
Abstract
Alterations in DNA methylation occur during development, but the mechanisms by which they influence gene expression remain uncertain. There are few examples where modification of a single CpG dinucleotide directly affects transcription factor binding and regulation of a target gene in vivo. Here, we show that the erythroid transcription factor GATA-1 — that typically binds T/AGATA sites — can also recognise CGATA elements, but only if the CpG dinucleotide is unmethylated. We focus on a single CGATA site in the c-Kit gene which progressively becomes unmethylated during haematopoiesis. We observe that methylation attenuates GATA-1 binding and gene regulation in cell lines. In mice, converting the CGATA element to a TGATA site that cannot be methylated leads to accumulation of megakaryocyte-erythroid progenitors. Thus, the CpG dinucleotide is essential for normal erythropoiesis and this study illustrates how a single methylated CpG can directly affect transcription factor binding and cellular regulation.
Similar content being viewed by others


Introduction
DNA methylation is an important epigenetic modification that is essential to mammalian development1. Co-ordinated changes in DNA methylation have been documented during cellular differentiation, for example, in haematopoiesis, where many gene promoters undergo demethylation as early progenitors differentiate into granulocyte-macrophage progenitors, and genome-wide demethylation occurs during the terminal stages of erythropoiesis2,3,4. However, the mechanisms by which the DNA methylation influences gene expression and differentiation are still not fully understood.
There are numerous examples where DNA methylation at multiple CpGs, for example in CpG islands in promoters and enhancers, is accompanied by a loss of transcription factor binding5,6,7,8,9. For example, NFR1 is a DNA methylation-sensitive transcription factor whose binding is abrogated when a broad region encompassing its recognition motif is hypermethylated8,9.
In this study, we explored how DNA methylation at a single CpG dinucleotide could interfere with binding and regulation by GATA-110,11,12,13, a critical transcription factor that modulates the expression of most if not all erythroid-specific genes14,15,16. We show that methylation of a CGATA element reduces GATA-1 binding and gene regulation in cell lines. We extend these observations by showing that a single point mutation that converts the CGATA element to a TGATA site in a regulatory region of c-Kit—which can still be bound by GATA-1 but that is no longer sensitive to methylation—interferes with normal haematopoiesis in mice.
Results
GATA-1 binds to CGATA motifs and is blocked by methylation
The previously defined GATA-1 recognition motif contains either AGATA or TGATA within a A/TGATAA/G consensus sequence (Fig. 1a)17,18,19. However, in vitro GATA-1 can also bind CGATA elements20. Using an electrophoretic mobility shift assay (EMSA) we confirmed that COS cell overexpressed and MEL cell endogenous GATA-1 is able to bind to CGATA, AGATA and TGATA, but less well to GGATA motifs, as previously reported20 (Fig. 1b and Supplementary Fig. 1a). We then focussed on the CGATA element, since this motif contains a CpG element that may be subject to methylation. Chemically synthesised probes containing unmethylated, methylated or hemi-methylated CGATA motifs were tested in EMSAs (Fig. 1c, d). This revealed that both full methylation and hemi-methylation of the CGATA motif inhibits GATA-1 binding. Probe cold competition assays support the hypothesis that GATA-1 preferentially binds to unmethylated CGATA probes (Supplementary Fig. 1b, c). In comparison with GATA-1, the related protein GATA-2 binds only weakly to probes containing CGATA motifs but this binding is also reduced by DNA methylation (Supplementary Fig. 2).

a GATA-1 canonical consensus motif from JASPAR19. Electrophoretic mobility shift assays (EMSAs) assessing the ability of full-length mouse GATA-1 overexpressed in COS cells to bind to probes containing CGATA, AGATA, TGATA and CGATA motifs b, unmethylated/methylated CGATA probes c or unmethylated/hemi-methylated CGATA probes d. Untransfected COS cells are shown as a control in each case. In b, c a GATA-1 antibody has been used to super-shift the GATA-1-probe complex. The gel shifts shown in b–d were repeated three times independently with similar results. Arrows indicate migration of super-shifted GATA1, GATA-1-DNA complexes and free probes respectively. Source data are provided as a Source data file.
Methylation levels change at GATA-1 bound CGATA sites
We interrogated existing GATA-1 ChIP-seq data from G1E-ER4, murine erythroleukemia (MEL) cells and erythroblasts21 (Fig. 2a). This identified 38799 AGATA motifs, 24126 TGATA motifs and 2708 CGATA motifs that lay near the centre of GATA-1 ChIP-seq peaks reflecting in vivo binding (Supplementary Figure 3). Among all CGATA sites, there were 1139 CGATA sites which co-occurred in the same peak with (A/T)GATA motifs. We then integrated these data with genome-wide bisulphite sequencing information4,22 to identify individual CGATA sites where methylation levels changed during blood cell differentiation (Fig. 2b, Supplementary Figure 4 and Supplementary Table 1). Among these genes, c-Kit was repressed by GATA-1 while Zfpm1 and Rhd were activated by GATA-1 (Supplementary Figure 5). The remaining genes, Rnf220, Abat, Ctdp1, Pbx1, Ulk4, Scrt1, Ncor2 and Camsap111, did not show evidence of being regulated by GATA-1 in this system (Supplementary Figure 5). We focussed our attention on a single CGATA element within an enhancer/silencer in intron 2 of the c-Kit gene, a gene that encodes an important cell surface receptor for the haematopoietic growth factor, stem cell factor.

a Bioinformatics analysis pipeline used to identify CGATA sites bound by GATA-1 where methylation decreases during mouse haematopoiesis4,22. b Heat-map of DNA methylation levels in blood differentiation at GATA-1 targets containing CGATA sites22. Genome-wide DNA methylation level has been investigated in hematopoietic stem cells (HSC), multipotent progenitor 1 (eMPP, Flk2 negative), multipotent progenitor 2 (MPP, Flk2 positive), common myeloid progenitor (CMP), megakaryocyte-erythroid progenitor (MEP) and nucleated erythroblasts (Eryth). All cells were purified from the bone marrow of adult C57BL/6 J mice22. c Schematic of mouse erythropoiesis showing changes in DNA methylation at the c-Kit CGATA22. d Chromatin status at CGATA site within Intron 2 of the mouse c-Kit gene. IGV peak tracks at CGATA (TATCG reverse complement) in GATA-1 ChIP-Seq (G1E cells)21, ATAC-Seq (megakaryocyte-erythroid progenitor cell)21, DNase-Seq (MEL cells)31, H3K4me1 ChIP-Seq (megakaryocyte embryo 14.5)21, H3K4me3 ChIP-Seq (MEL cells)21 and H3K27ac ChIP-Seq (MEL cells)21. The numbers in square brackets on the left side represent peak height. e Flow cytometry cell sorting was used to purify Lineage negative, Scal positive and c-Kit positive (LSK) cells and Ter119 positive and CD71 positive (erythroid; ERY) cells from mouse bone marrow. DNA methylation level at the c-Kit CGATA site in LSK and ERY cells was determined by pyrosequencing, n = 4 biologically independent animals, mean ± standard deviation. Measurements were taken from distinct samples (animals). Source data are provided as a Source data file.
c-Kit is a GATA-1 target gene with a regulatory CGATA site
c-Kit is broadly expressed in hematopoietic stem cells and progenitors, and its expression is downregulated as cell differentiation proceeds23,24,25. High expression of c-Kit in haematopoietic stem cells and progenitors is essential for their self-renewal and proliferation26,27,28, and the ultimate repression of c-Kit in the erythroid lineage is mediated in part via GATA-129,30 and is associated with terminal differentiation. Existing data suggest that DNA methylation of the CGATA motif in intron 2 of c-Kit is high in stem cells but declines as differentiation proceeds4,22 (Fig. 2c, Supplementary Table 2), potentially allowing binding and repression by GATA-1. Importantly, we noted that the intron 2 CGATA element resides in a small region that in erythroid and related cells is not only notable for its strong GATA-1 ChIP-Seq peak, but is also accessible to ATAC sequencing and DNase-I mapping, and carries histone marks consistent with it being part of an functional distal regulatory element (e.g. an enhancer and/or silencer) (Fig. 2d)21,31.
We first compared the levels of methylation at this element, in purified murine haematopoietic stem cells and cells that had differentiated down the erythroid lineage, to assess whether methylation declined as expected. Haematopoietic stem cells (LSK; Lineage−, Scal+, c-Kit+) and erythroblasts (ERY, Ter119+, CD71+) were collected through flow cytometry cell sorting and subjected to pyrosequencing (Fig. 2e). Consistent with previous genome-wide bisulphite sequencing data, the site is highly methylated in LSKs (~80%) but hypomethylated in the erythroid lineage (~10%) (Fig. 2e).
We next moved to cellular models to investigate whether GATA-1 could bind and regulate c-Kit expression from this element in living cells, and whether mutation of this motif, or alteration in its methylation status affected GATA-1 binding and c-Kit regulation. It should be noted that while GATA-1 can serve as an activator or repressor, it is known to repress c-Kit in maturing erythroid cells through multiple elements29,30. Therefore, we anticipated that disrupting the CGATA motif in intron 2 might de-repress c-Kit in erythroid cell lines.
To test whether the CGATA motif in c-Kit intron 2 is regulatory, we utilised MEL cells in which the CGATA element is essentially unmethylated (Supplementary Figure 6) and in which we therefore expected to observe GATA-1 binding (Fig. 3a). ChIP assays confirmed that GATA-1 binds this site in MEL cells (Fig. 3d). We then used CRISPR-mediated editing in MEL cells to mutate the c-Kit +33 kb CGATA element to TTATA to disrupt the motif (Fig. 3b) and tested whether this prevented GATA-1 binding. As expected, this mutation disrupted GATA-1 binding without affecting GATA-1 binding to the positive control site or at other bound GATA sites within the c-Kit gene locus (Fig. 3d). We also tested the impact of this mutation on c-Kit expression and saw a clear de-repression (Fig. 3c), consistent with the view that GATA-1 can bind and repress c-Kit at least in part via binding the intron 2 CGATA element that is unmethylated and able to be bound in mature erythroid cells.

a Schematic of GATA sites in the c-Kit gene locus21. b Sequencing confirms that a (CG > TT)ATA mutation was introduced at c-Kit intron 2. c The mRNA expression level of c-Kit in wild type and (CG > TT)ATA mutant clones, n = 11 biologically independent cells for WT, n = 4 biologically independent cells for mutant, mean ± standard deviation. Measurements were taken from distinct samples (separate clonal cell populations). d GATA-1 ChIP qPCR in wild type and mutant (CG > TT)ATA clones, n = 4 biologically independent cells, mean ± standard deviation. The promoter region of Klf1 served as a positive control (+ve). An upstream region distal to the c-Kit gene was used as a negative control (−ve). All GATA consensus binding sites with evidence of GATA-1 binding in ChIP-seq studies in the c-Kit gene locus have been tested (+5, +7, +33, +49.5, +58 kb; see a). All experiments were performed in undifferentiated MEL cells. Source data are provided as a Source data file.
Increasing c-Kit CGATA site methylation alters regulation
To investigate if DNA methylation of the CGATA element could block GATA-1 binding and whether it influenced the functional repression of c-Kit by GATA-1, we employed another murine erythroid cell line, G1E-ER4. This line has become a useful model for functional studies as it expresses an estrogen inducible GATA-1-ER fusion13,32,33,34. We tested for methylation of the CGATA element in G1E-ER4 cells and found the site was largely unmethylated (Fig. 4b, Supplementary Fig. 8). Reasoning that transient methylation might be being erased by demethylases of the Tet family35, we checked the expression of Tet1, Tet2 and Tet3. We found that Tet2 was highly expressed (Supplementary Fig. 7) and used CRISPR-mediated gene editing to knock out Tet2 in G1E-ER4 cells (Fig. 4a). This disruption was associated with ensuing methylation of the CGATA element (Fig. 4b, Supplementary Fig. 9). GATA-1 is inducible (Supplementary Fig. 10a) and leads to G1E-ER4 cell differentiation (Supplementary Fig. 10b) in both wild type and Tet2 knock out G1E-ER4 cells. Tet2 knock out did not influence G1E-ER4 cell survival or proliferation (Supplementary Fig. 10c, d).

a CRISPR/Cas9 was used to knock out Tet2 in G1E-ER4 cells. Western blot using an antibody to Tet2 identifies clones where Tet2 has been knocked out. A blot using an antibody to actin is shown as a loading control. Three Tet2 knock out clones and clonal wild-type controls were selected for further investigation. The experiment was repeated 3 times independently with similar results. b Methylation level by pyrosequencing of the c-Kit Intron 2 CGATA site in three clonal G1E WT and G1E Tet2 knock out cells (n = 3 biologically independent cells), mean ± standard deviation. c The expression level of c-Kit in G1E WT and G1E Tet2 KO cells by qPCR. Expressed relative to 18S expression, n = 3 biologically independent cells, mean ± standard deviation. Cells were induced with the addition of tamoxifen for 24 h to restore GATA-1-ER fusion proteins in cell nucleus (TAM). Cells incubated with ethanol were used as mock control (NC). d GATA-1 ChIP-qPCR in wild type and G1E Tet2 cells after 24 h of GATA-1 induction by incubating with tamoxifen. KLF1 promoter region was used a positive control (+ve). An upstream region distal to the KLF3 gene was used as a negative control (−ve), the CGATA site at c-Kit intron 2 is the +33 site, n = 3 biologically independent cells, mean ± standard deviation, ChIP-qPCR was repeated two times independently with similar results. Source data are provided as a Source data file.
We tested the binding of GATA-1 to the CGATA element using ChIP assays in wildtype and Tet2 knockout cells and as expected found reduced GATA-1 binding to the CGATA +33 kb site in the c-Kit promoter in knockout cells where methylation levels are higher (Fig. 4d). Interestingly, GATA-1 binding was also reduced at intron 4 of the c-Kit gene (49.5 kb from the TSS), suggesting the that binding to this site might be also influenced by DNA methylation possibly indirectly via chromatin conformation or some other mechanism since this is not a CGATA element (Fig. 4d). However, global hypermethylation did not affect the binding of GATA-1 at a positive control region or other GATA sites in c-Kit gene locus (Fig. 4d). This in vivo result is consistent with the in vitro EMSA assays (Fig. 1), suggesting that methylation at CGATA sites blocks GATA-1 binding. We then considered function. Importantly, the overall levels of c-Kit were comparable in the wildtype and the Tet2 knockout lines before GATA-1 induction with tamoxifen, suggesting that no major perturbation of the chromatin configuration at the locus had occurred (Fig. 4c). When GATA-1 is induced in the wildtype line c-Kit expression diminishes, consistent with previous results30. In Tet2 knockout cells, where the CGATA site is more heavily methylated, GATA-1-mediated repression was attenuated (Fig. 4c). In summary, our results confirm that methylation modulates GATA-1 binding in cell lines and suggest that c-Kit regulation is also attenuated in vivo.
Methylation at the c-Kit CGATA site is important in mice
To definitively determine whether this single CpG dinucleotide is required for c-Kit regulation during haematopoiesis in vivo we tested the importance of this site using CRISPR gene editing in mice. Rather than simply disrupting the site, we introduced a C > T mutation, to convert the CGATA element to a TGATA site (Fig. 5a). We reasoned that this site would now be bound constitutively, rather than only after the onset of de-methylation during haematopoiesis. Bone marrow samples were collected from wild type and homozygous mutant mice (Supplementary Fig. 11a). The overt phenotype of the mutant mice and blood counts were not significantly different from wildtype (Supplementary Fig. 11b). We investigated the proportion and absolute number of bone marrow precursors and proportion of erythroid populations using flow cytometry. There was no change in the proportion of hematopoietic stem cells, multipotent progenitor populations or erythroid populations between wild type mice and mutant mice (Supplementary Fig. 12). However, we detected a clear accumulation of megakaryocyte-erythroid progenitors (MEPs) in mutant mice (Fig. 5b). In addition, in mutant mice, the overall expression level of c-Kit in MEPs was significantly reduced in comparison to that of wild type mice (Supplementary Fig. 13). This result is consistent with this single CpG dinucleotide playing a role in haematopoietic differentiation in vivo.

a A (C > T)GATA mutation was introduced at c-Kit Intron 2 in C57BL/6J mice using CRISPR/Cas9. b Flow cytometry analysis of progenitor populations in mouse bone marrow from WT and homozygous (C > T)GATA mutant mouse littermates. CD16/32 low and CD34 negative cells are megakaryocyte-erythroid progenitors (MEPs). CD16/32 medium and CD34 positive cells are common-myeloid progenitors (CMPs). CD16/32 high and CD34 positive cells are granulocyte-monocyte progenitors (GMPs). All progenitors were derived from lineage negative, Scal negative, c-Kit positive (LSK) populations. c, d The percentage and absolute cell number of different populations in each mouse have been summarised in the histogram, n = 4 biologically independent animals, mean ± standard deviation. Mann–Whitney U, two-tailed statistical test, P-values shown. Measurements were taken from distinct samples (animals). Source data are provided as a Source data file.
Discussion
In vivo GATA-1 has been implicated in binding primarily to AGATA or TGATA elements, and not to CGATA sites, but this may be a consequence of genomic ChIP-Seq analyses under-appreciating the importance of CGATA sites, due to the rarity of CpG dinucleotides in mammalian genomes. In addition, since CpG dinucleotides are often methylated the subset of CGATA elements bound by GATA-1 will be further limited. Nevertheless, the fact that CGATA elements can be subjected to methylation, and that this influences binding of GATA-1, means that such elements may be instrumental in mediating the effects of alterations in methylation that have been observed to occur during haematopoietic differentiation.
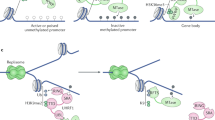
Our work provides a clear illustration that at least one CpG dinucleotide within a CGATA site, that is progressively demethylated during haematopoiesis, is involved in c-Kit regulation in vivo. Methylation of the site inhibits GATA-1 binding in vitro and in cellular assays, and modestly impairs repression by GATA-1. A single point mutation that converts the CGATA element to a TGATA site—which can still be bound by GATA-1 but that can no longer provide a methylation sensitive binding site—interferes with normal haematopoiesis in mice (Fig. 6). These results suggest that DNA methylation at the binding site inhibits GATA-1 binding and show that a single residue’s ability to be methylated can influence gene regulation in vivo.

In wild type mice, the CGATA site at intron 2 of c-Kit gene is highly methylated at early stages of haematopoiesis and gradually becomes de-methylated during haematopoiesis. GATA-1 binds to the CGATA site at intron 2 of the c-Kit gene when it is de-methylated and represses the expression of c-Kit. In mutant mice, the (C > T)GATA mutation is unable to be modified by DNA methylation and GATA-1 can bind to this site at earlier stages of haematopoiesis. We observe a decrease in c-Kit expression and an increase in MEP cell number in mice homozygous for this c-Kit (C > T)GATA mutation. Note that the diagram illustrates that methylation of the CGATA element is required for normal regulation of c-Kit but is not likely to be sufficient for the full regulation of the c-Kit gene. Other GATA elements in additional regulatory elements are also likely to be relevant but for brevity are not included here.
Methods
EMSA
EMSA were performed36. The sense oligonucleotide was labelled with [γ-32P] triphosphate before annealing with the antisense strand via slow cooling from 100 °C to room temperature. Labelled duplex DNA oligonucleotides were purified with Quick Spin Columns (Sigma-Aldrich, #11273922001). In all, 5 μg pMT3-GATA-1 (full length) was transfected into COS-7 cells (gift from Stu Orkin, Harvard Medical School Boston, USA) for 48 h using Fugene6 (Promega, #E2691). Nuclear extracts were harvested from COS-7 cells37. Nuclear extracts were incubated with radiolabelled probes at 4 °C for 10 min. The protein-probe mixtures were run at 4 °C on a 6% native polyacrylamide gel in TBE buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA). COS empty lane was included as control to show any background binding to the probe. Super-shifting of GATA-1 was achieved using an anti-GATA-1 antibody (Santa Cruz technology, #sc-256, 1:5000 dilution). Gels were run for 1 h and 30 min, at 250 volts. After running, gels were dried under vacuum. Dried gels were exposed to a FULJIFILM BAS CASSETTE2 2025 phosphor screen overnight followed by visualised using a FUJIFILM FLA-5100 Fluorescent Image Analyser. All probes used in EMSAs are listed in Supplementary Table 3.
Western blot
Nuclear extracts from cultured G1E-ER4 cells (gift from Stu Orkin, Harvard Medical School Boston, USA) were made37. To detect Tet2 expression, 60 μg of cell nuclear extract was loaded onto a NuPAGETM NovexTM 10% Bis-Tris Protein Gel (ThermoFisher Scientific, #NP0301BOX) and gels were run in MOPS buffer at 200 V for 60 min. Proteins were transferred onto a nitrocellulose membrane at 30 V for 70 min. Blots were blocked with 4% skim milk for 30 min. After blocking, blots were probed for Tet2 and Actin using an anti-Tet2 antibody (Abcam, #ab124297, 1:5000 dilution) and an anti-β-Actin antibody (Sigma, #A1978, 1:10000 dilution) respectively.
RNA extraction and cDNA synthesis
Total RNA was extracted with Tri-reagent according to the manufacturer’s protocol (Qiagen, #74004). RNA was treated with a DNA removal kit (Qiagen, #79254) to ensure the purity of RNA samples. RNA concentrations were determined by UV-light absorbance at 260 nm using a Nanodrop (ThermoFisher Scientific).
Approximately 500 ng of RNA was used for cDNA synthesis with SuperScript VILO cDNA synthesis kit (ThermoFisher Scientific, Cat#11755050) in which random hexamers were used as primers for generation of first strand cDNA. For each sample, a negative control was also set up in the absence of SuperScript III reverse transcriptase (RT) to check for genomic DNA contamination.
Quantitative real-time PCR
Quantitative real-time PCRs (qPCR) were set up with SYBR Green PCR Master Mix, forward and reverse primers targeting locus of interest and template of cDNA or ChIP DNA. All reactions were set up in duplicate and run on the Applied Biosystems ViiA7 Real-Time PCR system (Applied Biosystems). For transcript level analysis, the results were normalised to the 18S rRNA levels of the respective samples. For ChIP experiments, the results were normalised against the respective input samples containing total sonicated genomic DNA. Primers used for qPCR are listed in Supplementary Table 4.
Cell culture (G1E-ER4, MEL)
The murine erythroleukaemia cell line (MEL, gift from Stu Orkin, Harvard Medical School Boston, USA) was cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, #435847) supplemented with 10% (v/v) foetal calf serum (FCS) (Interpath, #SFBSF13) and 1% (v/v) penicillin/streptomycin/glutamine (PSG) solution (Life Technologies, #10378016)38. G1E-ER4 is a subclone of GATA-1 null erythroblasts which stably expresses a GATA-1-ER (estradiol receptor ligand binding domain) fusion protein32. G1E-ER4 cells were cultured in Iscove’s MDM medium (Life Technologies, #12440-061) supplemented with 15% heat-inactivated foetal calf serum (Interpath, #SFBSF13), 1% PSG (Life Technologies, #10378016), 62 μl of 10% monothioglycerol (Sigma-Aldrich, #M6145), 0.6% home-made Kit-ligand conditioned medium, 50ul mouse erythropoietin (Biolegend, #587606) in 500 ml IMDM medium39. Kit-ligand conditioned medium was made from CHO cells (gift from Stu Orkin, Harvard Medical School Boston, USA)32. Exponentially proliferating G1E-ER4 cells were induced with the addition of 4-hydroxytamoxifen (tamoxifen) (Sigma, #H7904) to a final concentration of 0.1 nmol for 24 h to restore GATA-1-ER fusion protein into cell nucleus32. G1E-ER4 cells incubated with ethanol (1 ul ethanol in 10 ml media) was used as mock (NC) control.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) experiments were performed40. In all, 7 × 107−1 × 108 cells were used for each ChIP. Cells were crosslinked with 1% formaldehyde for 10 min and the reaction was quenched by addition of 2.5 mM glycine. Fixed cells were sonicated at high voltage for 20 min (30 s on, 30 seconds off) using Bioruptor® (Diagenode) to obtain 200–300 bp DNA fragments. The fragmented chromatin was pulled down at 4 °C overnight using an antibody against GATA-1 (Santa Cruz Biotechnology, #sc-256, 1:5000 dilution). Chromatin cross-linking was then reversed at 65 °C overnight followed by DNA purification. Real-time qPCR was performed on ChIP DNA on an Applied Biosystems ViiA7 Real-time PCR System (Applied Biosystems). Primers used for ChIP-qPCR are listed in Supplementary Table 5.
Pyrosequencing
Genomic DNA was extracted and purified from cells using a PureLink Genomic DNA Mini Kit, according to the manufacturer’s instructions (Life Technologies, #K1820-02). Briefly, harvested cells were lysed at 55 °C for 10 min and then gDNA was precipitated by adding 100% ethanol and purified through the provided column. Genomic DNA was treated with bisulphite and purified following using a EpiTect Fast Bisulfite Conversion Kit, following manufacturer’s instructions (Qiagen, #59824). Target regions were amplified from bisulphite converted gDNA using MyTaq HS DNA polymerase (Bioline, #BIO-25045) with Biotin-labelled primers. Pyrosequencing of samples were performed by Australia Genome Research Facility (AGRF) using the PyroMark Q24 platform. All primers used for pyrosequencing are listed in Supplementary Table 6.
CRISPR/Cas9 genome editing in cell lines
Single guide RNAs (sgRNAs) were designed using the website [https://www.benchling.com/crispr/] and guides with low off-target scores were selected41. sgRNAs were cloned into pSpCa9(BB)−2A-GFP (px458) plasmid (Addgene, #48138)41. Tet2 knock out cells were generated by CRISPR/Cas9 using non-homologous end joining (NHEJ). For site specific mutations, a DNA donor template was used to drive homology-directed repair (HDR). In all, 150 bp DNA donor templates for genomic modification were synthesised by Integrated DNA Technologies (IDT), containing the mutation in the middle of the sequence. sgRNA plasmid and DNA donor were co-transfected into cells using Neon transfection system (ThermoFisher). In all, 48 h after transfection, cells were sorted with BD FACSAriaIII flow cytometer for those positive for GFP (indicating that they had taken up the pSpCa9(BB)−2A-GFP plasmid) and negative for the live-dead marker 7-AAD (BIO-RAD, #1351102, one drop per sample). After a further 48 h, GFP and 7-AAD negative cells were single sorted into 96-well plates. After a week, clonal populations derived from single cells were screened through Sanger sequencing and western blot. Primers and donors used for genome editing are listed in Supplementary Table 7.
BrdU cell proliferation assay
The proliferation of G1E-ER4 cells was assessed by using a BrdU cell proliferation assay kit, according to the manufacturer’s instructions (Cell Signaling Technology, #6813). We plated 1 × 104 G1E-ER4 cells into wells of a 96-well plate, added 10 μl 1x BrdU into each well and incubated for 4, 8 or 24 h. After the incubation, the cells were fixed and denatured for 30 min, then 100 μl/well BrdU detection antibody solution (Cell Signaling Technology, #94079, 1:100 dilution) was added and incubate at room temperature for 1 h. After the incubation, each well was washed three times with 100 μl wash buffer. Next 100 μl/well HRP-conjugated secondary antibody solution (Cell Signaling Technology, #34709, 1:100 dilution) was added and incubated at room temperature for 30 min. After the incubation, the cells were washed three times with 100 μl wash buffer. TMB substrate (100 μl) was added and incubated for 30 min before adding stopping buffer. After adding the stopping buffer, absorbance was read at 450 nm to detect cell proliferation.
Generation of c-Kit (C > T)GATA site mutant mice
Generation of mice was done with approval of the UNSW Animal Care and Ethics Committee (Approval No. 16/19A). The CRISPR/Cas9 system was utilised for c-Kit (C > T)GATA mouse generation by injecting sgRNA and Cas9 protein (EnGen® Spy Cas9 NLS) into fertilised oocytes42. Briefly, sgRNAs were designed using the website [https://www.benchling.com/crispr/]41. A linearised DNA template for each sgRNA was generated using a non-cloning method by virtue of a T7-conjugated PCR. The forward primer contains the T7 promoter minimal sequence, upstream of the 20 bp sgRNA sequence and a sequence complimentary to the 5′ end of the sgRNA scaffold of pSpCa9(BB)−2A-GFP. The reverse primer is complimentary to the 3′ end of the sgRNA scaffold of pSpCa9(BB)−2A-GFP. The linearised DNA was amplified within T7 forward primer and sgRNA scaffold reverse primer using Q5 polymerase (NEB, #M0491). The resulting linearised DNA was in vitro transcribed into sgRNA using a T7 Quick High Yield RNA synthesis kit, following the manufacturer’s instructions (NEB, #E2050S). sgRNAs were purified using NucAway Spin columns (ThermoFisher, #AM10070). The donor DNA template was synthesised from Integrated DNA Technologies (IDT). The sgRNA, Cas9 protein and DNA donor were microinjected into C57BL/6J mouse oocytes at 0.5 days post-coitum (dpc) and immediately transferred to foster mothers (Swiss strain). The C57BL/6J offspring were genotyped through Sanger sequencing. Briefly, mouse tails were digested in Direct PCR (tail) lysis reagent (Australian Biosearch, #AB-102-T) at 55 °C overnight and then heat inactivated at 85 °C for 45 min. In all, 1 μl of lysates were used as templates for a genotyping PCR using Q5 polymerase (NEB, #0491). PCR products were Sanger sequenced to check for the presence of the mutation. Mice that were heterozygous for the c-Kit (C > T)GATA mutation were backcrossed with wild type C57BL/6J mice for five generations to eliminate potential off-target effects. Primers used for in vitro transcription and genotyping are listed in Supplementary Table 8.
Animal husbandry
All animal work was carried out in accordance with approval from the UNSW Animal Care and Ethics Committee (Approval Nos. 16/5B and 18/156B). Animals were housed in a specific pathogen-free environment, at a constant ambient temperature of 22 °C, on a 12 h high-dark cycle and with ad libitum access to standard chow food and water.
Flow cytometry analysis of erythrocyte populations
Mouse bone marrow cells were stained with CD71 (Biolegend, #334104, 1:100 dilution), Ter119 (BD Bioscience, #553673, 1:200 dilution) and DAPI (Life Technologies, #62248) for 30 min. After staining, cells were washed with 3 ml FACS buffer and centrifuged at 300×g at 4 °C for 5 min. Cells were resuspended in 500ul FACS buffer. Stained samples were run on BD LSRFortessa SORP flow cytometer. Data was collected using BD FACSDiva software and analysed using FlowJo V10 software. Flow cytometry gating strategies are shown in Supplementary Fig. 14b.
Flow cytometry analysis of HSPCs
Haematopoietic stem cell and progenitor cells (HSPCs) were analysed by flow cytometry as described below. Mouse bone marrow was lysed with 9 ml RO water for 10 s followed by adding 1 ml 10x PBS to get rid of mature red blood cells since mature red blood cells do not have cell nucleus and will burst immediately under low osmotic pressure in water. Lysed bone marrow cells were stained with biotin-conjugated lineage marker antibody cocktail:CD3 monoclonal antibody (ThermoFisher Scientific, #13-0037-82, 1:200 dilution), CD4 monoclonal antibody (ThermoFisher Scientific, #13-0041-85, 1:100 dilution),CD11b monoclonal antibody (ThermoFisher Scientific, #13-0112-85, 1:100 dilution), CD5 monoclonal antibody (ThermoFisher Scientific, #13-0051-85, 1:200 dilution), CD8 monoclonal antibody (ThermoFisher Scientific, #13-0081-85, 1:200 dilution), CD45R (B220) monoclonal antibody (ThermoFisher Scientific, #13-0452-85, 1:200 dilution), Ly-6G monoclonal (ThermoFisher Scientific, #13-5931-85, 1:200 dilution),TER-119 monoclonal antibody (ThermoFisher Scientific, #13-5921-82, #1:200 dilution), at 4 °C for 30 min. Stained cells were washed with FACS buffer (1xPBS, 5% FCS, 2 mM EDTA) and centrifuged at 300 × g at 4 °C for 5 minutes. Cells were resuspended with FACS buffer and stained with PE-CF594 Streptavidin (BD Bioscience, #562318, 1:300 dilution),, LY-6A/E(Sca1)-V500 antibody (BD Bioscience, #561229, 1:200 dilution), CD117(cKit)-APC-H7 antibody(BD Bioscience, #560250, 1:200 dilution), CD34-FITC antibody (BD Bioscience, #553733, 1:100 dilution), CD16/32-PE-Cy7 antibody (ThermoFisher, #25-0161-81, 1:200 dilution) and DAPI (Life Technologies, #62248) at 4 °C for 30 min. After staining, cells were washed with 3 ml FACS buffer and centrifuged at 300 × g at 4 °C for 5 min. Cells were resuspended in 500 μl FACS buffer. Stained samples were run on BD LSRFortessa SORP flow cytometer. Data was collected using BD FACSDiva software and analysed using FlowJo V10 software. Flow cytometry gating strategies are shown in Supplementary Fig. 14a.
Peripheral blood count
Blood was collected by cardiac puncture from wild type and c-Kit (C > T)GATA homozygous mutant mice and added to 50 μl 100U ml−1 heparin (Sigma, #H3149-250KU) in BD vacutainer blood collection tubes (BD Bioscience, #367839). Blood counts were measured by Sysmex XN-1000RF. Original numbers of cells were calculated using the following dilution factor formula:
Bioinformatic analysis
GATA-1 ChIP-Seq datasets from different cell types were download from the ENCODE portal43 [https://www.encodeproject.org]. Broad peaks were first split into narrow peaks using PeakSplitter function in PeakAnalyzer software44 for improving individual subpeaks’ analysis. Different components from MEME SUITE was used to perform peak and motif analysis: Comprehensive motif analysis was performed using MEME ChIP45,46; FIMO45 was then used to scan the peaks containing ‘CGATA’ motifs. Finally peak annotation was performed using PeakAnnotator function in PeakAnalyzer44. Reduced representation bisulphite sequencing (RRBS) datasets from blood cells and foetal liver tissues were downloaded from GEO4,22. Single CpG methylation data from different stages of erythroid maturation was analysed using the Bioconductor package RnBeads47. DNA methylation levels of CGATA Peaks were measured by BEDtools48 from RnBeads output. Heatmaps of DNA methylation changes of CGATA Peaks were drawn using gplots library49 in R. All dataset accession numbers are listed in Supplementary Table 9.
Statistical analysis
The mean and standard deviation (SD) are shown for the data in each figure, except where n < 3. Two-tailed Mann–Whitney U (non-parametric) tests were performed to assess the significance of differences of proportions of common-myeloid progenitors (CMPs) megakaryocyte-erythroid progenitors (MEPs) in bone marrow between wild type and (C > T)GATA mutant mice. The statistical analyses for ChIP-Seq and RRBS experiments were performed by various software programs, details are provided above.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. All ChIP-Seq, ATAC-Seq and DNase-Seq data were downloaded from Encyclopedia of DNA Elements Database (ENCODE) (Supplementary Table 9). All Reduced Representation Bisulfite Sequencing (RRBS) data were downloaded from Gene Expression Omnibus (GEO) database (Supplementary Table 9). Publicly available DNA methylation dynamics data were open sources online (http://www.medical- epigenomics.org/papers/broad_mirror/invivomethylation/index.html). The source data underlying Figs. 1b–d, 2e, 3c, d, 4a–d and 5c, d and Supplementary Figs 1a, b, 2a, b, 5, 7, 10a–d, 11b, 12a–c and 13 are provided as a Source data file.
References
Smith, Z. D. & Meissner, A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204 (2013).
Bocker, M. T. et al. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood 117, e182–e189 (2011).
Calvanese, V. et al. A promoter DNA demethylation landscape of human hematopoietic differentiation. Nucleic Acids Res. 40, 116–131 (2011).
Shearstone, J. R. et al. Global DNA demethylation during mouse erythropoiesis in vivo. Science 334, 799–802 (2011).
Lal, G. et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 182, 259–273 (2009).
Medvedeva, Y. A. et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics 15, 119 (2014).
Polansky, J. K. et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J. Mol. Med. 88, 1029–1040 (2010).
Domcke, S. et al. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528, 575 (2015).
Zhu, H., Wang, G. & Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 17, 551–565 (2016).
Katsumura, K. R. & Bresnick, E. H. The GATA factor revolution in hematology. Blood 129, 2092–2102 (2017).
Pevny, L. et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349, 257–260 (1991).
Wakabayashi, A. et al. Insight into GATA1 transcriptional activity through interrogation of cis elements disrupted in human erythroid disorders. Proc. Natl Acad. Sci. USA 113, 4434–4439 (2016).
Welch, J. J. et al. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104, 3136–3147 (2004).
Hong, W. et al. FOG‐1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J. 24, 2367–2378 (2005).
Rodriguez, P. et al. GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J. 24, 2354–2366 (2005).
Wadman, I. A. et al. The LIM‐only protein Lmo2 is a bridging molecule assembling an erythroid, DNA‐binding complex which includes the TAL1, E47, GATA‐1 and Ldb1/NLI proteins. EMBO J. 16, 3145–3157 (1997).
Cheng, Y. et al. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 19, 2172–2184 (2009).
Yu, M. et al. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell 36, 682–695 (2009).
Khan, A. et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266 (2018).
Merika, M. & Orkin, S. H. DNA-binding specificity of GATA family transcription factors. Mol. Cell. Biol. 13, 3999–4010 (1993).
Yue, F. et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355 (2014).
Bock, C. et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 47, 633–647 (2012).
Broudy, V. C. Stem cell factor and hematopoiesis. Blood 90, 1345–1364 (1997).
Galli, S. J., Zsebo, K. M. & Geissler, E. N. The kit ligand, stem cell factor. Adv. Immunol. 55, 1–96 (1993).
Iemura, A., Tsai, M., Ando, A., Wershil, B. K. & Galli, S. J. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 144, 321–328 (1994).
Kimura, Y. et al. c-Kit-mediated functional positioning of stem cells to their niches is essential for maintenance and regeneration of adult hematopoiesis. PloS ONE 6, e26918 (2011).
Sharma, S. et al. Stem cell c-KIT and HOXB4 genes: critical roles and mechanisms in self-renewal, proliferation, and differentiation. Stem Cells Dev. 15, 755–778 (2006).
Shin, J. Y., Hu, W., Naramura, M. & Park, C. Y. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 211, 217–231 (2014).
Jing, H. et al. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol. Cell 29, 232–242 (2008).
Munugalavadla, V. et al. Repression of c-kit and its downstream substrates by GATA-1 inhibits cell proliferation during erythroid maturation. Mol. Cell. Biol. 25, 6747–6759 (2005).
The, E. P. C. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57 (2012).
Gregory, T. et al. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94, 87–96 (1999).
Pilon, A. M. et al. Genome-wide ChIP-Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood 118, e139–e148 (2011).
Weiss, M. J., Yu, C. & Orkin, S. H. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol. Cell. Biol. 17, 1642–1651 (1997).
Wu, X. & Zhang, Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18, 517 (2017).
Crossley, M. et al. Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol. Cell. Biol. 16, 1695–1705 (1996).
Andrews, N. C. & Faller, D. V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19, 2499–2499 (1991).
Friend, C., Scher, W., Holland, J. G. & Sato, T. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc. Natl Acad. Sci. USA 68, 378–382 (1971).
Rylski, M. et al. GATA-1-mediated proliferation arrest during erythroid maturation. Mol. Cell. Biol. 23, 5031–5042 (2003).
Schmidt, D. et al. ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods 48, 240–248 (2009).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Delerue, F. & Ittner, L. M. Generation of genetically modified mice through the microinjection of oocytes. J. Vis. Exp. 124, 55765 (2017).
Davis, C. A. et al. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2017).
Salmon-Divon, M., Dvinge, H., Tammoja, K. & Bertone, P. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinforma. 11, 415 (2010).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME suite. Nucleic Acids Res. 43, W39–W49 (2015).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Assenov, Y. et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184 (2009).
Acknowledgements
We acknowledge C. Brownlee and E. Johansson Beves from BRIL (Mark Wainwright Analytical Centre, UNSW Sydney, Sydney, NSW, Australia) for assistance with flow cytometry, D. Chandler and J. Jorritsma from the Australian Genome Research Facility (AGRF, Australia) for assistance with pyrosequencing and the Veterinary Pathology Diagnostic Service (VPDS, University of Sydney, Sydney, NSW, Australia) for mouse blood counts. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138; RRID:Addgene_48138). This work was supported by funding from the Australian Research Council to M.C. (DP150103503). M.R.W. acknowledges support from the Australian Government NCRIS scheme. L.Y. was supported by a China Scholarship Council scholarship granted by the Chinese Government. E.S.S. was supported by an Australian Government Research Training Program (RTP) Scholarship. K.G.R.Q. was supported by a Scientia Fellowship from UNSW Sydney. L.M.I. is a National Health and Medical Research Council Principal Research Fellow (GNT1136241).
Author information
Authors and Affiliations
Contributions
L.Y., M.C. and K.G.R.Q designed the study and experiments. L.Y. performed and analyzed most experiments and data. Z.C. and M.R.W performed the bioinformatic analysis to identify CGATA sites that are bound by GATA-1 in existing ChIP-seq datasets and where there is a change in methylation levels during haematopoiesis. F.D and L.M.I. generated the CRISPR/Cas9 gene edited mouse line. E.S.S. assisted with the animal studies and optimisation of the haematopoietic flow cytometry panel. L.Y., M.C. and K.G.R.Q. wrote the paper. K.G.R.Q. and M.C. supervised the study. All authors have read and approved the contents of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Emery Bresnick and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, L., Chen, Z., Stout, E.S. et al. Methylation of a CGATA element inhibits binding and regulation by GATA-1. Nat Commun 11, 2560 (2020). https://doi.org/10.1038/s41467-020-16388-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16388-1
This article is cited by
-
The mitoepigenome responds to stress, suggesting novel mito-nuclear interactions in vertebrates
BMC Genomics (2023)
-
Enzyme-free targeted DNA demethylation using CRISPR–dCas9-based steric hindrance to identify DNA methylation marks causal to altered gene expression
Nature Protocols (2022)
-
5-AZA-dC induces epigenetic changes associated with modified glycosylation of secreted glycoproteins and increased EMT and migration in chemo-sensitive cancer cells
Clinical Epigenetics (2021)
-
Activation of γ-globin gene expression by GATA1 and NF-Y in hereditary persistence of fetal hemoglobin
Nature Genetics (2021)
-
Genome-wide identification and function characterization of GATA transcription factors during development and in response to abiotic stresses and hormone treatments in pepper
Journal of Applied Genetics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
