
Abstract
Uveal melanoma (UM) is the most common intraocular tumour in adults and despite surgical or radiation treatment of primary tumours, ~50% of patients progress to metastatic disease. Therapeutic options for metastatic UM are limited, with clinical trials having little impact. Here we perform whole-genome sequencing (WGS) of 103 UM from all sites of the uveal tract (choroid, ciliary body, iris). While most UM have low tumour mutation burden (TMB), two subsets with high TMB are seen; one driven by germline MBD4 mutation, and another by ultraviolet radiation (UVR) exposure, which is restricted to iris UM. All but one tumour have a known UM driver gene mutation (GNAQ, GNA11, BAP1, PLCB4, CYSLTR2, SF3B1, EIF1AX). We identify three other significantly mutated genes (TP53, RPL5 and CENPE).
Similar content being viewed by others


Introduction
Uveal melanoma (UM) arises from melanocytes in the uveal tract, and though less common than cutaneous melanoma, a higher proportion of UM patients die from the disease1,2,3. Risk determination of metastatic spread can be obtained through assessment of specific chromosome copy number alterations (CNAs)4, gene expression profiles5 and mutation status of known UM driver genes6.
Previous genomic studies have pointed to the existence of four UM categories, strongly linked to prognosis7,8,9. Similarly we segregate our tumours into four categories based on CNAs: category 1 are chromosome 3 disomy (D3) tumours lacking chromosome 8q copy-number gain and frequently possessing EIF1AX mutations; category 2 are D3 UM with chromosome 6p and 8q gain and a high proportion of SF3B1 mutations; category 3 are chromosome 3 monosomy (M3) tumours lacking chromosome 8q gain dominated by BAP1 mutations; category 4 UMs are M3 with chromosome 8q gain and BAP1 mutations. These genomic stratifications, while prognostic, are not indicative of treatment responses once progression has occurred.
To improve knowledge of UM genomics and to identify potential therapeutic targets, we conduct a whole-genome sequencing (WGS) study of 103 UM, comprised of 91 primary tumours and 12 metastases, with matched germline DNA. Eighty-four tumours originate from the choroid, eight from the iris, four from the ciliary body, and seven without known primary uveal site (Supplementary Data 1).
Results
Recurrent copy number aberrations
In line with previous studies8,9,10,11, TMB was low in the majority of UM (median 0.50 mutations per megabase, range 248–42,669, Supplementary Data 2) and tumours generally displayed low counts of structural variations (SVs) (median: 13; range 0–213) (Fig. 1). One sample had noticeably more SVs, of which the majority (71%) were midsized (<100 kb) deletions, suggesting this was not due to chromothripsis. There were no additional notable features or known driver mutations in this sample. Commonly observed losses of chromosome 1p, 3, 8p, 6q and 16q were present, as were gains of 1q, 6p and 8q (Fig. 2)4,12. Two samples presented with whole-genome duplication (WGD). Tumours were grouped into the four categories described. Category 4 was predominant, with 55 samples (53%) displaying M3 and copy-number gain of chromosome 8q. In previous studies8,9, the vast majority (93–95%) of M3 tumours also displayed 8q gain, whereas in this cohort a notable proportion (13/68, 19%) of the M3 samples showed no 8q gain (category 3) (two-sided Fisher’s exact test, P = 0.04). Some D3 samples (11/35, 31%) had gain of 8q (category 2), as previously observed8,9. Chromosome 8p loss was only observed in samples with 8q gain (categories 2 and 4), reflecting the formation of isochromosome 8q, whereas the other alterations were spread across categories 2, 3 and 4. The majority of category 1 tumours lacked these common, large CNAs, instead displaying either few or more dispersed rearrangements.

a Tumour mutation burden (substitutions and indels) in 103 uveal melanomas. b Burden of structural variation in each tumour displayed as numbr of events. Variants are coloured according to their event category. One outlier was observed with >200 events, mostly mid-sized (<100 kb) deletions. c Spectrum of single base substitution (SBS) signatures shown as percentage. Most samples were dominated by signature 5, which has been observed in most cancers. Signatures SBS7a and SBS7b, only seen in iris melanomas here, are commonly observed in cutaneous melanoma and associated with exposure to ultraviolet radiation (UVR). SBS1 was predominantly observed in melanomas with loss of MBD4 and is associated with deamination of 5-methylcytosine. Spectrum of double base substitutions shown as number of substitutions in each tumour. DBS1 is characterised by CC>TT transition and associated with exposure to UVR.

Samples are arranged according to their category defined by status of chromosome arms 3p and 8q. a Total number gains/losses in each genomic region. b Gains (red) and losses (green) shown in each sample and region. c The fraction of the genome having aberrations in each sample. * Sample with whole-genome duplication. Source data are provided as a Source data file.
UV mutation signatures in iris tumours
Assessment of single base substitution (SBS) signatures revealed SBS5 to predominate in most cases, with strong representation of SBS3, SBS39 and SBS40 in some samples (Fig. 1)13. Two samples were dominated by mutation signature SBS1 (associated with spontaneous deamination) and had correspondingly high TMB (>3 mutations per Mb). As previously observed14,15, these features corresponded to the presence of germline loss-of-function (LOF) MBD4 mutations; this takes the published tally of germline MBD4 mutant UM cases to six14,15, strengthening its role as a UM predisposition gene. The two UMs with germline LOF BAP1 mutations displayed no unique features. Strikingly, all iris melanomas displayed the genomic features associated with UVR damage (mutation signatures SBS7a, SBS7b and DBS113 combined with a high TMB). While exposure to UVR has been suggested as a cause of the elevated UM risk among arc-welders16, no molecular evidence of UVR as an aetiological factor has yet been observed in UM sequencing studies. The iris is located anteriorly within the uveal tract and is directly exposed to sunlight that breaches the cornea; we now show that UVR-associated DNA damage results from this exposure and is a unique genomic feature of iris UM.
Patterns of driver mutations and chromosomal aberrations
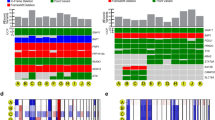
Assessment of known UM driver genes revealed an oncogenic driver mutation in 102 of 103 tumours: 51 in GNAQ (48 p.Q209P/L, two p.R183Q, one p.G48L), 46 in GNA11 (44 p.Q209L/P, two p.R183C), five in PLCB4 (three p.D630Y, two p.D630N) and two in CYSLTR2 (p.L129Q). These mutations were generally mutually exclusive except for two PLCB4 p.D630 mutations that co-occurred with GNAQ/GNA11 p.R183H mutations. This co-occurrence between PLCB4 mutation and the minor hotspot p.R183, rather than the stronger oncogenic p.Q209 hotspot mutations, has previously been described in the UM TCGA data8. Though not previously highlighted, GNAQ p.G48L mutations have been reported in two UM samples from two separate studies8,17, as well as in two hepatic small vessel neoplasms, which are driven by activating GNAQ/GNA14 mutations18. This suggests that GNAQ p.G48L is another minor UM oncogenic hotspot mutation. Similar to previous observations19,20, BAP1 was the most altered gene in M3 samples (75%), including eight splice site mutations, two germline and 32 somatic LOF mutations, and three cases with disrupted BAP1 due to SV breakpoints. In addition, two D3 tumours carried BAP1 mutations, indicating that although BAP1 inactivation typically occurs after M38, BAP1 aberration can also occur in D3 tumours, which may or may not later undergo loss of chromosome 3. Of note, one of these D3 tumours (MELA_0800) had a low BAP1 variant allele frequency (VAF = 9/80) suggesting it was only present in a subclone, and as copy number tools are not as sensitive as mutation callers, it is possible that the subclone had loss of heterozygosity (LOH) that was not detected by the algorithm. Five tumours had BAP1 mutations and copy-neutral LOH, suggesting that the mutations occurred before WGD in the two tetraploid UMs and before the LOH event in the three diploid UMs. SF3B1 mutations were present in 15 tumours, the majority occurring in category 2, in line with other studies7,8,9. EIF1AX hotspot mutations were observed in 19% of tumours. EIF1AX mutations were first discovered in D3 UMs21 and in the TCGA cohort they were restricted to category 1 tumours (D3 and no 8q gain)8, while in the cohort presented by Royer-Bertrand and colleagues two of seven mutations were seen in tumours with M3 and/or 8q gain9. Similarly, here six of the 20 EIF1AX mutations (30%) were seen in UM with M3 (n = 5) or 8q gain (n = 1) (Fig. 3a).

Samples are arranged in the same order as Fig. 2 by their category (1–4). a Alterations (substitutions, indels, SVs and CNAs) in significantly mutated genes and known UM driver genes. Nonsense mutations and frame-shift indels are labelled LOF. Loss-of-function breakpoints due to structural variants are labelled SV. b Copy number status of the commonly aberrant chromosomes 1, 3, 6 and 8, in UM. c Positions of mutations in encoded protein of significantly mutated genes, CENPE, TP53 and RPL5. Mutations are coloured black (LOF) and green (missense). Proteins domains are depicted wider and structurally important regions are coloured dark grey (disorder), light green (coiled-coil) and light blue (low complexity).
Significantly mutated genes
In addition to these known UM genes, three other statistically significantly mutated genes (SMGs) were identified (CENPE, TP53, RPL5; Supplementary Table 1). Three missense mutations and two LOF SVs were identified in CENPE, along with one LOF germline mutation (late truncating) (collectively ~5% of UM). An additional six samples were hemizygous for CENPE (Fig. 3a). CENPE is a plus end-directed kinetochore motor protein which plays a critical role in mitosis and chromosome segregation. Knockdown of CENPE has been shown to cause chromosome misalignment and lagging22,23. Two of the missense mutations (p.R14W and p.R251W) occurred in the kinesin motor domain (Fig. 3c) at highly evolutionarily conserved regions (ECRs) and the third (p.Q1098P) occurred in a reasonably conserved residue (Supplementary Fig. 1). An additional two missense mutations were identified in the UM TCGA cohort at p.I1038T (weak ECR) and p.K1821N (reasonable ECR), both also in a coiled-coil domain24. CENPE has been shown to interact with CENPF, BUB1B and Aurora B, the latter two being critical in the activation of the spindle assembly checkpoint25,26,27. In the UM cohort described here, one sample had a BUB1B missense substitution (p.R691H) within the region reported to directly interact with CENPE25; another had a p.D303E substitution in a highly ECR of the Aurora B catalytic domain. It is possible that disruption of this pathway is responsible for creating genomic instability allowing for chromosome aberrations to occur. Indeed, the twelve UM with CENPE alterations had significantly higher genome percentages with CNAs (Mann–Whitney, P = 0.028, median 23% vs 15%), though this association is confounded since tumours with high CNA generally have more genome-wide regions of LOH. Studies both in cell line and mouse models have demonstrated that CENPE/Cenpe functions in a haploinsufficient manner, with elevated levels of chromosome mis-segregation observed in heterozygous cells and animals28,29. Functional work on CENPE missense mutations is required to determine their impact in UM.
TP53 is commonly disrupted in uveal melanoma
High expression of p53 has been reported in some UM, often associated with histological and clinicopathological features correlated with poor prognosis, but the potential genetic basis of these observations was not assessed30,31,32. Somatic mutations in TP53 have been described in two UM. A hotspot mutation p.R175H (n = 1216 in IARC TP53 database) was observed in a hypermutated metastatic UM with deficient MBD433 and another hotspot mutation p.M237I (n = 196 in IARC TP53 database) was observed in a UM in a pan-cancer study of metastatic tumours17. Here we identified TP53 as an SMG and report six somatic TP53 mutations across four tumours in addition to eight cases of LOH (Figs. 3a, b). One LOH case overlapped with an LOF mutation (p.C277*) resulting in a double-hit in TP53. Another double-hit was seen in a sample with two missense mutations (p.H193R and p.T155I) confirmed as occurring on different alleles by assessing read pairs spanning both mutations. To evaluate the consequence of these mutations, we applied a computational prediction tool, FATHMM34, and assessed the results of two comprehensive characterisation studies of TP53 mutations (Table 1)35,36. p.H193R is a recurrent hotspot classified as pathogenic by PHANTM, RFS and FATHMM, while the consequence of p.T155I is more uncertain, as the variant is classified pathogenic by PHANTM but predicted to have neutral impact by RFS and FATHMM. Finally, one UM had a LOF mutation (p.R342*, COSM11073) and a missense p.R248Q mutation, both of which frequently occur in malignancies and are classified as pathogenic (Table 1). RNA-seq data revealed one read pair spanning both positions, which contained p.R248Q and was wildtype for p.R342; furthermore, there was a significantly lower VAF at p.R342 (4/78) than at p.R248 (25/68) (two-sided Fisher’s exact test, P = 2 × 10−6). These data suggest the two mutations occurred on different alleles, with the majority of the transcripts from the p.R342* allele undergoing nonsense mediated decay. TP53 is a tumour suppressor gene frequently deleted or mutated, resulting in either no production of p53 or the expression of a truncated and unstable protein. The spectrum of a few highly recurrent missense mutations, including p.R248Q, has, however, given rise to hypotheses that these hotspot mutations translate to mutant p53 with gained oncogenic functions37. For example, the p.R248Q mutation reported here has been shown to increase the migratory potential of cells in an in vitro model38.
RPL5 is significantly mutated in uveal melanoma
SMG analysis also identified RPL5, with truncating mutations in three cases and LOH in an additional 30 cases (Fig. 3a). No UM had a double hit in RPL5, in line with previous studies suggesting RPL5 is a haploinsufficient tumour suppressor, with heterozygous inactivation in glioblastoma (11%), breast cancer (34%) and cutaneous melanoma (28%)39. In UM the majority of RPL5 LOH occurred through loss of chromosome 1p, where the gene is located, but in four cases, focal loss occurred, suggesting that RPL5 may drive positive selection for 1p loss. Chromosome 1p loss in UM has been reported as a marker of poor prognosis, independent of M312. RPL5 encodes ribosomal protein L5, which complexes with 5S rRNA and forms an important part of the impaired ribosome biogenesis checkpoint (IRBC). Together with RPL11 and ARF, RPL5 binds to and inhibits MDM2, resulting in p53 stabilisation in response to blocks in ribosome biogenesis and nucleolar stress40,41,42. Of note, one truncating mutation in RPL11 was observed in the UM TCGA cohort8 further supporting the importance of this pathway in UM. Given the link between RPL5 and p53, we tested for an association between aberrations in RPL5 and TP53. Indeed, mutations in these genes were mutually exclusive, but when also considering copy-number loss there was no association. However, as p53 functions in multiple pathways, it may be inactivated in some tumours with defective RPL5 due to selective pressures outside the IRBC response. Interestingly, the IRBC is often triggered by oncogene-induced translational stress40,41,42, with oncogenic MYC being shown to significantly increase IRBC activation41,42,43,44. In addition to their IRBC role, RPL5 and RPL11 have also been shown to bind to MYC transcripts, mediating RNA-induced silencing and interfering with c-Myc driven transcription45,46. Overexpression of MYC is thought to be the driving force behind positive selection of chromosome 8q gains in UM. It is possible that c-Myc overexpression leads to ribosomal stress and IRBC activation, likely inhibiting tumour growth. To overcome this, it may be necessary to disrupt this pathway through mutation/loss of either RPL5, RPL11 or TP53. Supporting this notion, RPL5 and TP53 disruption (including mutations, SV breakpoints and chromosomal copy loss) was more common in cases with 8q gain (48%) than in cases without 8q gain (22%) (two-sided Fisher’s exact test, P = 0.01). Tumours with alterations in this pathway were predominantly M3, making it difficult to disentangle the prognostic impact; however, these individuals had poorer prognosis (Supplementary Fig. 2).
Genomic categories correlate with prognosis
To correlate UM genomic categories with prognosis, time from first presentation to metastasis was examined for all patients. Comparing the four TCGA categories, there was a trend that category 4 tumours had shorter relapse-free survival (RFS) (median: 2.5 years) than UMs in category 3 (median: 7.5 years) and similarly category 2 had shorter RFS (median: 7.0 years) than category 1 (median not reached), but the differences were not statistically significant (P3vs4 = 0.11, P1vs2 = 0.28) (Fig. 4a). However, M3 UM had significantly shorter RFS (median: 2.9 years) than D3 UM (median 7.0 years, log-rank test, P = 0.001, Fig. 4b), confirming the prognostic strength of M3/D3 status. Interestingly, while iris melanomas are associated with earlier detection and favourable prognosis, 4/8 iris cases had M3, with two already having progressed to metastatic disease. The high TMB in iris UM reported here suggests they are more likely to respond to immunotherapy, given the observations for MBD4 germline UM patients14,15, who have high TMB, a predictor of response to immunotherapy response in cutaneous melanoma and other cancers47. In combination with previous reports of metastatic iris UM48, these data suggest a subset of iris UM are at high risk for disease progression and will likely respond to immunotherapy in the event of progression and perhaps even in the adjuvant setting.

Higher risk categories have shorter RFS. UM patients with monosomy of chromosome 3 has significantly shorter RFS than patients with disomy 3 (blue). Difference between curves were assessed using a two-sided log-rank (Mantel-Cox) test; p-values were not adjusted for multiple testing.
Methods
Human melanoma samples
Fresh-frozen tissue and matched normal samples were obtained from the Terrace Eye Centre (Brisbane, Australia), Rigshospitalet (Copenhagen, Denmark), Melanoma Institute Australia (Sydney, Australia), Lions Eye Institute (Perth, Australia), Royal Perth Hospital (Perth, Australia), St John of God Hospital (Subiaco, Australia), and Ludwig Institute for Cancer Research (Brussels, Belgium). All tumour and blood/saliva samples were accrued with written informed patient consent following institutional review board approval from the Human Research Ethics Committees of the QIMR Berghofer Medical Research Institute, the Sydney Local Health District RPAH zone, the University of Western Australia, the Capitol Region of Denmark and the Ludwig Institute for Cancer Research. Patient information was stored in Progeny 10.
DNA extractions
Tumour DNA was extracted using the AllPrep DNA/RNA Kit (80204, Qiagen Ltd, Hilden, Germany), blood DNA using standard salting out methods, and saliva DNA was collected and extracted using the Oragene DNA kit (OG-500, DNA Genotek, Ottawa, Canada) according to the manufacturer’s instructions. All samples were quantified using a NanoDrop (ND1000; Thermo Fisher Scientific, Waltham, Massachusetts, USA) and Qubit dsDNA HS Assay Q32851; Life Technologies, Carlsbad, California, USA).
Whole-genome sequencing
Sequencing libraries were constructed using TruSeq DNA Sample Preparation kits (Illumina, San Diego, California, USA) according to the manufacturer’s instructions. WGS was performed on Novaseq or HiSeq X Ten instruments (Illumina) by Macrogen (Seoul, South Korea). Sequence data were adapter trimmed using Cutadapt v1.949 and aligned to the GRCh37 assembly using BWA-MEM v0.7.12 and SAMtools v1.850,51. Duplicate reads were marked with Picard MarkDuplicates v1.129 (https://broadinstitute.github.io/picard).
Similarly, RNA sequence reads were adapter adapter trimmed using Cutadapt and aligned using STAR v2.5.2a to the GRCh37 assembly with the gene, transcript, and exon features of Ensembl gene model v7052.
Somatic mutations
Somatic SNV and indels were detected using an established pipeline53 in which SNVs were called with qSNP54 and GATK HaplotypeCaller and indels were detected with GATK55. The contribution from different mutation signatures was inferred by approximating (minimising the squared error) the distribution of mutations as a linear combination of COSMIC signature v313 with the constraint that contributions were non-negative. SMGs were identified using MutSigCV 1.3.5 (via GenePattern) as well as Oncodrive-fm and OncodriveClust (via IntOGen)56. A Benjamini-Hochberg adjusted p-value (q-value) below 0.05 was considered significant. To avoid false negatives in hotspots of known UM genes, these regions (GNAQ p.48, p.183, and p.209; GNA11 p.183 and p.209; SF3B1 codons p.625, p.666 and p.700; EIF1AX codons p.1-20, PLCB4 p630; and CYSLTR2 p.129) were called with higher sensitivity. For each sample and genomic position the variant and reference read counts were compared with the variant and reference read counts in the pool of all 103 normal/germline samples at that specific position. Fisher’s exact test was used to identify somatic mutations and a Bonferroni corrected p-value below 0.001 was considered statically significant.
Classification of TP53 mutations
To evaluate the TP53 mutations, they were compared with two comprehensive characterisation studies. The Phenotypic Annotation of TP53 Mutations (PHANTM) score v1.0 is a weighted sum of z-scores for which common (i.e. benign) germline variants have values around 0 and recurrent somatic hotspot mutations have scores around 135. The relative fitness score (RFS) is on average −2.50 for synonymous variants, while the average score for protein truncating variants is 0.4236. FATHMM is an in silico tool predicting the probability that variants are deleterious34. Scores above 0.5 are considered deleterious. The IARC TP53 mutation database (R20, July 2019) was used to evaluate how frequent mutations are57.
Copy number aberrations and SV
Copy number aberrations were identified using ascatNgs58. Copy number of at least 6 was considered high gain. The underlying model of ascatNgs assumes the data come from two clones of cells: the tumour and normal contamination. For a heterogeneous tumour it may therefore overestimate copy numbers; to distinguish heterogeneity from WGD, the distribution of VAF for somatic mutations within regions with allelic balance were used. For a tetraploid tumour, two peaks in VAF are expected corresponding to mutations occurring before and after the copy number gain, with the latter having half the VAF of the former. Statistical models for the data coming from a homogeneous tetraploid tumour and from a heterogeneous diploid tumour, respectively, were inferred using maximum-likelihood and if the heterogeneity was significantly more likely (P < 0.001), copy numbers were adjusted.
Structural variants were identified using an in-house tool, qSV, as previously described53. Gene truncating breakpoints and consequence of the SVs were determined using in-house scripts and transcript annotation from Ensembl.
Survival analysis
Relapse-free curves were estimated using the Kaplan–Meier method. Difference between curves was assessed with the log-rank (Mantel-Cox) test.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The BAM files are deposited in the European Genome-phenome Archive ([https://www.ebi.ac.uk/ega/]) with accession number EGAS00001001552. The source data underlying Fig. 2 are provided as a data source file. All other data are available in the Article, Supplementary Information or available from the author upon reasonable request.
Code availability
Tools used in this publication that were developed in-house are available from the SourceForge public code repository under the AdamaJava project ([http://sourceforge.net/projects/adamajava/]). Updated versions of software are available at [https://github.com/AdamaJava].
References
Lane, A. M., Kim, I. K. & Gragoudas, E. S. Long-term risk of melanoma-related mortality for patients with uveal melanoma treated with proton beam therapy. JAMA Ophthalmol. 133, 792–796 (2015).
Damato, B. E., Heimann, H., Kalirai, H. & Coupland, S. E. Age, survival predictors, and metastatic death in patients with choroidal melanoma: tentative evidence of a therapeutic effect on survival. JAMA Ophthalmol. 132, 605–613 (2014).
Zbytek, B. et al. Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 3, 569–585 (2008).
Damato, B. et al. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Investigative Ophthalmol. Vis. Sci. 50, 3048–3055 (2009).
Onken, M. D., Worley, L. A., Ehlers, J. P. & Harbour, J. W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 64, 7205–7209 (2004).
Yavuzyigitoglu, S. et al. Uveal melanomas with SF3B1 mutations: a distinct subclass associated with late-onset metastases. Ophthalmology 123, 1118–1128 (2016).
Vichitvejpaisal, P. et al. Genetic analysis of uveal melanoma in 658 patients using the cancer genome atlas classification of uveal melanoma as A, B, C, and D. Ophthalmology 126, 1445–1453 (2019).
Robertson, A. G. et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 32, 204–220.e215 (2017).
Royer-Bertrand, B. et al. Comprehensive genetic landscape of uveal melanoma by whole-genome sequencing. Am. J. Hum. Genet. 99, 1190–1198 (2016).
Furney, S. J. et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 3, 1122–1129 (2013).
Johansson, P. et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 7, 4624–4631 (2016).
Kilic, E. et al. Clinical and cytogenetic analyses in uveal melanoma. Investigative Ophthalmol. Vis. Sci. 47, 3703–3707 (2006).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Johansson, P. A. et al. Prolonged stable disease in a uveal melanoma patient with germline MBD4 nonsense mutation treated with pembrolizumab and ipilimumab. Immunogenetics 71, 433–436 (2019).
Rodrigues, M. et al. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat. Commun. 9, 1866 (2018).
Shah, C. P., Weis, E., Lajous, M., Shields, J. A. & Shields, C. L. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology 112, 1599–1607 (2005).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Joseph, N. M. et al. Frequent GNAQ and GNA14 mutations in hepatic small vessel neoplasm. Am. J. surgical Pathol. 42, 1201–1207 (2018).
van de Nes, J. A. et al. Comparing the prognostic value of BAP1 mutation pattern, chromosome 3 status, and BAP1 immunohistochemistry in uveal melanoma. Am. J. Surgical Pathol. 40, 796–805 (2016).
Harbour, J. W. et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–1413 (2010).
Martin, M. et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 45, 933–936 (2013).
Tame, M. A., Raaijmakers, J. A., Afanasyev, P. & Medema, R. H. Chromosome misalignments induce spindle-positioning defects. EMBO Rep. 17, 317–325 (2016).
Ciossani, G. et al. The kinetochore proteins CENP-E and CENP-F directly and specifically interact with distinct BUB mitotic checkpoint Ser/Thr kinases. J. Biol. Chem. 293, 10084–10101 (2018).
Chang, K. T., Guo, J., di Ronza, A. & Sardiello, M. Aminode: identification of evolutionary constraints in the human proteome. Sci. Rep. 8, 1357 (2018).
Chan, G. K., Schaar, B. T. & Yen, T. J. Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J. cell Biol. 143, 49–63 (1998).
Yao, X., Abrieu, A., Zheng, Y., Sullivan, K. F. & Cleveland, D. W. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol. 2, 484–491 (2000).
Mao, Y., Abrieu, A. & Cleveland, D. W. Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell 114, 87–98 (2003).
Weaver, B. A., Silk, A. D., Montagna, C., Verdier-Pinard, P. & Cleveland, D. W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36 (2007).
Weaver, B. A. & Cleveland, D. W. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res. 67, 10103–10105 (2007).
Liu, H. & Zhou, M. Evaluation of p53 gene expression and prognosis characteristics in uveal melanoma cases. OncoTargets Ther. 10, 3429–3434 (2017).
Brantley, M. A. Jr. & Harbour, J. W. Deregulation of the Rb and p53 pathways in uveal melanoma. Am. J. Pathol. 157, 1795–1801 (2000).
Hussein, M. R. The relationships between p53 protein expression and the clinicopathological features in the uveal melanomas. Cancer Biol. Ther. 4, 57–59 (2005).
Rodrigues, M. et al. Evolutionary routes in metastatic uveal melanomas depend on MBD4 alterations. Clin. Cancer Res. 25, 5513–5524 (2019).
Shihab, H. A., Gough, J., Cooper, D. N., Day, I. N. & Gaunt, T. R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29, 1504–1510 (2013).
Giacomelli, A. O. et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387 (2018).
Kotler, E. et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell 71, 178–190 e178 (2018).
Oren, M. & Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2, a001107 (2010).
Yoshikawa, K. et al. Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomed. Res. 31, 401–411 (2010).
Fancello, L., Kampen, K. R., Hofman, I. J., Verbeeck, J. & De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 8, 14462–14478 (2017).
Dai, M. S. & Lu, H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279, 44475–44482 (2004).
Sloan, K. E., Bohnsack, M. T. & Watkins, N. J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 5, 237–247 (2013).
Sun, X. X., Wang, Y. G., Xirodimas, D. P. & Dai, M. S. Perturbation of 60 S ribosomal biogenesis results in ribosomal protein L5- and L11-dependent p53 activation. J. Biol. Chem. 285, 25812–25821 (2010).
van Riggelen, J., Yetil, A. & Felsher, D. W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309 (2010).
Morcelle, C. et al. Oncogenic MYC induces the impaired ribosome biogenesis checkpoint and stabilizes p53 independent of increased ribosome content. Cancer Res. 79, 4348–4359 (2019).
Liao, J. M., Zhou, X., Gatignol, A. & Lu, H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 33, 4916–4923 (2014).
Challagundla, K. B. et al. Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol. Cell. Biol. 31, 4007–4021 (2011).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Shields, C. L. et al. Iris melanoma: risk factors for metastasis in 169 consecutive patients. Ophthalmology 108, 172–178 (2001).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. https://doi.org/10.14806/ej.17.1.200 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Hayward, N. K. et al. Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180 (2017).
Kassahn, K. S. et al. Somatic point mutation calling in low cellularity tumors. PLoS ONE 8, e74380 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Gonzalez-Perez, A. et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10, 1081–1082 (2013).
Bouaoun, L. et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 37, 865–876 (2016).
Van Loo, P. et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA 107, 16910–16915 (2010).
Acknowledgements
We are indebted to the patients and their families for their participation and support of this study, and the many clinicians and allied health professionals involved in their management. We gratefully acknowledge the participation of Sharon Morris, Nicholas O’Rourke, David Cavallucci Thomas O’Rourke, Helen Marfan and Rachel Susman. As well, we thank Kevin Whitehead, Gary Quagliotto, and Sullivan Nicolaides Pathology staff for processing the Queensland samples. This project was funded by the National Health and Medical Research Council (NHMRC; 1093017), the Walking On Sunshine Foundation, Anne Stanton, Nicola Laws and Lloyd Owen in Memorial and Civic Solutions. This study was also funded by Fight for Sight, Denmark. A.L.P. is supported by Highland Island Enterprise (HMS9353763). This work was supported by an NHMRC Program Grant (G.V.L., G.J.M., R.A.S. and N.K.H.). G.V.L. is supported by an NHMRC Practitioner Fellowship and The University of Sydney, Medical Foundation. R.A.S. is supported by an NHMRC Practitioner Fellowship. Support from Melanoma Institute Australia and The Ainsworth Foundation is also gratefully acknowledged. J.S.W. is supported by a NHMRC early career fellowship (1111678). N.W. is supported by an NHMRC Senior Research Fellowship (1139071). N.K.H. is supported by an NHMRC Senior Principal Research Fellowship (1117663).
Author information
Authors and Affiliations
Contributions
P.A.J. and K.B. were responsible for data interpretation and writing the manuscript. N.K.H., R.A.S. and G.J.M. conceptualised the study. P.A.J., F.N., C.L., S.W., N.B., L.T.K., J.V.P. and N.W. performed data analysis. K.B. N.B., V.N., C.W.S., R.D. and A.B.B. were responsible for sample processing and extraction. R.A.S., N.K.H., R.A.S., G.V.L., H.R., E.G. acted in a supervisory capacity. A.L.P., J.M.P., J.S.W., M.S.C., M.H., H.R., G.V.L., H.H., J.J.P., O.J.R., J.F.K., T.I., N.vB., K.W.W., L.A.M., A.S., S.K.W., W.G. were responsible for patient data curation, recruitment and tumour acquisition. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.V.P. and N.W. are founders and shareholders of genomiQa Pty Ltd, and members of its Board. K.W.W. participated in one Advisory board meeting for MSD and AstraZeneca. R.A.S. receives fees for professional services from Merck Sharp & Dohme, GlaxoSmithKline Australia, Bristol-Myers Squibb, Dermpedia, Novartis Pharmaceuticals Australia Pty Ltd, Myriad, NeraCare and Amgen. G.V.L. is consulant advisor for Aduro, Amgen, Array, BMS, MERCK MSD, Novartis, Pierre-Fabre, Roche. None of these relationships involve the work described in this manuscript. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johansson, P.A., Brooks, K., Newell, F. et al. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat Commun 11, 2408 (2020). https://doi.org/10.1038/s41467-020-16276-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16276-8
This article is cited by
-
Ambient ultraviolet radiation and ocular melanoma incidence in the United States, 2000−2019
Eye (2024)
-
Landscape of enhancer disruption and functional screen in melanoma cells
Genome Biology (2023)
-
Advances in the clinical management of uveal melanoma
Nature Reviews Clinical Oncology (2023)
-
Iris melanocytic tumours in New Zealand/Aotearoa: presentation, management and outcome in a high UV exposure environment
Eye (2023)
-
The journey from melanocytes to melanoma
Nature Reviews Cancer (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
