 124, 257402 (2020).
Selig, M. et al. Quenching of intervalley exchange coupling in the presence of momentum-dark states in TMDCs. Phys. Rev. Research 2, 023322 (2020).
Li, X. et al. Intrinsic electrical transport properties of monolayer silicene and MoS2 from first principles. Phys. Rev. B 87, 115418 (2013).
Jin, Z. et al. Intrinsic transport properties of electrons and holes in monolayer transition-metal dichalcogenides. Phys. Rev. B 90, 045422 (2014).
Takayama, R., Kwong, N. H., Rumyantsev, I., Kuwata-Gonokami, M. & Binder, R. T-matrix analysis of biexcitonic correlations in the nonlinear optical response of semiconductor quantum wells. Eur. Phys. J. B Condens. Matter Complex Syst. 25, 445–462 (2002).
Schumacher, S., Czycholl, G. & Jahnke, F. Coherent propagation of polaritons in the nonlinear optical regime. Phys. Rev. B 73, 035318 (2006).
Acknowledgements
We thank Dominik Christiansen for many stimulating discussions. We gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft via the Projects No. 420760124 (KN 427/11-1, F.K., A.K.) as well as No. 182087777-SFB 951 (B12, M.S., A.K.). We also acknowledge support of the European Union’s Horizon 2020 research and innovation program under Grant Agreement No. 734690 (SONAR, A.K. and F.S.). This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skodowska-Curie (grant agreement no. 734690) and from the European Research Council (ERC, grant agreement no. 816313- F.S. and no. 850875- I.K.). G.C. and S.D.C. acknowledge support by the European Union Horizon 2020 Programme under Grant Agreement No. 881603 Graphene Core 3. S.D.C. and C.T. acknowledge financial support from MIUR through the PRIN 2017 Programme (Prot. 20172H2SC4). P.J.S. and K.Y. acknowledge support from Programmable Quantum Materials, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under award DE-SC0019443. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05-CH11231. This research was supported in part by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under Contract No. DE-AC02-05-CH11231, within the van der Waals Heterostructures Program (KCWF16), which provided for sample growth, and under the sp2-bonded Materials Program (KC2207), which provided for SEM sample characterization (A.Z. and A.Y.). Support was also provided by the U.S. National Science Foundation under Grant No. DMR-1807233 which provided for additional TEM sample characterization (A.Z. and A.Y.).
Author information
Authors and Affiliations
Contributions
N.J.B., S.D.C., G.C., and P.J.S. conceived the experiment. C.T. and S.D.C. designed the experiment and performed the measurements. R.B.V., I.K., and F.S. contributed to the experimental work. F.K., M.S., and A.K. performed the theoretical calculations. K.Y., A.Y., and A.Z. prepared and characterized the sample. All the authors wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trovatello, C., Katsch, F., Borys, N.J. et al. The ultrafast onset of exciton formation in 2D semiconductors. Nat Commun 11, 5277 (2020). https://doi.org/10.1038/s41467-020-18835-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18835-5
This article is cited by
-
Spin-polarized spatially indirect excitons in a topological insulator
Nature (2023)
-
Radiative suppression of exciton–exciton annihilation in a two-dimensional semiconductor
Light: Science & Applications (2023)
-
Anomalous non-equilibrium response in black phosphorus to sub-gap mid-infrared excitation
Nature Communications (2022)
-
Blue light polymeric emitters for the development of OLED devices
Journal of Materials Science: Materials in Electronics (2022)
-
Efficient phonon cascades in WSe2 monolayers
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
