
Abstract
Efficient broadband infrared (IR) light-emitting diodes (LEDs) are needed for emerging applications that exploit near-IR spectroscopy, ranging from hand-held electronics to medicine. Here we report broadband IR luminescence, cooperatively originating from Eu2+ and Tb3+ dopants in CaS. This peculiar emission overlaps with the red Eu2+ emission, ranges up to 1200 nm (full-width-at-half-maximum of 195 nm) and is efficiently excited with visible light. Experimental evidence for metal-to-metal charge transfer (MMCT) luminescence is collected, comprising data from luminescence spectroscopy, microscopy and X-ray spectroscopy. State-of-the-art multiconfigurational ab initio calculations attribute the IR emission to the radiative decay of a metastable MMCT state of a Eu2+-Tb3+ pair. The calculations explain why no MMCT emission is found in the similar compound SrS:Eu,Tb and are used to anticipate how to fine-tune the characteristics of the MMCT luminescence. Finally, a near-IR LED for versatile spectroscopic use is manufactured based on the MMCT emission.
Similar content being viewed by others


Introduction
There is an urgent technological demand for broadband infrared (IR) phosphors to build phosphor-converted IR light-emitting diodes (pc-LEDs) with a broad spectral output. Broadband near-IR LEDs are a promising energy-efficient alternative for incandescent lamps for applications that rely on near-IR spectroscopy1,2. A large market that is presently explored is IR LEDs for the use in smart devices (e.g., mobile phones) to analyze food: information about its freshness, caloric value, or allergen content can be derived from the built-in spectrometer and IR LED3. Proof of concepts are demonstrated, but current state-of-the-art materials do not reach the desired energy efficiency, mostly due to the low absorption strength of the parity-forbidden 3d–3d transitions of the Cr3+ or Mn2+ ions that are used4,5,6,7,8,9,10. Alternatively, near-IR parity-allowed Eu2+ 5d–4f emission has been recently proposed11. Higher external quantum efficiencies can thus be achieved; however, at the cost of a smaller IR bandwidth. Material technologies and design strategies to obtain broadband IR phosphors that have a high absorption strength in the visible are hence highly desired to get this technology off the ground.
Other applications follow from the transparency of biological tissue for near-IR light and are hence situated in medicine12. This comprises imaging without the use of radioisotopes13,14,15, analyzing tissue during biopsy or endoscopy16, and IR therapy17. Also here, activators such as Cr3+ or Nd3+ feature substandard low excitation efficiencies due to the forbidden character of the used 3d–3d or 4f–4f transitions13,15. Circumventing this limitation can be done by incorporating Eu2+ in a suitable host, where the allowed 5d–4f emission is shifted towards longer wavelengths. A promising host for Eu2+ is CaS, where emission ~650 nm was shown in nanoparticles18,19. Upon co-doping with Dy3+, trapping can be induced upon ex situ UV excitation, after which red emission can be obtained upon in situ IR stimulation. As a downside, the red Eu2+ emission in this compound lies only partly in the first optical window of human skin tissue12, and would hence benefit from a further shift to the IR.
Large efforts have already been undertaken to optimize IR luminescent materials for the above-mentioned applications, starting from the well-known luminescence transitions such as 3d–3d, 4f–4f, or 5d–4f 4,5,6,7,8,9,10,13,20. On the contrary, charge transfer (CT) states have remained below the radar because they often cause luminescence quenching, rather than generating luminescence themselves. Nonetheless, the so-called anomalous emission in several phosphors has recently been attributed to intervalence CT (IVCT) transitions21,22,23,24,25,26, that is, electron transfers between two lanthanide dopants that differ only in oxidation state. CT between two different lanthanide elements, that is, metal-to-metal CT (MMCT) states, has not been reported as such. The only exception is the tentative assignment of a direct lanthanide-to-lanthanide CT absorption by Poolton et al.27 and concerns a Ce3+ + Sm3+ → Ce4+ + Sm2+ MMCT in YPO4.
CT states between dopants remain often unnoticed because their absorption bands are very weak, yet they are important28. The multivalent nature of lanthanide and transition metal ions induces numerous CT states at low energy, intercalating with the excited states that are typically responsible for the luminescence of the individual ions. Therefore, they are expected to significantly alter the excited state dynamics by quenching existing or by generating new luminescent levels.
Here, broadband IR emission is reported upon addition of Tb to the red phosphor CaS:Eu2+. This broadband IR emission can be efficiently pumped with long-wavelength visible light, substantiating an effective improvement of existing IR phosphors for pc-LED applications and for in vivo biomedicine without the need for prior ex situ charging nor expensive detection in the second or third optical window.
CaS:Eu2+,Tb3+ and SrS:Eu2+,Tb3+ are investigated in detail in a combined experimental–theoretical study that evidences that the observed IR emission is due to the radiative decay of MMCT states of Eu2+–Tb3+ pairs, a type of luminescence that was hitherto not described to the best of our knowledge. The ab initio multiconfigurational calculations explain why the MMCT luminescence is found for CaS:Eu2+,Tb3+, but not for SrS:Eu2+,Tb3+. This knowledge is utilized to show that MMCT emission can only be obtained when several conditions in terms of the electronic structure of the lanthanide pair, and the structural rigidity of the host crystal are fulfilled. This goes beyond the mere location of energy levels, but comprises vibrational frequencies and lanthanide–ligand bond lengths as well. The reported IR phosphor is finally applied to construct a broadband near-IR LED with a radiant output that surpasses the current state of the art.
Results and discussion
Photoluminescence spectra
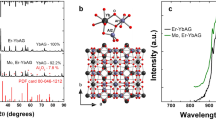
The photoluminescence (PL) emission spectra for various MS:Eu,Tb (M = Ca,Sr) singly and codoped powders are shown in Fig. 1. Singly Eu-doped MS shows the characteristic broadband emission, peaking ~650 and 615 nm for CaS and SrS, respectively. This band is attributed to the radiative de-excitation of the 4f65d1 states of Eu2+ towards the 4f7(8S) ground state29,30,31. The associated excitation spectrum as shown in Fig. 2 consists of a very broad band, ranging from 410 to 610 nm, composed of numerous transitions towards the dense \(4{f}^{6}5d{t}_{{\rm{2}}g}^{1}\) manifold31,32. No trace of Eu3+ line emission is found. This does however not exclude its presence, because IVCT states are known to quench the Eu3+ emission in case it pairs with Eu2+ ions28. However, high-energy-resolution fluorescence-detected X-ray absorption near-edge structure (HERFD-XANES33,34) spectra show that no Eu3+ is present in the prepared samples within the detection limits (see Supplementary Fig. 2).

Photoluminescence emission spectra of Eu2+- and Tb3+-codoped CaS (a) and SrS (b) phosphors. The spectra were collected upon 285 nm excitation for the Eu- and Eu/Tb-codoped powders and upon 305 nm excitation for the Tb-doped powders. The spectral shape of the infrared metal-to-metal charge transfer (MMCT) emission is highlighted in yellow as a guide to the eye. All spectra were measured at room temperature.

Photoluminescence excitation spectra of Eu2+- and Tb3+-codoped CaS phosphors. Spectra were collected for the Tb3+ 4f–4f emission (545 nm, green solid lines), the Eu2+ 5d–4f emission (650 nm, red and gold solid lines) and the Eu–Tb metal-to-metal charge transfer (MMCT) emission (790 nm, dashed line). The estimated spectral profile of the Eu–Tb MMCT excitation band is highlighted in yellow. The Eu2+ 5d–4f and Eu-Tb MMCT emission spectral profiles are shown in addition (gray dotted lines). All spectra were measured at room temperature.
Singly Tb-doped MS shows characteristic Tb3 + 5D4 → 7FJ line emission across the visible range (green curve in Fig. 1), most notably in the green (545 nm, J = 5). This intraconfigurational 4f8 emission can be excited by a relatively high-lying 4f8 → 4f75d1 excitation band in the near-UV (Fig. 2)35,36,37. In case of CaS and SrS, the fundamental absorption of the host lies in the same energy range, and the near-UV absorption and excitation bands are likely the result of a mixture of host- and dopant-related transitions30,31.
When Eu and Tb are combined in the same sample, an additional IR emission band emerges in case of CaS, but not in case of SrS. This band is highlighted in Fig. 1. It peaks at 810 nm (12,345 cm−1) and is very broad, with a full-width at half-maximum of 195 nm (2960 cm−1). On the high-energy side, it overlaps with the Eu2+4f65d1 → 4f7 luminescence around 650 nm and extends up to 1100–1200 nm. Its excitation spectrum (dashed curve in Fig. 2) is very similar as for the Eu2+ luminescence, where the \(4{f}^{7}\to 4{f}^{6}5d{t}_{{\rm{2}}g}^{1}\) band can be identified, with a small redshift of 20 nm, corresponding to about 500 cm−1. In addition to this band, some excitation intensity can be found in the region around 370 nm where no allowed transitions for Eu2+ are found31, suggesting the presence of additional excited states when Eu and Tb are codoped. These features are also visible at low temperature, as well as in the diffuse reflectance spectra (see Supplementary Figs. 6 and 7).
Lifetime measurements (see Supplementary Fig. 9 and Supplementary Table 1) indicate that the IR emission bands feature a very similar decay behavior than the Eu2+ 5d → 4f emission, with time constants around 500 ns at room temperature38.
In order to exclude that this previously unseen emission band is due to the used precursors, codoped powders from different precursor batches were prepared, using fluorides, oxides, and sulfides as lanthanide precursors39,40,41,42. All syntheses where Eu and Tb were both present as dopants resulted in the same IR emission band, while this IR band was always absent in case that only one dopant was used (see Supplementary Fig. 4) . The similarity of the IR emission regardless of the synthesis conditions suggests that the details of the charge compensation mechanism for Tb3+ do not directly affect the IR emission. Explicit compensation of Tb3+ by adding a monovalent codopant such as Na+ is shown to be detrimental for the luminescence properties, leading to a efficiency drop of a factor 10 (see Supplementary Fig. 8). The reason is that, when Tb3+ is extrinsically compensated, the formation of Eu3+ will also be favored29, generating undesired Eu2+–Eu3+(–Na+) centers in addition to the intended Eu2+–Tb3+(–Na+) centers. The former centers are responsible for the luminescence efficiency drop by IVCT quenching28,43.
This check, along with the demonstrated phase purity (from X-ray diffraction (XRD), see Supplementary Fig. 1) and the presence of only one oxidation state for both dopants in the absence of extrinsic charge compensation, that is, Eu2+ and Tb3+ (from HERFD-XANES, see Supplementary Fig. 2) strongly suggests that this IR emission band is a physical effect that emerges due to an interaction between the Eu2+ and Tb3+ centers in the calcium sulfide crystal.
To acquire more information about this peculiar luminescence, its properties are investigated as a function of the Eu and Tb doping concentrations. The PL emission spectra indicate that the Tb3+ intraconfigurational 4f8 emission is firmly diminished upon the addition of Eu2+ ions until it completely vanishes (Fig. 1). This is not surprising due to the large overlap between the Tb3+ emission and Eu2+ excitation spectra, enabling an efficient energy transfer where the Tb3+ ion sensitizes the Eu2+ luminescence44,45. When a small amount of Eu2+ is added to a Tb3+-doped sample (CaS:Eu0.001Tb0.01), or vice versa (CaS:Eu0.01Tb0.001), the IR emission shows up, but with a limited intensity. The relative intensities are comparable in both cases (see Fig. 1). For higher concentrations, 1% for both dopants (CaS:Eu0.01Tb0.01), the IR emission stands out, featuring a larger integrated intensity than the Eu2+5d → 4f emission. Upon increasing the doping concentrations even more (CaS:Eu0.03Tb0.03), the IR emission dominates the entire emission spectrum.
The emergence of the IR emission upon Tb addition to CaS:Eu is accompanied by a decrease in PL quantum efficiency (QE), which decreases from 35% for CaS:Eu0.01 to 10% for CaS:Eu0.01Tb0.01 (see Supplementary Fig. 8). This relatively low internal QE is partly compensated by the efficient excitability of the IR emission (see Fig. 2), where >90% of the incident visible (400–600 m) light is absorbed (see Supplementary Fig. 7). It is hence clear that practical applications require a trade-off between conversion efficiency and the fraction of IR in the emission spectrum (see further)46.
Concentration dependence
The concentration-dependent PL study indicates that the intensity of the IR emission scales with the product of the concentrations of both dopants; however, drawing quantitative conclusions is hindered by the limited number of concentrations that can be prepared and does not account for uncontrollable microscopic concentration differences47,48,49,50. To get a more detailed picture, a microscopic study is performed.
For this, two grains with extreme doping inhomogeneity are explored51,52,53. It should be stressed that these grains were selected for this purpose and that they do not represent the global doping homogeneity of the phosphors, which is much better. As shown in Supplementary Fig. 3 and the accompanying discussion, variations in local concentrations are limited to less than a percent, corresponding to a decent doping homogeneity.
The microscopic study of the inhomogeneous grains is shown in Fig. 3. One grain exhibits predominantly red emission (lower left), while the other grain shows a strong IR emission (upper right). From the elemental analysis by energy-dispersive X-ray spectroscopy (EDX), it is clear that the doping is indeed inhomogeneous and that the local Eu and Tb concentrations range from 0 to roughly 4%54. As a direct consequence, the cathodoluminescence (CL) spectrum shows strong variations across the sample because the intensity of the IR emission depends strongly on the Eu and Tb concentrations. As an illustration, five local spectra are shown in Fig. 3c. It is clear that the smaller grain on the bottom of the image shows negligible IR emission (Fig. 3c, d), which is compatible with the elemental analysis that suggests that Eu and Tb are well separated in this grain, indicated by a limited amount of yellow in Fig. 3b. For the larger grain, Eu and Tb clearly congregate (there is more yellow in Fig. 3b) and intense IR emission is found (Fig. 3c, d).

Scanning electron microscope (SEM) study on a CaS:Eu0.01Tb0.01 powder. Backscattered electron image (a), along with energy-dispersive X-ray (EDX) maps for terbium and europium (b). The colors encode the local terbium (green) and europium (red) concentrations. Simultaneous detection leads to a yellow color. c Cathodoluminescence (CL) spectra for a few selected points that are indicated in d. d False color image, displaying the total integrated CL intensity. e shows how the relative contributions of the red Eu2+ (integrated from 600 to 650 m) and IR (integrated from 760 to 815 m) luminescence band to the total CL spectrum evolve as a function of the local Eu and Tb concentrations, [Eu] ⋅ [Tb].
From the PL study, it is clear that both Eu and Tb are required to induce IR emission; therefore, the product of both local concentrations, [Eu] ⋅ [Tb], determined per pixel in the scanning electron microscope (SEM)-EDX map, is used as dependent variable to correlate the luminescence properties to the CL spectrum, measured for the same pixel49,55. The red and IR contributions to the local CL spectrum are integrated and subsequently analyzed as a function of [Eu] ⋅ [Tb]. Figure 3e was obtained by averaging data points along the abscissa. At low [Eu] ⋅ [Tb] values, the IR emission increases linearly as a function of [Eu] ⋅ [Tb], while the red Eu2+ emission decreases accordingly. After the linear increase/decrease, the relative intensities stabilize and the spectrum does not change appreciably upon increasing the product of the doping concentrations above roughly 3 × 10−4, which corresponds to a symmetric doping concentration, \(\sqrt{[{\rm{Eu}}]\cdot [{\rm{Tb}}]}\), of 1.7%. No spot is found where the red Eu2+ emission completely disappears, not in the area displayed in Fig. 3, nor in any other grain that was investigated, nor in the emission spectra of phosphors with doping concentrations >5% (see Supplementary Fig. 5). This equilibrium between red and IR emissions implies that the IR emission is presumably not the result of an energy transfer from Eu2+ to another emitting center because it would then be expected for the Eu2+ emission to vanish completely if the concentrations are stretched sufficiently44,56. Yet, the Eu2+ absorption bands are clearly present in the excitation spectrum of the IR band. This means that the IR emission stems directly from a Eu2+-containing defect cluster.
Temperature dependence
Measurement of the PL intensities as a function of temperature, that is, a thermal quenching (TQ) experiment can reveal more information about the equilibrium between the red and IR emissions. The result is shown in Fig. 4. The red Eu2+ emission follows a rather standard behavior, which resembles the shape of a single-barrier model57,
Using this phenomenological model to fit the data yields a barrier height of ΔET,red = 1484 cm−1 (A = 1.07 × 103). This TQ performance is comparable to the one of singly doped CaS:Eu0.01 phosphors, which contain sufficient Eu for concentration quenching to be noticeable29,58,59.

Thermal quenching curves for the red and infrared (IR) emission bands of CaS:Eu0.01Tb0.01, measured upon 470 nm excitation. The black curves are the result of fitting Eqs. (1) and (2) for the red and IR emissions, respectively. The small shoulder ~340 K is likely due to thermoluminescence (TL) and is not explored further here. The gray curve shows the thermal quenching of singly doped CaS:Eu0.01 for comparison.
In contrast to the red emission, which shows the expected TQ behavior, the IR emission looks more complicated as a function of temperature, with an increase in intensity between 100 and 225 K. This indicates that the IR emission is to some extent thermally activated. However, a substantial fraction of the IR emission, ~77% of the maximal output at 225 K, is also being emitted at low temperature. This is reminiscent of the temperature dependence of internal conversion (IC) and inter-system crossing (ISC) in molecular chromophores where rate constants are typically written as the sum of a temperature-dependent and temperature-independent term60,61. The IR TQ curve can hence be modeled by combining the ISC rate constant with a single-barrier model for the TQ behavior,
where I1 and I2 represent the temperature-independent and temperature-dependent rate constants, respectively. ΔEISC is the associated barrier height for the latter. Fitting Eq. (2) to the TQ profile of the IR emission yields ΔEISC = 476 cm−1 and ΔET,IR = 1500 cm−1 as barriers (I1/I2 = 9.5 and A = 1.61 × 103). The TQ at higher temperature is roughly the same as for the red emission, indicated by the similar energy barriers for quenching.
The intensities of the red and IR emission of the Eu,Tb-codoped phosphor already decrease at lower temperature with respect to the red emission of singly Eu-doped CaS (see gray curve in Fig. 4), featuring ΔET,red = 1989 cm−1 (A = 2.04 × 103, Eq. (1)). This indicates that the addition of Tb opens an additional non-radiative decay channel that is active around room temperature. At higher temperatures, the TQ of the singly Eu-doped and Eu,Tb-codoped phosphors coincide, reaching 50% of the initial intensity at T0.5 ≈ 375 K. This value is in correspondence with prior studies58,59,62, even though values of 475 K have been reported for single crystals58, suggesting that efficiency gains are still feasible by optimizing the synthesis.
As shown by a model calculation by Struck and Fonger and subsequent surveys of experimental literature by various authors, the physical meaning of the above-determined energy barriers is rather limited due to tunneling effects and the importance of the details of the electron-vibrational structure on the non-radiative transition probabilities63,64,65,66. For that reason, Eqs. (1) and (2) should be regarded as strictly empirical prescriptions.
Qualitative interpretation of the above analysis suggests that an excited Eu2+ ion in the Eu2+, Tb3+-codoped material has two radiative decay possibilities, the standard red luminescence, and the IR luminescence which is achieved after some kind of internal transition towards another energy level. If both emissions would originate from the same initial level and differ in final level, the temperature-induced intensification of the IR emission would not be expected.
MMCT model
The experimental findings suggest that MMCT states might be involved in the complicated luminescence of this material. Given the oxidation states of the dopants in this compound, Eu2+ and Tb3+, the most probable scenario to be investigated are the Eu3+-Tb2+ MMCT states, that is, those where an electron is transferred from Eu2+ to Tb3+. To this means, ab initio embedded cluster calculations are employed (see Fig. 5).

Graphical representation of the simulated system and how it is divided in the (LnS6M6)2+/3+ (M = Ca, Sr and Ln = Eu, Tb) cluster, which is treated at the highest level of theory (right), subject to the embedding potential composed of the full-ion ab initio model potentials (AIMP) of the 316 ions in the next four coordination shells (middle) and 24,389 point charges at the crystal lattice sites (left). The indicated distances are for CaS and are slightly larger for SrS.
Diabatic potential energy surfaces and configurational diagrams of Eu2+-Tb3+ pairs are obtained from the results of independent embedded cluster calculations23,67. This approach has proven its reliability by explaining the anomalous emission of several Ce- and Yb-doped phosphors23,25,26,68, predicting the existence of absorption bands due to IVCT states in Eu-doped phosphors28 and by showing the role of MMCT states at quenching luminescent levels of Pr3+69.
The configurational coordinate diagrams along the breathing mode for Eu2+, Tb3+, Eu3+, and Tb2+ are the ingredients for the electron transfer diagrams that describe the Eu-to-Tb MMCT transitions according to the recipe in refs. 22,23. Calculations are performed for CaS and SrS hosts. Details, intermediate and final results are collected in Supplementary Tables 2–7 and in Supplementary Figs. 10–15.
The excited state landscape of Eu2+ was previously discussed in detail in ref. 31 and is practically defined by a single ground state level, 4f7(8S7/2), separated from a very dense 4f65d1 manifold by ~16,000–17,000 cm−1. Eu3+ and Tb3+ feature conjugate ground state configurations with 4f6(7F0) and 4f8(7F6) multiplets, respectively. The higher-lying 4f6(5D0) and 4f8(5D4) states are the main 4f–4f emitting levels. The 4fN − 15d1 configurations feature high excitation energies for these trivalent lanthanides and are hence unnecessary for the current calculation. For Tb2+, the 4f85d1 and 4f9 configurations are close in energy, the former constituting the ground state in CaS (1Γ7g), while a reversed order is found for SrS (1Γ7u). More details about the electronic structures of Tb3+ and Tb2+ in CaS and SrS are given in the Supplementary Discussion, in particular regarding the relative energy of the 4f85d1 and 4f9 manifolds of Tb2+. The equilibrium Eu–S and Tb–S bond lengths and breathing mode vibrational frequencies are summarized in Table 1. Prior comparisons with experimental results for Eu2+ and Eu3+ proved an excellent quantitative agreement of <300 cm−1 for excitation energies and at most a few % for equilibrium bond lengths and vibrational frequencies31.
Within the diabatic approximation, the energy level scheme of a lanthanide pair can be constructed by combining all the levels of the individual ions, the resulting energy is given by addition of the individual energies with the Coulomb and exchange energies between the two lanthanides. The last two contributions are assumed to be state independent67. The resulting energy levels are uniquely labeled by combining both individual labels. As an example, the ground state of a Eu2+–Tb3+ pair is denoted as 4f7(8S7/2)–4f8(1A1g). Diabatic potential energy curves have proven their use by successfully explaining qualitative trends of CT processes upon chemical substitutions in host compounds22,23,28,43.
For every energy level of the Eu–Tb pair, a two-dimensional potential energy surface is obtained, spanned by the breathing modes of the EuS6 and TbS6 moieties. Every point in this two-dimensional space hence corresponds to a unique pair of (dTb−S,dEu−S). The equilibrium point of the Eu2+–Tb3+ states correspond to a relatively large dEu−S value (divalent ion) and relatively small dTb−S value (trivalent ion). The Eu2+–Tb3+4f65d1(1Γ8g)–4f8(1A1g) potential energy surface is represented in the top panels of Fig. 6 for CaS and SrS (green contours), along with the lowest MMCT level, Eu3+–Tb2+4f6(1A1g)–4f8(1Γ7g) (black contours), where dEu−S decreased (trivalent ion) and dTb−S increased (divalent ion). The intersection of both potential energy surfaces is a curved line (dashed red line in the contour plots of Fig. 6).

Contour plots of selected potential energy surfaces in the two-dimensional configurational space (dEu–S, dTb–S) (a, b) and configurational coordinate diagrams along the electron transfer reaction coordinate (Qet) (c, d) for Eu–Tb pairs in CaS (a, c) and SrS (b, d). The configurational coordinate diagrams display the following levels for the Eu2+–Tb3+ configuration: 4f7(8S)–4f8(7F) (black), \(4{f}^{6}5d{t}_{{\rm{2}}g}^{1}-4{f}^{8}(1\,{A}_{{\rm{1}}g})\) (green for high-spin states, red for low-spin states) and 4f7(8S)–4f8(5D, L, G) (gold), and following levels for the Eu3+–Tb2+ configuration: 4f6(7F)–4f85d1(1Γ7g) (black), 4f6(1A1g)–4f85dt2g (green for high-spin states, red for low-spin states), and 4f6(1A1g)–4f9 (gray). The quasi-continuum formed by all other levels of the Eu–Tb pair, where none of both ions is in its ground state are represented by the green-colored background. The contour plots display the isolines corresponding to the Eu2+4f65d1(1Γ8g)–Tb3+4f8(1A1g) and Eu3+4f6(7F)–Tb2+4f85d1(1Γ7g) (lowest metal-to-metal charge transfer (MMCT) state) surfaces. The reaction coordinate is indicated in red, along with the intersection between both surfaces (dashed red line).
The lower panels of Fig. 6 display the configurational coordinate diagrams for Eu2+–Tb3+ pairs in both host compounds along the electron transfer reaction coordinate, Qet. This coordinate is shown in the contour plots and is defined here as the piecewise straight line that connects the equilibria of the Eu2+–Tb3+4f65d1(1Γ8g)−4f8(1A1g) and Eu3+–Tb2+4f6(1A1g)−4f85d1(1Γ7g) potential energy surfaces in the two-dimensional (dEu−S, dTb−S) configurational space with their saddle point, that is, the minimum of the intersection of both surfaces. This one-dimensional diagram is a simplification of the two-dimensional space that is probed for convenient visualization. Reported data such as location of minima, crossing points, barriers, and transition energies are however obtained from the two-dimensional surfaces.
The configurational coordinate diagrams show that the Eu3+–Tb2+ MMCT configuration gives rise to low-lying states, lying around 20,000 cm−1 above the ground state (Table 1). The presence of these MMCT states alters the excited state dynamics after 4f7 → 4f65d1 excitation of the Eu2+ ion. The excited electron can be non-radiatively transferred to the Tb3+ ion, forming a transient Eu3+–Tb2+ pair. Shortly, the Tb2+5d electron is transferred back to the Eu3+4f orbital, leading to a decay of the MMCT state.
Different decay channels are found in the case of Eu,Tb-codoped CaS or SrS, evoked by their structural differences. In case of CaS, the minimum of the lowest MMCT state (4f6(1A1g)−4f85d1(1Γ7g)) is metastable. Therefore, radiative decay of this state can be expected to the structurally stressed 4f7(8S7/2)−4f8(6FJ) ground state (red arrows in Fig. 6c). In SrS, the minimum of the lowest MMCT state is crossed by the branches of the stressed 4f7(8S7/2)−4f8(6FJ) states, enabling efficient non-radiative decay due to fast bottom crossover (red arrows in Fig. 6d) that impedes any radiative decay. The associated diabatic energy barrier is 94 cm−1, and will likely disappear or be of negligible size in an adiabatic calculation. This is indeed the behavior, which is experimentally found for Eu,Tb-codoped CaS and SrS.
Due to the large horizontal offset between ground and MMCT states, the resulting emission band in case of CaS:Eu,Tb is expected to be broad. The MMCT emission is predicted to start around 11,000 cm−1 (900 nm), which is at slightly lower energy than the experimental IR emission, which starts ~14,000–15,000 cm−1. This quantitative discrepancy between experimental and computed transition energies is in line with what can be expected from the diabatic approximation23,67.
The low-lying MMCT states do not only cause an additional emission band. Vertical excitation from the ground state towards the MMCT states is possible starting from about 28,000 cm−1 (360 nm), which coincides with the energy range where the low-spin \(4{f}^{6}5d{t}_{{\rm{2}}g}^{1}\) levels are found. The latter are spectroscopically invisible by direct excitation from the 4f7(8S7/2) ground state because of the spin selection rule31. The presence of the Eu–Tb pairs and the MMCT states induces transition probability in this otherwise forbidden energy region as evidenced by the excitation band ~370 nm in the experimental spectrum (Fig. 2). An estimated spectral shape is highlighted as a guide to the eye. This MMCT absorption is also visible in the excitation spectrum of the regular Eu2+ red emission at 650 nm for the CaS:Eu0.01,Tb0.01 sample, indicating that a substantial fraction of the dopants can already interact in the studied concentration range.
The PL excitation spectrum of the MMCT emission (dashed line in Fig 2) indicates that it can be excited with the same wavelengths as the regular Eu2+ emission, even when the photon energy of the excitation light is insufficient to reach an MMCT branch by vertical excitation from the ground state. This can be explained by considering the double-well shape of the potential energy surface that is formed by the Eu2+(1Γ8g)–Tb3+(1A1g) red-emitting and the Eu3+(1A1g)–Tb2+(1Γ7g) IR-emitting MMCT state. Even at low temperature, when the barrier cannot be thermally overcome, quantum mechanical tunneling will partially populate the MMCT state. This does not only explain why both emissions always appear together, but also why the IR emission is intensified when sufficient thermal energy is available to overcome the barrier and why the luminescent lifetimes for both emission bands are comparable.
Broadband IR LED
The broadband IR MMCT emission is now applied to construct an IR pc-LED that can directly be used for the numerous above-mentioned spectroscopic applications. Because of the high absorption strength of the parity-allowed 4f−5d transitions of Eu2+, higher external quantum efficiencies can be achieved than with the current state-of-the-art, Cr3+-based phosphors4,5,6,8,9,10. Furthermore, the extremely broad MMCT emission extends the covered spectral range by several hundreds of nanometers to the IR compared to red/near-IR single Eu2+5d−4f emission11.
Figure 7 displays the spectrum, expressed in mW nm−1, for the obtained LED, where a blue 450 nm pumping LED was used. The LED features a width of 430 nm in the IR, ranging from 620 to 1050 nm. The operation of the IR LED is illustrated by the pictures in Fig. 7b–e. Here, a high-pass cut-off filter of 780 nm is used to filter out the transmitted blue pumping light in order to appreciate the brightness of the IR emission. The pictures were taken with a camera that is also sensitive to near-IR light, as the emission is barely visible by the naked eye. The total radiant flux of the IR part of the emission amounts to 38 mW. This value surpasses the current Cr3+-based state-of-the-art, having IR radiant fluxes in the range of 20–25 mW4,5,6,8,9,10, by 50%.

a Spectral output of the constructed broadband infrared (IR) light-emitting diode (LED). The inset shows the LED design, comprising a blue pumping LED (b), combined with the CaS:Eu,Tb broadband infrared phosphor (d), under operation below a 780-nm high-pass cut-off filter without (c) and with (e) the infrared phosphor present, captured by a camera with a near-IR responsivity.
It is clear that this broadband IR MMCT emission has a huge application potential. The next step comprises further optimization of its luminescence to arrive at higher efficiencies and to enable some spectral tuning to optimally meet the requirements of the different applications. As the luminescence mechanism in CaS:Eu,Tb was resolved in detail by our ab initio calculations, some prospects and further insights can be given based on these findings.
As shown, SrS is not a suitable host to obtain a luminescent MMCT state. There are two main differences between CaS and SrS that are crucial in this regard. First, the vibrational frequency is smaller by a few tens of cm−1 in case of SrS (242 cm−1, compared to 292 cm−1 for CaS, see Table 1), which causes a slight opening of the branches that cross the MMCT state. This effect is not large because the vibrational frequency differs only by a small amount between CaS and SrS. Second, the offset in equilibrium geometry for the ground and MMCT states is larger for SrS (Qet = 0.548 Å, compared to 0.428 Å for CaS). This is the direct consequence of the larger bond length difference between divalent and trivalent lanthanides with respect to CaS (see Table 1) and causes the MMCT minimum to be pushed into the 4f7(8S7/2)−4f8(6FJ) branches, causing quenching of the MMCT state. The latter parameter is the dominant one for the different behavior of CaS:Eu,Tb and SrS:Eu,Tb.
The above analysis can be expanded to devise guidelines for finding other phosphors that exhibit MMCT luminescence. To achieve this, the ground state potential energy surface should cross the emitting MMCT level at a sufficiently large Qet value. For this, small curvatures are required, which translates into small vibrational frequencies, a property that is not only typical for sulfide hosts30,70, but also for selenides30, nitrides71, chlorides72, bromides72, and iodides73. Additionally, the crossing between the ground state and the MMCT level can be shifted away from the MMCT minimum by decreasing the horizontal offset between both parabolas, that is, by decreasing the equilibrium Qet value. This value is proportional to the change in lanthanide–ligand bond length upon CT and to the square of the coordination number of the lanthanide67. MMCT luminescence will hence be more probable in hosts with a small site for the lanthanide as these experience smaller bond length changes. Ca-based hosts, preferably with a low coordination number, are hence more desirable with respect to Sr- or Ba-based hosts.
When the chemical composition of host compounds is engineered to accomplish MMCT luminescence, also the vertical offset, and hence the MMCT emission energy, is expected to be affected. Indeed, the vertical offset is given by the difference of the ionization potential (IP) of the donor and the electron affinity (EA) of the acceptor, supplemented with the Coulomb and exchange interaction between both ions67, and these parameters are host dependent. A more radical manipulation of the vertical offset can be achieved by substituting the lanthanide ions. A rough idea on the effect of host modification and lanthanide substitution on the vertical offset can be acquired by consultation of the lengthy empirical data on IP’s and EA’s of lanthanide ions and their systematic behavior74,75.
In summary, the presence or absence of MMCT luminescence, and the emission energy is affected by three parameters: the local vibrational frequency, bond length change, and the selected lanthanide pair. A perfect balance between these parameters is required to achieve MMCT luminescence as in the case of CaS:Eu2+,Tb3+.
In this combined experimental–theoretical study, broadband IR emission in CaS:Eu2+,Tb3+ is reported, characterized, and explained. The emission spectrum overlaps with the regular red Eu2+5d−4f luminescence and ranges up to 1200 nm. Importantly, it can be efficiently pumped with long-wavelength visible light.
Concentration-dependent and microscopy experiments showed that the IR emission is caused by a cooperative effect between Eu and Tb. Ab initio multiconfigurational calculations support that the IR-emitting state is a Eu3+–Tb2+ MMCT state whose local structure differs significantly from the Eu2+–Tb3+ ground state.
The type of host has a critical influence on the properties of the MMCT luminescence, as shown by the fact that the IR emission is quenched in the similar compound SrS. This behavior was explained by the ab initio calculations, which show that the location of MMCT states, and hence their luminescence properties can be fine-tuned by tweaking a few parameters. These are experimentally accessible by altering the anions and cations in the host, or the lanthanide pair, namely the local vibrational frequencies and structural rearrangement upon MMCT, as well as the IP and electron affinities of the dopants.
The CaS:Eu,Tb phosphor was used to construct a broadband IR pc-LED for spectroscopic applications in smart electronics, food safety, and medicine. The IR emission of the LED covers a 430-nm-wide spectral range in the red and near-IR. Moreover, this is achieved with an IR output radiant flux of 38 mW, surpassing current state of the art.
Methods
Experimental method
Powders of CaS and SrS, doped with Eu and/or Tb were prepared by a solid-state synthesis, using high-purity CaS (Alfa Aesar, 99.9%), SrS (Alfa Aesar, 99.9%), EuF3 (Alfa Aesar, 99.95%), and TbF3 (Alfa Aesar, 99.9%) as precursors. Stoichiometric quantities were weighed, mixed, and subsequently heat treated in a tube furnace for 2 h at 1000 °C under a constant flow of H2S gas. After the heat treatment, the samples were allowed to cool naturally. Finally, the samples were slightly ground and stored in an inert atmosphere. All samples were phase pure, as verified by powder XRD (see Supplementary Fig. 1). Doping concentrations that were reported are molar concentrations with respect to the cation, for example, CaS:Eu0.010Tb0.001 is used for Ca0.989Eu0.010Tb0.001S.
PL emission and excitation spectra were measured on an Edinburgh FS920, using a 450 W xenon arc lamp as excitation source and equipped with a Hamamatsu R928P red-sensitive photomultiplier (wavelength range from 200 to 850 nm) and a Ge IR detector (700–1600 nm). Temperature-dependent PL was measured with the same spectrometer, equipped with a cryostat (Oxford Instruments Optistat CF).
The microscopy results were obtained with a Hitachi S-3400N SEM. A Thermo Scientific Noran System 7 EDX was used for chemical analysis and an optical fiber to collect the CL, which was subsequently analyzed by an Acton SP2300 monochromator and detected by a ProEM 1600 EMCCD (both Princeton Instruments). All shown spectra were properly calibrated for the spectral sensitivity of the various detectors.
An IR pc-LED was constructed using a blue pumping LED. For this a Xicato XTM LED module was used, operated at a constant current of 130 mA, corresponding to a voltage of 16.7 V. Its spectral radiant flux was obtained using a Thorlabs S401C thermal power meter.
Computational method
Diabatic potential energy surfaces and derived configurational coordinate diagrams were calculated for metal-to-metal electron transfer states of Eu2+/Tb3+ mixed valence pairs in CaS and SrS, using the results of independent embedded cluster calculations as proposed in refs. 22,23.
The electronic structures of the electron donor (Eu) and acceptor (Tb) octahedral embedded clusters (LnS6M6)2+ and (LnS6M6)3+ (M = Ca, Sr) were obtained with the suite of programs MOLCAS,76 using D2h symmetry, in two-step spin–orbit coupling state-average restricted-active-space self-consistent-field (SA-RASSCF)/multi-state second-order perturbation theory (MS-RASPT2)/restricted-active-space state-interaction spin–orbit (RASSI-SO) DKH calculations. In a first step, the spin–orbit-free many-electron relativistic second-order DKH Hamiltonian77,78 was used to perform all-electron calculations using the same type of basis sets as in ref. 31: Gaussian atomic natural orbital relativistic basis sets ANO-RCC for S79, Eu, and Tb80, with respective contractions (17s12p5d)/[6s5p3d] (quadruple-zeta with polarization without f-functions quality) and (25s22p15d11f4g2h)/[9s8p5d4f3g2h] (quadruple-zeta with polarization quality). In addition, the six-electron valence was explicitly added to the six alkaline earth metal ions next to the sulfur ligands in the [100] directions using adapted ANO-RCC basis sets, Ca(20s16p6d)/[3s4p1d] and Sr(23s19p12d)/[3s4p1d]. The inner shells of these ions were frozen, using a [Mg] and [Zn] core for Ca and Sr, respectively.
First, SA-RASSCF81,82,83 calculations were performed, allowing all possible occupations in the Ln 4f shells and up to four electrons in the Ln 5d, 6s, and 5f shells, in order to account for the so-called double-shell effect84. Following states were obtained: those of the 4f6 and 4f8 configurations of Eu3+ and Tb3+ for which 2S + 1 = 7, 5, those of the 4f7, 4f65d1, and 4f85d1 of Eu2+ and Tb2+ for which 2S + 1 = 8, 6 and those of the Tb2+ 4f9 configuration for which 2S + 1 = 6. States with lower multiplicities were not considered because they are not expected to influence the lowest part of the energy spectrum that is of interest. Subsequently, MS-RASPT285,86,87,88 calculations allowed to correlate all cluster valence electrons, except the 4d electrons of the lanthanides. A standard IPEA value (0.25 a.u.)89 and an imaginary shift of 0.15 a.u. (Eu) or 0.50 a.u. (Tb) was used. Second, the AMFI approximation of the DKH spin–orbit coupling operator was added to the Hamiltonian90 and RASSI-SO91,92 calculations were performed. Here, all states of a given cluster computed in the first step were allowed to interact.
In all calculations, the clusters were embedded in ab initio model potentials (AIMPs)93 that include Coulomb, exchange, and Pauli repulsion interactions from the CaS and SrS host lattices obtained in ref. 31 from self-consistent embedded ions94 Hartree–Fock calculations76. Figure 5 shows how the small cluster is embedded in AIMPs and point charges.
Data availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Multiconfigurational ab initio calculations were performed using MOLCAS.
References
Tessler, N., Medvedev, V., Kazes, M., Kan, S. & Banin, U. Efficient near-infrared polymer nanocrystal light-emitting diodes. Science 295, 1506–1508 (2002).
Gong, X. et al. Highly efficient quantum dot near-infrared light-emitting diodes. Nat. Photonics 10, 253–257 (2016).
Halper, M. Osram’s latest infrared LED identifies what’s in your food (UPDATED). LEDs Magazine (2018).
Hayashi, D., van Dongen, A. M., Boerekamp, J., Spoor, S., Lucassen, G. & Schleipen, J. A broadband LED source in visible to short-wave-infrared wavelengths for spectral tumor diagnostics. Appl. Phys. Lett. 110, 233701 (2017).
Zhang, L. et al. A high efficiency broad-band near-infrared Ca2LuZr2Al3O12:Cr3+ garnet phosphor for blue led chips. J. Mater. Chem. C 6, 4967–4976 (2018).
Rajendran, V. et al. Super broadband near-infrared phosphors with high radiant flux as future light sources for spectroscopy applications. ACS Energy Lett. 3, 2679–2684 (2018).
Song, E. et al. Heavy Mn2+ doped MgAl2O4 phosphor for high-efficient near-infrared light-emitting diode and the night-vision application. Adv. Opt. Mater. 7, 1901105 (2019).
Zeng, H., Zhou, T., Wang, L. & Xie, R.-J. Two-site occupation for exploring ultra-broadband near-infrared phosphor-double-perovskite La2MgZrO6:Cr3+. Chem. Mater. 31, 5245–5253 (2019).
Wang, C. et al. An ultra-broadband near-infrared Cr3+-activated gallogermanate Mg3Ga2GeO8 phosphor as light sources for food analysis. ACS Appl. Electron. Mater. 1, 1046–1053 (2019).
Zhang, L. et al. Cr3+-doped broadband NIR garnet phosphor with enhanced luminescence and its application in NIR spectroscopy. Adv. Opt. Mater. 7, 1900185 (2019).
Qiao, J., Zhou, G., Zhou, Y., Zhang, Q. & Xia, Z. Divalent europium-doped near-infrared-emitting phosphor for light-emitting diodes. Nat. Commun. 10, 5267 (2019).
Smith, A. M., Mancini, M. C. & Nie, S. M. Second window for in vivo imaging. Nat. Nanotechnol. 4, 710–711 (2009).
Maldiney, T. et al. The in vivo activation of persistent nanophosphors for optical imaging of vascularization, tumours and grafted cells. Nat. Mater. 13, 418–426 (2014).
Eggebrecht, A. T. et al. Mapping distributed brain function and networks with diffuse optical tomography. Nat. Photonics 8, 448–454 (2014).
Xu, J. & Tanabe, S. Persistent luminescence instead of phosphorescence: history, mechanism, and perspective. J. Lumin. 205, 581–620 (2019).
Evers, D. J. et al. Diffuse reflectance spectroscopy: a new guidance tool for improvement of biopsy procedures in lung malignancies. Clin. Lung Cancer 13, 424–431 (2012).
Ly, T. K. et al. Near-infrared organic light-emitting diodes with very high external quantum efficiency and radiance. Nat. Photonics 11, 63–68 (2017).
Rodriguez Burbano, D. C., Rodriguez, E. M., Dorenbos, P., Bettinelli, M. & Capobianco, J. A. The near-IR photo-stimulated luminescence of CaS:Eu2+/Dy3+ nanophosphors. J. Mater. Chem. C 2, 228–231 (2014).
Rodriguez Burbano, D. C., Sharma, S. K., Dorenbos, P., Viana, B. & Capobianco, J. A. Persistent and photostimulated red emission in CaS:Eu2+,Dy3+ nanophosphors. Adv. Opt. Mater. 3, 551–557 (2015).
Xu, J., Murata, D., Ueda, J., Viana, B. & Tanabe, S. Toward rechargeable persistent luminescence for the first and third biological windows via persistent energy transfer and electron trap redistribution. Inorg. Chem. 57, 5194–5203 (2018).
van Schaik, W., Lizzo, S., Smit, W. & Blasse, G. Influence of impurities on the luminescence quantum efficiency of (La, Ce, Tb)PO4. J. Eur. Ceram. Soc. 140, 216–222 (1993).
Barandiarán, Z. & Seijo, L. Intervalence charge transfer luminescence: interplay between anomalous and 5d–4f emissions in Yb-doped fluorite-type crystals. J. Chem. Phys. 141, 234704 (2014).
Seijo, L. & Barandiarán, Z. Intervalence charge transfer luminescence: the anomalous luminescence of cerium-doped Cs2LiLuCl6 elpasolite. J. Chem. Phys. 141, 214706 (2014).
Kulesza, D., Cybińska, J., Seijo, L., Barandiarán, Z. & Zych, E. Anomalous red and infrared luminescence of Ce3+ ions in SrS:Ce sintered ceramics. J. Phys. Chem. C 119, 27649–27656 (2015).
Strek, W. et al. Broadband anti-stokes white emission of Sr2CeO4 nanocrystals induced by laser irradiation. Phys. Chem. Chem. Phys. 18, 27921–27927 (2016).
Hughes-Currie, R. B. et al. X-ray excitation triggers ytterbium anomalous emission in CaF2:Yb but not in SrF2:Yb. J. Phys. Chem. Lett. 8, 1175–1178 (2017).
Poolton, N. R. J., Bos, A. J. J., Jones, G. O. & Dorenbos, P. Probing electron transfer processes in YPO4:Ce,Sm by combined synchrotron-laser excitation spectroscopy. J. Phys. Condens. Matter 22, 185403 (2010).
Joos, J. J., Seijo, L. & Barandiarán, Z. Direct evidence of intervalence charge-transfer states of Eu-doped luminescent materials. J. Phys. Chem. Lett. 10, 1581–1586 (2019).
Yamashita, N., Fukumoto, S., Ibuki, S. & Ohnishi, H. Photoluminescence of Eu2+ and Eu3+ centers in CaS:Eu,Na phosphors. Jpn. J. Appl. Phys. 32, 3135–3139 (1993).
Yamashita, N., Harada, O. & Nakamura, K. Photoluminescence spectra of Eu2+ centers in Ca(S,Se)Eu and Sr(S,Se)Eu. Jpn. J. Appl. Phys. 34, 5539–5545 (1995).
Joos, J. J., Smet, P. F., Seijo, L. & Barandiarán, Z. Insights into the complexity of the excited states of Eu-doped luminescent materials. Inorg. Chem. Front. 7, 871–888 (2020).
Weakliem, H. A. Electronic interactions in 4f65d configuration of Eu2+ in crystals. Phys. Rev. B 6, 2743–2748 (1972).
Gauthier, C., Sole, V. A., Signorato, R., Goulon, J. & Moguiline, E. The ESRF Beamline ID26: X-ray absorption on ultra dilute sample. J. Synchrotron Radiat. 6, 164–166 (1999).
Glatzel, P. et al. Reflections on hard X-ray photon-in/photon-out spectroscopy for electronic structure studies. J. Electron Spectrosc. 188, 17–25 (2013).
Lehmann, W. Activators and co-activators in calcium sulfide phosphors. J. Lumin. 5, 87–107 (1972).
Tripathi, R. N. & Sivaraman, S. Fluorescence spectra of CaS:(Tb, Eu) phosphors under X-ray excitation. Phys. Stat. Sol. A 55, K123–K126 (1979).
Leskelä, M., Niinistö, L., Nykänen, E., Soininen, P. & Tiitta, M. Electroluminescent thin-films containing terbium-activated strontium sulfide. J. Less Common Met. 153, 219–222 (1989).
Poort, S. H. M., Meijerink, A. & Blasse, G. Lifetime measurements in Eu2+-doped host lattices. J. Phys. Chem. Solids 58, 1451–1456 (1997).
Kato, K. & Okamoto, F. Preparation and cathodoluminescence of CaS:Eu and Ca1−xSrxS:Eu phosphors. Jpn. J. Appl. Phys. 22, 76–78 (1983).
Joos, J. J., Meert, K. W., Parmentier, A. B., Poelman, D. & Smet, P. F. Thermal quenching and luminescence lifetime of saturated green Sr1−xEuxGa2S4 phosphors. Opt. Mater. 34, 1902–1907 (2012).
Zhang, L. et al. The evolution and role of NH4Cl flux used to synthesize Sr2SiO4:Dy3+ phosphor by solid-state reaction method. J. Am. Ceram. Soc. 95, 3871–3877 (2012).
Liu, L. H., Xie, R.-J., Zhang, C. N. & Hirosaki, N. Role of fluxes in optimizing the optical properties of Sr0.95Si2O2N2:0.05Eu2+ green-emitting phosphor. Materials 6, 2862–2872 (2013).
Neefjes, I., Joos, J. J., Barandiarán, Z. & Seijo, L. Mixed-valence lanthanide-activated phosphors: invariance of the intervalence charge transfer (IVCT) absorption onset across the series. J. Phys. Chem. C 124, 2619–2626 (2020).
Dexter, D. L. A theory of sensitized luminescence in solids. J. Chem. Phys. 21, 836–850 (1953).
Tagiev, O. B. & Ganbarova, K. B. Energy transfer from Tb3+ to Eu2+ in Ga2S3:(Eu2+, Tb3+) crystals. Semiconductors 49, 448–451 (2015).
Leyre, S. et al. Absolute determination of photoluminescence quantum efficiency using an integrating sphere setup. Rev. Sci. Instrum. 85, 123115 (2014).
Poelman, D., Doerschel, J. & Smet, P. F. Cathodoluminescence study of the activator distribution in SrS:Cu,Ag thin luminescent films. Spectrochim. Acta B 59, 1775–1780 (2004).
Noto, L. L. et al. Photoluminescence and phase related cathodoluminescence dynamics of Pr3+ doped in a double phase of ZnTa2O6 and ZnAl2O4. Ceram. Int. 42, 5497–5503 (2016).
Martin, L. I. D. J., Poelman, D., Smet, P. F. & Joos, J. J. Microscopic study of dopant distribution in europium doped SrGa2S4: impact on thermal quenching and phosphor performance. ECS J. Solid State Sci. Technol. 7, R3052–R3056 (2018).
Li, W. Y., Smet, P. F., Martin, L. I. D. J., Pritzel, C. & Auf der Günne, J. S. Doping homogeneity in Co-doped materials investigated at different length scales. Phys. Chem. Chem. Phys. 22, 818–825 (2020).
Hirosaki, N., Takeda, T., Funahashi, S. & Xie, R.-J. Discovery of new nitridosilicate phosphors for solid state lighting by the single-particle-diagnosis approach. Chem. Mater. 26, 4280–4288 (2014).
Takeda, T., Hirosaki, N., Funahshi, S. & Xie, R.-J. Narrow-band green-emitting phosphor Ba2LiSi7AlN12:Eu2+ with high thermal stability discovered by a single particle diagnosis approach. Chem. Mater. 27, 5892–5898 (2015).
Wang, X. J., Funahashi, S., Takeda, T., Suehiro, T., Hirosaki, N. & Xie, R. J. Structure and luminescence of a novel orange-yellow-emitting Ca1.62Eu0.38Si5O3N6 phosphor for warm white LEDs, discovered by a single-particle-diagnosis approach. J. Mater. Chem. C 4, 9968–9975 (2016).
Packwood, R. H. & Brown, J. D. A gaussian expression to describe ϕ(ρz) curves for quantitative electron probe microanalysis. X-Ray Spectrom. 10, 138–146 (1981).
Smet, P. F., Botterman, J., Parmentier, A. B. & Poelman, D. Thermal quenching at the microscopic level in multi-phase thiosilicate phosphors. Opt. Mater. 35, 1970–1975 (2013).
Bettinelli, M., Speghini, A., Piccinelli, F., Ueda, J. & Tanabe, S. Energy transfer processes in Sr3Tb0.90Eu0.10(PO4)3. Opt. Mater. 33, 119–122 (2010).
Mott, N. F. On the absorption of light by crystals. Proc. R. Soc. Lond. Ser. A 167, 384–391 (1938).
Ando, M. & Ono, Y. A. Temperature effects in the emission characteristics of CaS:Eu thin-film electroluminescent devices. J. Cryst. Growth 117, 969–974 (1992).
Nyenge, R. L. et al. Thermal quenching, cathodoluminescence and thermoluminescence study of Eu2+ doped CaS powder. J. Alloy. Compd. 657, 787–793 (2016).
Meyer, B., Phillips, L. F. & Smith, J. J. Temperature dependence of intersystem crossing—lifetime and intensity of SO2 phosphorescence in low-temperature solids. Proc. Natl Acad. Sci. USA 61, 7–11 (1968).
Stacy, W. T. & Swenberg, C. E. Temperature dependence of intersystem crossing in crystalline anthracene. J. Chem. Phys. 52, 1962–1965 (1970).
Yang, L., Zhang, N., Zhang, R. Y., Wen, B., Li, H. L. & Bian, X. B. A CaS:Eu based red-emitting phosphor with significantly improved thermal quenching resistance for LED lighting applications. Mater. Lett. 129, 134–136 (2014).
Struck, C. W. & Fonger, W. H. Thermal quenching of Tb3+, Tm3+, Pr3+, and Dy3+ 4fn emitting states in La2O2S. J. Appl. Phys. 42, 4515–4516 (1971).
Struck, C. W. & Fonger, W. H. Unified model of temperature quenching of narrow-line and broad-band emissions. J. Lumin. 10, 1–30 (1975).
Struck, C. W. & Fonger, W. H. Temperature quenching of luminescence for linear and derivative nuclear operators. J. Lumin. 14, 253–279 (1976).
Koepke, C., Lempicki, A. & Wojtowicz, A. J. Luminescence quenching of strongly coupled systems. Phys. Stat. Sol. B 179, 233–240 (1993).
Barandiarán, Z., Meijerink, A. & Seijo, L. Configuration coordinate energy level diagrams of intervalence and metal-to-metal charge transfer states of dopant pairs in solids. Phys. Chem. Chem. Phys. 17, 19874–19884 (2015).
MacKeen, C. et al. The complexity of the CaF2:Yb system: a huge, reversible, X-ray-induced valence reduction. J. Phys. Chem. C 121, 28435–28442 (2017).
Barandiarán, Z., Bettinelli, M. & Seijo, L. Color control of Pr3+ luminescence by electron-hole recombination energy transfer in CaTiO3 and CaZrO3. J. Phys. Chem. Lett. 8, 3095–3100 (2017).
Parmentier, A. B., Joos, J. J., Smet, P. F. & Poelman, D. Luminescence of Ytterbium in CaS and SrS. J. Lumin. 154, 445–451 (2014).
Pust, P. et al. Narrow-band red-emitting Sr[LiAl3N4]:Eu2+ as a next-generation LED-phosphor material. Nat. Mater. 13, 891–896 (2014).
Suta, M. & Wickleder, C. Spin crossover of Yb2+ in CsCaX3 and CsSrX3 (X = Cl, Br, I)—a guideline to novel halide-based scintillators. Adv. Funct. Mater. 27, 1602783 (2017).
Suta, M., Urland, W., Daul, C. & Wickleder, C. Photoluminescence properties of Yb2+ ions doped in the perovskites CsCaX3 and CsSrX3 (X = Cl, Br, and I)—a comparative study. Phys. Chem. Chem. Phys. 18, 13196–13208 (2016).
Dorenbos, P. A review on how lanthanide impurity levels change with chemistry and structure of inorganic compounds. ECS J. Solid State Sci. Technol. 2, R3001–R3011 (2013).
Joos, J. J., Poelman, D. & Smet, P. F. Energy level modeling of lanthanide materials: review and uncertainty analysis. Phys. Chem. Chem. Phys. 17, 19058–19078 (2015).
Karlström, G. et al. MOLCAS: a program package for computational chemistry. Comput. Mater. Sci. 28, 222–239 (2003).
Douglas, M. & Kroll, N. M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. (N.Y.) 82, 89–155 (1974).
Hess, B. A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 33, 3742–3748 (1986).
Roos, B. O., Lindh, R., Malmqvist, P. Å, Veryazov, V. & Widmark, P. O. Main group atoms and dimers studied with a new relativistic ANO basis set. J. Phys. Chem. A 108, 2851–2858 (2004).
Roos, B. O., Lindh, R., Malmqvist, P. Å, Veryazov, V., Widmark, P. O. & Borin, A. C. New relativistic atomic natural orbital basis sets for lanthanide atoms with applications to the Ce diatom and LuF3. J. Phys. Chem. A 112, 11431–11435 (2008).
Roos, B. O., Taylor, P. R. & Siegbahn, P. E. M. A complete active space SCF method (CASSCF) using a density-matrix formulated super-CI approach. Chem. Phys. 48, 157–173 (1980).
Siegbahn, P. E. M., Heiberg, A., Roos, B. O. & Levy, B. Comparison of the super-CI and the Newton–Raphson scheme in the complete active space SCF method. Phys. Scr. 21, 323–327 (1980).
Siegbahn, P. E. M., Heiberg, A., Almlöf, J. & Roos, B. O. The complete active space SCF (CASSCF) method in a Newton–Raphson formulation with application to the HNO molecule. J. Chem. Phys. 74, 2384–2396 (1981).
Barandiarán, Z. & Seijo, L. Radial correlation effects on interconfigurational excitations at the end of the lanthanide series: a restricted active space second order perturbation study of Yb2+ and SrCl2:Yb2+. J. Chem. Phys. 138, 074102 (2013).
Andersson, K., Malmqvist, P.-Å, Roos, B. O., Sadlej, A. J. & Wolinski, K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 94, 5483–5488 (1990).
Andersson, K., Malmqvist, P.-Å & Roos, B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 96, 1218–1226 (1992).
Zaitsevskii, A. & Malrieu, J.-P. Multi-partitioning quasidegenerate perturbation theory. A new approach to multireference Møller–Plesset perturbation theory. Chem. Phys. Lett. 233, 597–604 (1995).
Finley, J., Malmqvist, P.-Å, Roos, B. O. & Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 288, 299–306 (1998).
Ghigo, G., Roos, B. O. & Malmqvist, P.-Å A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2). Chem. Phys. Lett. 396, 142 (2004).
Hess, B. A., Marian, C. M., Wahlgren, U. & Gropen, O. A mean-field spin–orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 251, 365–371 (1996).
Malmqvist, P.-Å, Roos, B. O. & Schimmelpfennig, B. The RASSI approach with spin–orbit coupling. Chem. Phys. Lett. 357, 230–240 (2002).
Paulovic, J., Nakajima, T., Hirao, K., Lindh, R. & Malmqvist, P.-A. Relativistic and correlated calculations on the ground and excited states of ThO. J. Chem. Phys. 119, 798–805 (2003).
Barandiarán, Z. & Seijo, L. The ab initio model potential representation of the crystalline environment. Theoretical study of the local distortion on NaCl:Cu+. J. Chem. Phys. 89, 5739–5746 (1988).
Seijo, L. & Barandiarán, Z. Ab initio model potential study of local distortions around Cr+ and Cr3+. J. Chem. Phys. 94, 8158 (1991).
Acknowledgements
J.J.J. and D.V.d.H. acknowledge the UGent Special Research Fund (BOF/PDO/2017/002101 and BOF16/DOC/327). This work was partially supported by Ministerio de Economía y Competitividad, Spain (Dirección General de Investigación y Gestión del Plan Nacional de I + D + i, MAT2017-83553-P). We acknowledge the Fund for Scientific Research-Flanders (FWO) for financial support (project I002418N) and a travel grant (V416818N) and the European Synchrotron Radiation Facility (ESRF) for the allocation of beamtime (beamline ID26).
Author information
Authors and Affiliations
Contributions
J.J.J.: Conceptualization, methodology, software, validation, formal analysis, investigation, data curation, writing original draft, writing review and editing, visualization, supervision, project administration, and funding acquisition. D.V.d.H.: Methodology, validation, investigation, writing review and editing. L.I.D.J.M.: Investigation, writing review and editing. L.A.: Investigation, writing review and editing. P.F.S.: Methodology, resources, data curation, writing review and editing, supervision, funding acquisition. Z.B.: Methodology, software, resources, data curation, writing review and editing, supervision, funding acquisition. L.S.: Methodology, software, resources, data curation, writing review and editing, supervision, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Eszter Borbas, and other, anonymous, reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joos, J.J., Van der Heggen, D., Martin, L.I.D.J. et al. Broadband infrared LEDs based on europium-to-terbium charge transfer luminescence. Nat Commun 11, 3647 (2020). https://doi.org/10.1038/s41467-020-17469-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-17469-x
This article is cited by
-
Lanthanide doped lead-free double perovskites as the promising next generation ultra-broadband light sources
Light: Science & Applications (2022)
-
Compact ultrabroadband light-emitting diodes based on lanthanide-doped lead-free double perovskites
Light: Science & Applications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
