
Abstract
Extending photoresponse ranges of semiconductors to the entire ultraviolet–visible (UV)–shortwave near-infrared (SWIR) region (ca. 200–3000 nm) is highly desirable to reduce complexity and cost of photodetectors or to promote power conversion efficiency of solar cells. The observed up limit of photoresponse for organic-based semiconductors is about 1800 nm, far from covering the UV–SWIR region. Here we develop a cyanide-bridged layer-directed intercalation approach and obtain a series of two viologen-based 2D semiconductors with multispectral photoresponse. In these compounds, infinitely π-stacked redox-active N-methyl bipyridinium cations with near-planar structures are sandwiched by cyanide-bridged MnII–FeIII or ZnII–FeIII layers. Radical–π interactions among the infinitely π-stacked N-methyl bipyridinium components favor the extension of absorption range. Both semiconductors show light/thermo-induced color change with the formation of stable radicals. They have intrinsic photocurrent response in the range of at least 355–2400 nm, which exceeds all reported values for known single-component organic-based semiconductors.
Similar content being viewed by others


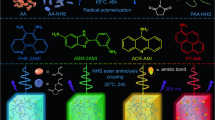
Introduction
Photoresponse range is a metric that significantly affects the potentials of semiconductors for photodetection, solar energy conversion and other applications1,2,3,4. As for photodetection, detection ranges of commercial Si-based photodetectors and InGaAs photodetectors are mostly in the 200–1100 nm and 900–3000 nm regions, respectively1,5. To realize photodetection in the ultraviolet (UV)–shortwave infrared (SWIR) region (ca. 200–3000 nm), either military or civil photodetecting devices usually have to combine the above two photodetectors. This will increase cost and complexity of the desirable device. As for solar energy conversion, photoresponse band of the widely used commercial photovoltaic material silicon is about 300–1200 nm6,7. This range far from covers the whole solar spectrum (∼295–2500 nm), resulting in limited energy conversion fields of Si-based solar cells2, 8. Constructing multiple blend systems or multi-junction device structures was demonstrated to be effect methods to fully utilize the solar radiation, but complexity, manufacture cost, and sometimes stability of the device should be concerned before stepping into the market9,10,11. The above issues for both photodetection and solar energy conversion devices are promisingly avoided, if a single semiconductor material with a photoresponsive range covering the entire UV–SWIR region is applied10,12. As reported, some single-component inorganic photoelectronic semiconductors showed strikingly wide detection ranges, for example, the photoresponse range of SnTe covers 254–4650 nm13. However, practical application of them is still greatly limited by material rigidity, as well as complex and expensive manufacturing processes14,15. Organic-based photoelectronic semiconductors are characteristic of flexibility and facile preparation, but the observed up limit for photoresponse is about 1800 nm10,12,15,16,17,18. It is still of importance to develop effective and general design methods for single-component organic-based semiconductors with intrinsic photoresponse in the entire UV–SWIR range.
Viologen (N,N′-disubstituted bipyridinium) compounds are good candidates for single-component organic-based semiconductors with broadband photoresponse. Firstly, strong cation···π interactions between viologen components favor the construction of organic semiconductors, and conductance and photoconductance of viologen-based semiconductors may dramatically increase after photoinduced electron transfer (PET) and generation of free radical products19,20,21. Secondly, single viologen cation usually has a red-shifted absorption band after forming a radical species. When viologen radicals are further closely π-stacked, radical–π interactions that are stronger than cation–π, and π–π interactions will make energy gap narrower and correspondingly absorption band much broader22,23,24. Even so, improving the following two properties is still needed for viologen compounds. Firstly. photoconversion rate in bulk media is usually low, owing to low penetrability of UV–Visible light25,26. Thermal treatment may avoid this problem, because heat easily conducts to the whole media. Many examples have indicated viologen compounds are probable to undergo heat-induced electron transfer (HET) when they tend to be planar27,28. Therefore, thermo-active viologen semiconductors are prospectively constructed with near-planar/planar viologen cations. Secondly, the radical products are highly active and easily faded in an oxygen atmosphere with the presence of heat or not29,30. Several examples have demonstrated that, the stability of viologen radicals can be clearly improved when the radicals are loaded in a porous framework31,32 or sandwiched by inorganic layers33 to avoid contact with oxygen.
In this work, we present a cyanide-bridged layer-directed intercalation approach to realize all above points and obtain single-component viologen-based semiconductors with intrinsic UV–SWIR photoresponse ability, photo/thermo activeness, and very long lifetime of radical products. Each cyanide-bridged layer in Prussian blue or its analogs with perovskite-like structures has periodically arranged hexacoordinated metal sites and limited metal-to-metal distances (Fig. 1)34. The periodic arrangement of metal coordination sites facilitates the orderly and infinite accumulation of axial ligands. The closest and non-contact metal-to-metal distances are usually 7.6 Å34, which are suitable to support two π stacking interactions35. As shown in Fig. 1, a sandwiched inorganic–organic hybrid structure with infinitely stacked organic supramolecular layers will be formed, when metals with these distances are all coordinated by one viologen ligand or its analogs and then the layers are intercalated. Coexistence of π···π and cation···π interactions in the organic supramolecular layer is predicable since viologen and its analogs are aromatic cations. As mentioned above, this case favors the construction of a semiconductor. In addition, the viologen ligand or its analogs will become planarization owing to the close stacking of adjacent ligands, which may bring thermo activeness as stated above. Based on these considerations, we integrated the redox photoactive N-methyl bipyridinium (MQ+) cation into cyanide-bridged MII–FeIII (MII=MnII or ZnII) layers as axial ligands, and obtained a series of two viologen-based 2D semiconductors, [{MII(MQ)2}{FeIII(CN)6}]Cl·3H2O (1, M=Mn; 2, M=Zn). These semiconductors are both thermo and light active in the crystalline state. After PET or HET, they generate long-lived radical products, and show intrinsic photoresponsive bands covering the UV–SWIR region (at least 355–2400 nm, monitored using our lasers).

δ and dπ–π denote the interannular angle and common separation for π–π interactions, respectively.
Results
Directed synthesis and structural characterization
Single crystals of 1 and 2 were all obtained from the diffuse reaction of MnCl2·5H2O, MQCl·H2O, and K3[FeIII(CN)6] in a molar ratio of 1:2:1 in water. Powder X-ray diffraction (PXRD; Supplementary Fig. 1) and elemental analysis demonstrated phase purity of the obtained crystalline samples. Only the crystal structure of 1 is described here since 1 and 2 are isostructural. As can be seen from Fig. 2a, cyanide-bridged layers of 1 are intercalated through π···π and cation···π interactions between two adjacent MQ+ ligands. Each FeIII atom coordinates to six cyano groups (four, bridged, two, mono-coordinated), while each MnII atom is ligated by two MQ+ ligands and four bridged cyano groups from four [Fe(CN)6]3− units. The interannular angle of each MQ+ cation is 0.44°, close to be planar (Fig. 2b). Centroid (pyridyl)···centroid (pyridyl) and N···centroid (pyridyl) distances between two adjacent π-stacked MQ+ cations are 3.88 and 3.71 Å, respectively (Fig. 2b). Every π-stacked MQ+ layer is sandwiched by two cyanide-bridged layers, which offers a chance to shield the air (Fig. 2c). After standing in the dark under the 100% relative humidity for 12 h, the samples still kept the same PXRD patterns to the as-prepared crystalline samples (Supplementary Fig. 1), illustrating high wet stability for both compounds. Thermogravimetric (TG) analyses indicated that both compounds contain water molecules (Supplementary Fig. 2). In the electron absorption spectrum (Fig. 3), a series of infrared (IR) overtone peaks for H2O can be observed, such as at 1412, 1916, and 2158 cm−1. Crystal structures of both compounds can be retained after thermal annealing at 180 °C and removing all the water molecules, as evidenced by the similar PXRD patterns between as-prepared and thermally annealed samples (Supplementary Fig. 1). Therefore, the following mentioned thermal annealing processes were all operated at 180 °C.

a Side view of the 3-D packing structure; b two π-stacked MQ+ cations; c infinitely π-stacked MQ+ cations between two perovskite-like cyanide-bridged layers (cyano groups are drawn as vertexes of octahedra). Dash lines depict π stacking interactions.

Time-dependent electron absorption spectra of 1 measured in the diffuse reflectance mode and color change upon irradiation (a) or thermal annealing at 180 °C (b).
Photo/thermo-induced coloration
Both 1 (Fig. 3) and 2 (Supplementary Fig. 3) may undergo photo/thermo-induced coloration. Also, only compound 1 is described in detail. The as-prepared crystalline sample (1A) of 1 changed its color from brown to black upon irradiation by a Xe lamp (∼200 mW cm−2) at room temperature (Fig. 3a). The photoinduced black sample (1B-P) appeared a broad electron absorption band around 620 nm and a much broader band in the range of ∼900–2500 nm (Fig. 3a). There was no further clear variation of the electron absorption spectrum when the sample was irradiated beyond 100 min. Upon thermal annealing under 180 °C in air, the 1A sample also turned black (Fig. 3b). The yielded black sample (1B-T) generated similar but stronger electron absorption bands (Fig. 3b) and electron spin resonance (ESR) signals (Fig. 4b) to those of 1B-P. If UV/Visible/NIR spectra and IR spectra are combined, we can see that the absorption band of 1B-T extends to 3000 nm (Fig. 4a and Supplementary Fig. 4), that is to say, the absorption spectrum of 1B-T covers the whole UV–SWIR region. The absorption spectrum did not change again after annealing for about 150 min (Fig. 3b). 1B-T was considerably stable because its absorption spectrum almost retained after standing in the dark in air at room temperature for six months (Fig. 4a). Similar to 1B-T, 2B-T also shows broad absorption in the UV–SWIR region and high stability in air (Supplementary Fig. 5a).

a Stability test for the thermo-induced sample 1B-T monitored through the combination of UV/Visible/NIR spectra (200–2500 nm) and IR spectra (2500–3000 nm). The sample was placed in air in the dark at room temperature for 6 months. The minor difference between 1B-T and 1B-T@6month was attributed to systematic errors caused by instrumental instability or shift of the sample holder. b ESR patterns for 1A, 1B-P, and 1B-T. Note: 1A is the as-prepared crystalline sample; 1B-P is the sample that was irradiated by an Xe lamp for 70 min; 1B-T is the sample that was thermally annealed at 180 °C for 150 min.
Electrical studies
Electron absorption (Fig. 3 and Supplementary Fig. 3) and ESR (Fig. 4b, Supplementary Fig. 5b, Supplementary Note 1, and Supplementary Table 2) data of 1 and 2 revealed that thermal annealing triggered higher conversion rate than the irradiation method. So, both thermo-induced samples 1B-T and 2B-T were selected to perform electric tests. A well-known two-probe method using silver paste for a pellet sample was adopted36. The current–voltage (I–V) characteristic curves before and after coloration for 1B-T showed a symmetrical nearly linear relationship at room temperature, which indicated that the sample formed an Ohmic contact and the carriers derived from intrinsic thermal excitation. After HET, the conductivity increased ∼4-folds (Supplementary Fig. 6), which is accordance with the decrease of activation energy (Supplementary Fig. 7). This phenomenon is consistent with the well-established conclusion that receiving electrons and forming a radical species is beneficial to improve the conductivity of one π aggregate18,20. As mentioned above, the 1B-T sample had an intrinsic absorption spectrum covering the whole UV–SWIR region (Supplementary Fig. 4). Owing to limited testing equipment, we selected one 355 nm diode pumped solid-state laser and the other OPO laser with tunable wavelength ranging from 410 to 2400 nm to monitor the photocurrent response in the UV and visible–SWIR regions, respectively. Photocurrent gain can be expressed by the relative magnitude of the current change, (Iirr − Idark)/Idark. Figure 5 illustrates that the 1B-T sample had photocurrent response in the range of 355–2400 nm. The other two samples showed the similar photocurrent curves and photocurrent gains, indicating that the photoresponsive behavior of 1B-T is repeatable (Supplementary Fig. 8). Meanwhile, the greater the illumination power, the greater the photocurrent gain under laser irradiation (Supplementary Fig. 9). Bulk electric tests of 2B-T indicated that 2B-T was also an intrinsic semiconductor and showed photocurrent response in the range of 355–2400 nm (Supplementary Figs. 10 and 11). The photoresponse ranges for both 1B-T and 2B-T exceed all known reported values for single-component organic-based semiconductors5,10,12,37,38.

A Newport Co. Pulseo GKNQL-355-3-30 diode pumped solid-state laser (70 kHz, 39 ns pulse width) and an OPO laser (10 Hz, 10 ns pulse width) are used for 355 nm and 410–2400 nm light, respectively. Powers for each wavelength: 355 nm, 2.1 W; 410 nm, 7.5 mW; 600 nm, 5.0 mW; 800 nm, 15 mW; 1000 nm, 25 mW; 1500 nm, 23 mW; 1800 nm, 22 mW; 2000 nm, 25 mW; 2400 nm, 6.0 mW.
Discussion
The cyanide-bridged inorganic layers in 1 are similar to those of the room-temperature phase of RbMnII[FeIII(CN)6]·H2O. The latter has a CN → FeIII charge-transfer band at ∼410 nm (3.02 eV), a d–d transition band of FeIII at ∼520 nm (2.38 eV), and a MMCT band at ∼680 nm (1.82 eV)39. As illustrated in Fig. 3, the 1A sample also contains these electron-transition bands. The photoresponsive range of 1A is in ∼280–420 nm, wherein the optimal wavelength is around 320 nm (3.88 eV). Calculations of band structure and partial density of states showed that the absorption around 320 nm can be assigned to Cl−/CN−/FeIII → MQ+ electron transitions (Supplementary Fig. 12). The newly emerged bands around 620 nm (2.00 eV) for 1B-P and 1B-T are characteristic of single MQ• radical40, indicating that the MQ+ ligand received an electron upon irradiation or thermal annealing. ESR signal of the MQ• radical was not clear for 1B-P, but was identifiable for 1B-T (at g = 2.005; Fig. 4b). Low penetration of light in the crystal resulted in the generation of a small number of radicals, whose ESR signals were covered by MnII signals. In comparison, the heating mode led to the generation of more radicals. As for 2, ESR signals for both photo/thermo-induced samples were clear without the shielding of MnII signals (Supplementary Fig. 5b).
All photo/thermo-induced samples of 1 showed PXRD patterns similar to that of the as-prepared sample (Supplementary Fig. 1). Therefore, the framework of 1 did not undergo large isomerization or decomposition during its coloration. The lattice water molecules in 1 can be excluded to be an electron donor for MQ+, because 1B-T contained no water and also displayed electron transfer (Supplementary Fig. 2 and Fig. 3). In the crystal structure of 1 (Supplementary Fig. 13), each free Cl− ion forms four C(MQ+)–H···Cl hydrogen bonds (H···Cl, 2.67 Å) with adjacent four MQ+ ligands, and each mono-coordinated CN− group connects to two MQ+ ligands through C(MQ+)–H···N(CN) hydrogen bonds (H···N, 2.48 Å). According to the Marcus electron transfer theory41, the shorter the separation between an electron donor and an electron acceptor is, the faster the electron transfer process. It seems that the electron transfer rate through the C(MQ+)–H···N(CN) path should be faster than the C(MQ+)–H···Cl path. However, as evidenced by the references, both Cl−32,42,43,44 and CN−45,46 ions may act as effective electron donors for viologen cations. X-ray photoelectron spectroscopy is a common tool to prove the direction of electron transfer, but the paramagnetic metals in 1 are easily reduced by X-ray-induced electrons, which brings the difficulty for analysis. Searching other applicable methods is still underway.
The multispectral photocurrent response of 1B-T and 2B-T originates from the ultrabroad electron absorption (Supplementary Figs. 3 and 4). Time-dependent density functional theoretical (TD-DFT) calculations (Supplementary Fig. 14)47, using simplified structural models truncated form the crystal structure, indicates that the emerged broad bands in the range of ∼900–3000 nm for all photo/thermo-induced samples can be assigned to the contribution of π-stacked MQ aggregates, which in the form of [(HMQ2+)(HMQ+•)]n or [(HMQ+•)(HMQ+•)]n (n refers to the number). In summary, aiming to achieve single-component semiconductors with intrinsic UV–SWIR photoresponse, a cyanide-bridged layer-directed intercalation approach has been firstly demonstrated. The obtained two viologen-based 2D semiconductors have intrinsic absorption bands covering the whole UV–SWIR region after photo/thermo activation. They show photocurrent response at least in the wavelength range of 355–2400 nm (monitored using our limited lasers), which makes a record for single-component organic-based semiconductors. The design strategy explored in this work may inspire the synthesis of new organic-based semiconductors for broadband photodetectors and solar cells. In the following work, we will further improve electrical properties (conductivity, photocurrent gain, etc.) and photoresponse bands (extending to the mid-IR region), and explore effective methods to construct optical devices.
Methods
Materials
MnCl2·5H2O, ZnCl2 and K3[Fe(CN)6] in AR grade were purchased commercially. They were directly used without further purification. Water was deionized and distilled before use. MQCl·H2O (MQ+=N-methyl-4,4′-bipyridinium) was synthesized according to the same procedure reported in the literature48.
Syntheses of [{MnII(MQ)2}{FeIII(CN)6}]Cl·3H2O (1)
Typically, a 50 mL small beaker was placed in a 300 mL big one, which was filled with distilled water to approximately 0.5 cm above the top of the small beaker. A frozen 2 mL aqueous solution of MQCl·H2O (899 mg, 4 mmol) and MnCl2·5H2O (432 mg, 2 mmol) was thrown into the bottom of the small beaker, while the other frozen 2 mL aqueous solution of K3[Fe(CN)6] (659 mg, 2 mmol) was put into the bottom of the big beaker. The big beaker was sealed with a plastic wrap and allowed to stand in the dark at room temperature for one week to yield dark brown cubic crystals. The crystals were filtered, washed with water and ethanol, and finally dried in air for 1 day. Yield based on K3[Fe(CN)6]: 40% for 1. All crystal samples for tests were carefully selected under microscope. The phase purity of all as-synthesized crystalline samples was checked via PXRD (Supplementary Fig. 1) and elemental analyses. Anal. Calcd (%) for C28H28ClFeMnN10O3: C, 48.12; H, 4.04; N, 20.04; Fe, 7.99; Mn, 7.86. Found: C, 48.14; H, 3.78; N, 20.57; Fe, 7.61; Mn, 7.26.
Syntheses of [{ZnII(MQ)2}{FeIII(CN)6}]Cl·3H2O (2)
Typically, a 50 mL small beaker was placed in a 300 mL big one, which was filled with distilled water to approximately 0.5 cm above the top of the small beaker. A frozen 2 mL aqueous solution of MQCl·H2O (899 mg, 4 mmol) and ZnCl2 (273 mg, 2 mmol) was thrown into the bottom of the small beaker, while the other frozen 2 mL aqueous solution of K3[Fe(CN)6] (659 mg, 2 mmol) was put into the bottom of the big beaker. The big beaker was sealed with a plastic wrap and allowed to stand in the dark at room temperature for one week to yield yellow plate crystals. The crystals were filtered, washed with water and ethanol, and finally dried in air for 1 day. Yield based on K3[Fe(CN)6]: 25% for 2. All crystal samples for tests were carefully selected under microscope. The phase purity of all as-synthesized crystalline samples was checked via PXRD (Supplementary Fig. 1) and elemental analyses. Anal. Calcd (%) for C28H28ClFeZnN10O3: C, 47.42; H, 3.98; N, 19.75; Fe, 7.87; Zn, 9.22. Found: C, 45.54; H, 3.38; N, 19.75; Fe, 7.43; Zn, 10.18.
Measurements
IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer using KBr pellets in the range of 4000–450 cm−1. Thermogravimetric analysis was conducted on a Mettler TOLEDO simultaneous TGA/DSC apparatus. Elemental analyses of C, H and N were measured on an Elementar Vario EL III microanalyzer, while those of Fe, Zn, and Mn were measured on an ULTIMA 2 ICP Optical Emission Spectrometer. Electron spin resonance (ESR) spectra were recorded on a Bruker ER-420 spectrometer with a 100 kHz magnetic field in the X band at room temperature. Powder X-ray diffraction (PXRD) patterns were collected on a Rigaku Desktop MiniFlexII diffractometer using Cu Kα radiation (λ = 1.54056 Å) powered at 30 kV and 15 mA. Diffuse reflectance spectra were recorded at room temperature in the wavelength range of 200–2600 nm on a PerkinElmer Lambda 900 UV/vis/NIR spectrophotometer equipped with an integrating sphere. A BaSO4 plate was used as the reference (100% reflection), on which the finely ground powder of the sample was coated. Photoirradiation for coloration was carried out with a PLS-SXE300D 50-W xenon lamp system, wherein an IR filter was applied. I–V curves were measured in a Keithley 4200-SCS semiconductor parameter analyzer using pellet samples by the two-probe method using silver paste. An OPOTEK Vibrant laser (10 Hz; 10 ns pulse width; spot size, ca. 1–2 cmφ) and a Newport Co. Pulseo GKNQL-355-3-30 diode pumped solid state (DPSS) laser (355 nm; 70 kHz; 39 ns pulse width; spot size, ca. 1.5 cmφ) were used for photocurrent tests.
Single-crystal X-ray crystallographic study
Single-crystal X-ray diffraction measurements of 1 and 2 were performed on a Rigaku SATURN70 CCD diffractometer, using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Intensity data sets were collected using scan techniques, and corrected for Lp effects. The primitive structures were solved by the direct method using the Siemens SHELXTLTM Version 5 package of crystallographic software49. Difference Fourier maps based on these atomic positions yielded other non-hydrogen atoms. The final structures were refined using a full-matrix least-squares refinement on F2. All non-hydrogen atoms were refined anisotropically. H atoms on N-substituted C atoms were not included for their symmetrical disorder, and those of lattice water molecules were also not added for weak diffraction. Other H atoms were generated geometrically. Supplementary Table 1 shows the crystal and structure refinement data.
Calculations of electron absorption spectra
All calculations were performed with the time-dependent density functional theory (TD-DFT) method at the pbe1pbe/6-31 g* level using the Gaussian 09 software package50. Calculation models were truncated from the crystal structure of 1 and modified with H atoms replacing metal atoms.
Calculations of band structures and partial density of states
All calculations were executed using the Cambridge Sequential Total Energy Package (Castep)51. A plane-wave energy of 400 eV and a 3 × 3 × 2 Monkhorst-Pack grid of k-points were selected. The exchange-correlation energy was described by the Perdew–Burke–Eruzerhof (PBE) functional within the generalized gradient approximation (GGA)52. The norm-conserving pseudopotentials were chosen to modulate the electron–ion interaction53. Pseudo atomic calculations were performed for C 2s22p2, H 1s1, N 2s22p3, O 2s22p4, Cl 3s23p5, Fe 3d64s2, and Mn 3d54s2. Other parameters used in the calculations were set by the default values of the CASTEP code.
Data availability
The X-ray crystallographic data (1 and 2) reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition number CCDC 1958300–1958301. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, U.K. Fax: (Internet) +44-1223/336-033. E-mail: deposit@ccdc.cam.ac.uk. We declare that the main data supporting the findings of this study are available within the article and its Supplementary Information files. All relevant source data are also available from the corresponding author upon reasonable request.
References
García de Arquer, F. P., Armin, A., Meredith, P. & Sargent, E. H. Solution-processed semiconductors for next-generation photodetectors. Nat. Rev. Mater. 2, 16100 (2017).
Zhuang, T. T. et al. Integration of semiconducting sulfides for full-spectrum solar energy absorption and efficient charge separation. Angew. Chem. Int. Ed. 55, 6396–6400 (2016).
Dong, X. Y. et al. A crystalline copper(II) coordination polymer for the efficient visible-light-driven generation of hydrogen. Angew. Chem. Int. Ed. 55, 2073–2077 (2016).
Yang, M. Q. et al. Disorder engineering in monolayer nanosheets enabling photothermic catalysis for full solar spectrum (250–2500nm) harvesting. Angew. Chem. Int. Ed. 58, 3077–3081 (2019).
Wang, H. & Kim, D. H. Perovskite-based photodetectors: materials and devices. Chem. Soc. Rev. 46, 5204–5236 (2017).
Xu, Y., Xuan, Y. & Yang, L. Full-spectrum photon management of solar cell structures for photovoltaic-thermoelectric hybrid systems. Energy Convers. Manag. 103, 533–541 (2015).
Einzinger, M. et al. Sensitization of silicon by singlet exciton fission in tetracene. Nature 571, 90–94 (2019).
Wang, F. et al. Full-spectrum liquid-junction quantum dot-sensitized solar cells by integrating surface plasmon-enhanced electrocatalysis. Adv. Energy Mater. 8, 1800136 (2018).
Gasparini, N., Salleo, A., McCulloch, I. & Baran, D. The Role of the third component in ternary organic solar cells. Nat. Rev. Mater. 4, 229–242 (2019).
Feng, G. et al. Thermal-driven phase separation of double-cable polymers enables efficient single-component organic solar cells. Joule 3, 1765–1781 (2019).
Swierk, J. R. & Mallouk, T. E. Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem. Soc. Rev. 42, 2357–2387 (2013).
He, Y. et al. Evidencing excellent thermal-and photostability for single-component organic solar cells with inherently built-in microstructure. Adv. Energy Mater. 9, 1900409 (2019).
Yang, J. et al. Ultra-broadband flexible photodetector based on topological crystalline insulator snte with high responsivity. Small 14, 1802598 (2018).
Liu, Y. et al. High performance MoO3-x/Si heterojunction photodetectors with nanoporous pyramid si arrays for visible light communication application. J. Mater. Chem. C. 7, 917–925 (2019).
Yang, D. & Ma, D. Development of organic semiconductor photodetectors: from mechanism to applications. Adv. Optical Mater. 7, 1800522 (2019).
Wang, G. E. et al. From lead iodide to a radical form lead-iodide superlattice: high conductance gain and broader band for photoconductive response. Angew. Chem. Int. Ed. 58, 2692–2695 (2019).
Christians, J. A., Miranda Herrera, P. A. & Kamat, P. V. Transformation of the excited state and photovoltaic efficiency of CH3NH3PbI3 perovskite upon controlled exposure to humidified air. J. Am. Chem. Soc. 137, 1530–1538 (2015).
Hou, J., Inganas, O., Friend, R. H. & Gao, F. Organic solar cells based on non-fullerene acceptors. Nat. Mater. 17, 119–128 (2018).
Sun, C. et al. Design strategy for improving optical and electrical properties and stability of lead-halide semiconductors. J. Am. Chem. Soc. 140, 2805–2811 (2018).
Sun, C., Yu, X. Q., Wang, M. S. & Guo, G. C. A smart photochromic semiconductor: breaking the intrinsic positive relation between conductance and temperature. Angew. Chem. Int. Ed. 58, 9475–9478 (2019).
Roy, S., Mondal, S. P., Ray, S. K. & Biradha, K. A Photoswitchable and photoluminescent organic semiconductor based on cation-π and carboxylate-pyridinium interactions: a supramolecular approach. Angew. Chem. Int. Ed. 51, 12012–12015 (2012).
Quiñonero, D. et al. Interplay between cation-π, anion-π and π-π interactions. ChemPhysChem 7, 2487–2491 (2006).
Matsunaga, Y., Goto, K., Kubono, K., Sako, K. & Shinmyozu, T. Photoinduced color change and photomechanical effect of naphthalene diimides bearing alkylamine moieties in the solid state. Chem. Eur. J. 20, 7309–7316 (2014).
Wei, Y. Q., Sun, C., Chen, Q. S., Wang, M. S. & Guo, G. C. SignificanT enhancement of conductance of a hybrid layered molybdate semiconductor by light or heat. Chem. Commun. 54, 14077–14080 (2018).
Harada, J., Nakajima, R. & Ogawa, K. X-ray diffraction analysis of photochromic reaction of fulgides: crystalline state reaction induced by two-photon excitation. J. Am. Chem. Soc. 13, 7085–7091 (2008).
Harada, J., Uekusa, H. & Ohashi, Y. X-ray analysis of structural changes in photochromic salicylideneaniline crystals solid-state reaction induced by two-photon excitation. J. Am. Chem. Soc. 121, 5809–5810 (1999).
Sun, C., Wang, M. S., Li, P. X. & Guo, G. C. Conductance switch of a bromoplumbate bistable semiconductor by electron-transfer thermochromism. Angew. Chem. Int. Ed. 56, 554–558 (2017).
Chen, H. J. et al. Photo- and thermo-activated electron transfer system based on a luminescent europium organic framework with spectral response from UV to visible Range. Chem. Commun. 50, 13544–13546 (2014).
Monk, P. M. S. The Viologens: Physicochemical Properties, Synthesis and Applications of the Salts of 4,4’-Bipyridine (J. Wiley and Sons, Chichester, 1998).
Sun, J. K., Yang, X. D., Yang, G. Y. & Zhang, J. Bipyridinium derivative-based coordination polymers: from synthesis to materials applications. Coord. Chem. Rev. 378, 533–560 (2019).
Xing, X. S. et al. An electron-transfer photochromic metal-organic framework (MOF) compound with a long-lived charge-separated state and high-contrast photoswitchable luminescence. RSC Adv. 6, 24190–24194 (2016).
Liu, Q. K., Ma, J. P. & Dong, Y. B. Adsorption and separation of reactive aromatic isomers and generation and stabilization of their radicals within cadmium(II)-triazole metal-organic confined space in a single-crystal-to-single-crystal fashion. J. Am. Chem. Soc. 132, 7005–7017 (2010).
Vermeulen, L. A. & Thompson, M. E. Stable photoinduced charge separation in layered viologen compounds. Nature 358, 656–658 (1992).
Zakaria, M. B. & Chikyow, T. Recent advances in prussian blue and prussian blue analogues: synthesis and thermal treatments. Coord. Chem. Rev. 352, 328–345 (2017).
Janiak, C. A critical account on π-π stacking in metal complexes with aromatic nitrogen-containing ligands. Dalton Trans. 21, 3885–3896 (2000).
Sun, L., Park, S. S., Sheberla, D. & Dinca, M. Measuring and reporting electrical conductivity in metal-organic frameworks: Cd2(TTFTB) as a case study. J. Am. Chem. Soc. 138, 14772–14782 (2016).
Cole, J. M., Pepe, G., Al Bahri, O. K. & Cooper, C. B. Cosensitization in dye-sensitized solar cells. Chem. Rev. 119, 7279–7327 (2019).
Zhang, Z., Zhu, Y., Chen, X., Zhang, H. & Wang, J. A Full-spectrum metal-free porphyrin supramolecular photocatalyst for dual functions of highly efficient hydrogen and oxygen evolution. Adv. Mater. 31, 1806626 (2019).
Ohkoshi, S., Tokoro, H. & Hashimoto, K. Temperature and photo-induced phase transition in rubidium manganese hexacyanoferrate. Coord. Chem. Rev. 249, 1830–1840 (2005).
Lv, X. Y. et al. Photochromic metal complexes of N-methyl-4,4′-bipyridinium: mechanism and influence of halogen atoms. Inorg. Chem. 51, 4015–4019 (2012).
Marcus, R. A. Electron transfer reactions in chemistry. theory and experiment. Rev. Mod. Phys. 65, 599–610 (1993).
Cai, L. Z. et al. Photochromism and photomagnetism of a 3d-4f hexacyanoferrate at room temperature. J. Am. Chem. Soc. 137, 10882–10885 (2015).
Ebbesen, T. W., Levey, G. & Patterson, L. K. Photoreduction of methyl viologen in aqueous neutral solution without additives. Nature 298, 545–548 (1982).
Zhang, J., Matsushita, M. M., Kong, X. X., Abe, J. & Iyoda, T. Photoresponsive coordination assembly with a versatile logs-stacking channel structure based on redox-active ligand and cupric ion. J. Am. Chem. Soc. 123, 12105–12106 (2001).
Wang, M. S., Xu, G., Zhang, Z. J. & Guo, G. C. Inorganic-organic hybrid photochromic materials. Chem. Commun. 46, 361–376 (2010).
Nishikiori, S., Yoshikawa, H., Sano, Y. & Iwamoto, T. Inorganic-organic hybrid molecular architectures of cyanometalate host and organic guest systems: specific behavior of the guests. Acc. Chem. Res. 38, 227–234 (2005).
Burke, K. & Gross, E. K. U. A Guided Tour of Time-Dependent Density Functional Theory (Burke) 116–146 (Springer, Berlin, Heidelberg, 1998).
Yang, C. et al. Crystal structures and visible-light excited photoluminescence of N-methyl-4,4′-bipyridinium chloride and its Zn(II) and Cd(II) complexes. Inorg. Chem. Commun. 13, 1021–1024 (2001).
Siemens. SHELXTLTM Version 5 Reference Manual (Siemens Energy & Automation Inc., Madison, Wisconsin, USA, 1994).
Frisch, M. J. et al. Gaussian 09, Revision D.01. (Gaussian, Inc., Wallingford, CT, 2009).
Clark, S. J. et al. First principles methods using CASTEP. Z. Krist. -Cryst. Mater. 220, 567–570 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Hamann, D. R., Schluter, M. & Chiang, C. Norm-conserving pseudopotentials. Phys. Rev. Lett. 43, 1494 (1979).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (91545201, 21827813), Key Research Program of Frontier Science, CAS (QYZDB-SSW-SLH020, QYZDJ-SSW-SLH028), and the Youth Innovation Promotion Association of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
M.S.W. conceived the idea and designed the experiments. X.Q.Y. did the synthesis, TGA, FT-IR, UV–Vis, PXRD, ESR, elemental analysis, electrical measurements, solved the crystal structures and performed the DFT calculations. C.S. analyzed the data. B.W.L. assisted X.Q.Y. in testing photocurrent measurements. M.S.W. and G.C.G. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Zhiyong Fu and Shuang-Quan Zang for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, XQ., Sun, C., Liu, BW. et al. Directed self-assembly of viologen-based 2D semiconductors with intrinsic UV–SWIR photoresponse after photo/thermo activation. Nat Commun 11, 1179 (2020). https://doi.org/10.1038/s41467-020-14986-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14986-7
This article is cited by
-
Preparation and Room Temperature Phosphorescent Properties of Cd(II) Homo- and Zn(II)Cd(II) Heterometallic Coordination Polymers Microstructures
Journal of Inorganic and Organometallic Polymers and Materials (2024)
-
Synthesis, structures, and characterizations of four uranyl coordination polymers constructed by mixed-ligand strategy
Journal of Radioanalytical and Nuclear Chemistry (2023)
-
Achieving large thermal hysteresis in an anthracene-based manganese(II) complex via photo-induced electron transfer
Nature Communications (2022)
-
Optically actuating ultra-stable radicals in a large π-conjugated ligand constructed photochromic complex
Science China Chemistry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
