
Abstract
Stem rust is an important disease of wheat that can be controlled using resistance genes. The gene SuSr-D1 identified in cultivar ‘Canthatch’ suppresses stem rust resistance. SuSr-D1 mutants are resistant to several races of stem rust that are virulent on wild-type plants. Here we identify SuSr-D1 by sequencing flow-sorted chromosomes, mutagenesis, and map-based cloning. The gene encodes Med15, a subunit of the Mediator Complex, a conserved protein complex in eukaryotes that regulates expression of protein-coding genes. Nonsense mutations in Med15b.D result in expression of stem rust resistance. Time-course RNAseq analysis show a significant reduction or complete loss of differential gene expression at 24 h post inoculation in med15b.D mutants, suggesting that transcriptional reprogramming at this time point is not required for immunity to stem rust. Suppression is a common phenomenon and this study provides novel insight into suppression of rust resistance in wheat.
Similar content being viewed by others


Introduction
Wheat (Triticum aestivum) is one of three major cereal crops that supply the majority of calories worldwide, with over 600 million tonnes harvested annually1. Wheat is an allopolyploid that includes genomes from three different grass species. A series of sequential hybridization events occurred, with the first at ~0.8 Mya between diploid T. urartu (A genome) and an unknown diploid species related to Aegilops speltoides (B genome) to form the progenitor of wild allotetraploid emmer wheat (T. turgidum subsp. dicoccoides; A and B genomes), and the second between emmer wheat and diploid goatgrass (Ae. tauschii; D genome) approximately 8000 ya to generate the allohexaploid progenitor of modern common wheat (T. aestivum; A, B, and D genomes)2,3,4. Importantly, the three genomes of common wheat (A, B, and D) originate from species that evolved independently from one another for 2–3 million years5.
Crop diseases are a major limitation to food production. Developing cultivars carrying effective disease resistance genes is a sustainable and environmentally responsible approach to disease management. However, pathogens can evolve virulence to resistance genes emphasizing the need to identify and utilize new resistance genes that are found within a crop and related species. Wheat stem rust (caused by Puccinia graminis Pers. f. sp. tritici Eriksson & Henning; Pgt) is a devastating fungal disease of wheat6 that has re-emerged as a worldwide threat to wheat production with the evolution of highly virulent races of Pgt in Africa, including the Ug99 race group7,8,9. Races of Pgt are defined by the combination of host resistance genes for which a given Pgt isolate displays either virulence or avirulence10.
Interaction between the genomes in allopolyploid wheat is associated with the reduction or loss (suppression) of resistance to fungal plant pathogens causing stem rust11,12,13, leaf rust11,14,15, stripe rust16, and powdery mildew17,18. One constraint in wheat breeding is the suppression of stem rust resistance (Sr) genes when transferred from diploid and tetraploid ancestors to hexaploid wheat11,12,13,14,15,19,20,21,22. Suppression of stem rust resistance was first reported in 1980 when it was discovered that the loss of the D genome chromosomes activated resistance to several Pgt races that were virulent to the hexaploid wheat cultivar “Canthatch” (CTH)23. Initially the D-genome chromosomes were removed from CTH leaving behind the tetraploid (A and B genomes) component, called Tetra CTH24. This tetraploid was resistant to several races of Pgt that were virulent to hexaploid CTH. Reintroduction of the D-genome by crossing Tetra CTH with Ae. tauschii to create a synthetic hexaploid (A, B, and D genomes) resulted in a loss of resistance that was the same as CTH23. This example is analogous to instances of Sr genes from tetraploid wheat being suppressed when transferred to hexaploid wheat11,12,13. Further analysis of CTH nullisomic and ditelosomic stocks and EMS-derived mutants showed that the suppression of stem rust resistance was conditioned by SuSr-D1 (Suppressor of Stem rust resistance1, D-genome), a single dominant gene on wheat chromosome arm 7DL23,25,26.
We have a limited understanding of the genes underlying suppression or the resistance genes that are being suppressed. Here, we establish, through genetic mapping, sequenced flow-sorted chromosomes, and mutational analysis that SuSr-D1 encodes Med15b.D, a subunit of the conserved Mediator complex that regulates transcription of protein-coding genes in eukaryotic organisms. Our findings reinforce the complexity of wheat, in particular, the interaction of the sub-genomes and its contribution to unique regulatory processes that impact transcription. As suppression of disease resistance is frequent, the isolation of SuSr-D1 and understanding its mechanistic role will contribute to developing approaches to understand how the sub-genomes of wheat interact, and how this contributes to suppressing immunity. Based on this knowledge, future efforts may unlock additional reservoirs of resistance genes for the promotion of sustainable agriculture and food security.
Results
Phenotypes of wild-type and mutant stocks
The key genetic stocks used in these experiments included Canthatch (CTH), Thatcher (TC), CTH mutant lines NS1 and NS225, and CTH mutant lines W01, W02, W03, W06, W07, W10, W11, and W1226, where all ten mutant lines lost SuSr-D1 activity through EMS mutagenesis. Seedlings of TC, CTH, and NS1 and NS2 showed clear phenotypic differences when inoculated with Pgt race QTHJC (C25), with the resistant response of the mutant lines showing no sporulation whereas the wildtype CTH showed a susceptible response with large sporulating pustules at the seedling stage (Fig. 1a). Inoculation of an extended panel of mutants (CTH-W series) with race QTHJC also displayed strong levels of resistance (Supplementary Fig. 1). Inoculating with Pgt races from the Ug99 group allowed some rust pustules to develop on mutants, whereas Pgt race TRTTF-elicited a susceptible response on CTH, NS1, and NS2 indicating that the resistance expressed in SuSr-D1 mutant lines was race-specific (Fig. 1b; Supplementary Table 1). Field tests in Kenya on adult plants confirmed seedling test results by showing increased resistance against Pgt race TTKSK (Ug99) in all CTH mutants when compared with wild type CTH (Supplementary Table 2).

a Pgt race QTHJC (C25) showed a qualitative differential response on susr-d1-NS1 and susr-d1-NS2 compared to wild-type (Canthatch; CTH) and Thatcher (TC) which showed susceptible responses characterized by large uredinia. Little Club (LC) is the susceptible control. b Pgt race TTKSK (Ug99) shows differential infection responses with large uredinia on CTH and low to intermediate infection types on susr-d1-NS1 and susr-d1-NS2. All images were taken 14 days post inoculation.
Mapping and isolating SuSr-D1 using enriched DNA sequencing
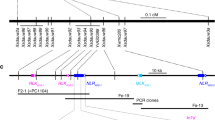
Two parallel approaches were used to isolate SuSr-D1. In the first approach, chromosome 7D was isolated using flow cytometric sorting from CTH and mutants NS1 and NS2 and sequenced to identify EMS-induced single nucleotide variations (SNVs) (Supplementary Figs. 2–4; Supplementary Table 3)27,28. Several SNVs were converted into DNA markers and genetically mapped to identify a minimum physical interval of 1079 kb on chromosome 7D in the Chinese Spring wheat reference sequence that corresponded to the region encompassing SuSr-D1 (Fig. 2a–c and Supplementary Figs. 5, 6; Supplementary Tables 4–7)29,30. Only one gene within the interval carried point mutations in both NS1 and NS2, generating nonsense mutations in Med15b.D, a gene encoding Mediator15, a subunit of the Mediator complex (Supplementary Fig. 7). In the second approach, de novo assembly of flow-sorted chromosomes from eight mutants identified two mutants with large chromosome deletions and six mutants with nonsense mutations in Med15b.D (Fig. 2d, Supplementary Figs. 7, 8). The nonsense mutations in the predicted protein were at several positions from amino acid 33 (W06) to 820 (W10 and W11) (Supplementary Figs. 7, 9). A common mutation in W10 and W11 suggests that the two mutants are sib lines as both possess common SNVs across many contigs26. The gene structure of Med15b.D spans 10.6 kb with 10 exons that encode a protein of 1301 amino acid residues (Fig. 2d, e). Taken together, independent mutations confirm that inactivation of Med15b.D was responsible for the activation of suppressed stem rust resistance.

a SuSr-D1 is located in the distal region of the long arm on chromosome 7D. b Genetic maps of SuSr-D1 in the NS1 and NS2 DH populations. The values between loci represent the number of recombinants observed in that interval on which the genetic maps in Supplementary Fig. 5 were based. c Physical interval encompassing SuSr-D1 with annotated high (HC) and low (LC) confidence genes (green). d Structure of the Med15b.D gene encoded by SuSr-D1. Exons are represented by thicker segments and introns are represented by thin lines (yellow). NS1 (orange) and NS2 (sky blue) each carry a mutation within an intron as well as the loss-of-function mutations labeled as Q435* NS1 and Q817* NS2. Additional CTH mutations (vermillion) shown are: Q221* W01, Q256* W02, W33* W06, Q461* W07, Q820* W10, and Q820* W11. e Med15b.D encodes a 1301 aa protein with KIX (CREB-binding domain93; vermillion), Gln-rich (yellow), coiled coil (green), and Armadillo-like helical (blue) domains.
Med15 duplication and expression in wheat
A duplication of Med15 present on the short (Med15a) and long (Med15b) arms of group 7 chromosomes in wheat and chromosome 7H in barley (Hordeum vulgare) occurred after its divergence from the ancestor of rice (Oryza sativa) and Brachypodium distachyon (Fig. 3a and Supplementary Fig. 10; Supplementary Table 8). While B. distachyon has a single copy of Med15, rice has two copies31. We identified six Med15 wheat homologs encompassing two families, Med15a and Med15b, with three homeologs each30. Homeolog-specific RNAseq analysis showed that all six wheat Med15 homologs are expressed in the first leaf (Fig. 3b and Supplementary Fig. 11); therefore, all six Med15 proteins could participate in forming the multi-protein Mediator complex. This recent duplication may form the basis of neo-functionalization of Med15 in Triticeae species. We performed branch-specific tests for variable levels of selective pressure for the Med15, Med15a, and Med15b clades (Supplementary Fig. 12). Purifying selection for non-Triticeae species was strongest (ω0 = 0.24), whereas both Med15a and Med15b experienced relaxed purifying selection (ωa = 0.38, ωb = 0.37; Supplementary Table 9). Although both ωa and ωb were below 1.0, indicating a lack of recurrent positive selection, the relaxation in purifying selection may impact a subset of Med15 codons.

a Rice and Brachypodium distachyon have a single copy of Med15, whereas barley and wheat have two and six copies of Med15, respectively. Rice contains an additional Med15 gene (Med15c; LOC_Os04g03860) that is highly divergent in protein identity and not present in B. distachyon, barley, or wheat31. Homeologous Med15a and Med15b gene families exhibit 98 and 97% protein identity, whereas there was 90% protein identity between the families. b All six copies of Med15 in wheat were expressed in leaf tissue. RNAseq profiling was performed in triplicate, with individual data points representing a single experiment.
Differential gene expression in wild-type and mutant lines
The role of Med15 in transcriptional regulation suggests that steady-state or pathogen responsive transcription might contribute to suppression of stem rust resistance. To identify putative steady-state transcriptional targets of Med15b.D, we performed RNAseq profiling on uninoculated leaves of CTH, NS1, and NS2. A total of 229 differentially expressed genes were identified using a false discovery rate of 5% (Fig. 4a and Supplementary Table 10). The physical distribution of differentially expressed genes was overlaid on the wheat genome, uncovering two regions located on chromosomes 1A and 1D that were associated with 31 downregulated genes and 169 upregulated genes, respectively (Fig. 4b). These regions are homeologous and share approximately half of the gene content (242 genes) between these regions in the individual sub-genomes. Characterization of the physical intervals found that almost all genes within the region were segmentally coregulated, with the chromosome 1A and 1D regions experiencing suppression and induction, respectively, whereas the homeologous region on chromosome 1B was not modified (Fig. 4c, d and Supplementary Table 11).

a Venn diagrams showing RNAseq-based differentially expressed (DE) genes between wild-type (CTH) and SuSr-D1 mutants NS1 and NS2 using a false discovery rate (FDR) of 5%. Top, upregulated genes (higher expression in mutant relative to wild-type). Bottom, downregulated genes (lower expression in mutant relative to wild-type). b Physical distribution of DE genes across the chromosome of wheat. The majority (87%) of DE genes localize to homeologous regions on chromosomes 1A and 1D. Upregulated and downregulated genes are shown in white and black filled circles, with A, B, and D genomes shown in orange, blue, and green, respectively. Below, Triticeae chromosome groups. c Pair-wise plots of leaf expression levels for homeologous chromosome group 1 regions. Black circles represent differentially expressed genes (FDR 5%), whereas gray circles are not differentially expressed. Diagonal lines indicate equal expression levels. x-axis (wild-type, SuSr-D1) and y-axis (mutant, SuSr-D1-NS1) are log10-transformed based on transcripts per million. d Histograms of fold change in gene expression for all genes shown in Fig. 4c with non-zero expression in both wild-type and mutant NS1.
To understand the impact on the transcriptional response in Med15b.D mutants relative to wild-type in the presence of Pgt, we performed RNAseq experiments on Pgt-inoculated and mock-inoculated seedlings of CTH, NS1, and NS2 with samples collected at 0 and 24 h post-inoculation (hpi). One hundred and thirty-nine, 19, and 56 genes were differentially expressed between Pgt and mock inoculated at 0 h post inoculation in seedlings of CTH, NS1, and NS2, respectively (Fig. 5a). After 24 h post inoculation, 8389 genes were differentially expressed in CTH, whereas NS2 had 2199 differentially expressed genes and NS1 had 55 using a false discovery rate of 5% (Fig. 5a, Supplementary Table 12). There was considerable overlap in differentially expressed genes in CTH versus NS2 (1435 genes) and NS1 (16 genes) (Fig. 5b). This suggests that differential gene expression at 24 hpi was not required for immunity to Pgt and that loss of function in Med15b.D severely impacted the transcriptional response of CTH to Pgt.

a Number of differentially expressed genes (DE genes) between Pgt-inoculated and mock-inoculated wild-type SuSr-D1 (CTH) and SuSr-D1 mutants (NS1 and NS2) based on FDR of 5%. b, c Venn diagrams showing overlap of DE genes between wild-type and SuSr-D1 mutants NS1 and NS2, respectively.
Many of the genes within the regions experiencing segmental co-regulation on chromosomes 1A and 1D in non-inoculated plants were also identified in both mock and Pgt-inoculated experiments. In the mock-inoculated experiment, comparison of CTH and NS1 found 188 of 284 (66%) and CTH and NS2 with 179 of 227 (79%) differentially expressed genes overlapping with non-inoculated differentially expressed genes on chromosomes 1A and 1D. At 24 hpi, CTH and NS1 had 10,035 differentially expressed genes after Pgt-inoculation, whereas CTH and NS2 had 668 differentially expressed genes. The high number of differentially expressed genes between CTH and NS1, and low number in CTH and NS2 reflected a lack of transcriptional response in NS1 after Pgt-inoculation. A total of 317 genes were shared between these two comparisons, with 113 genes (36%) in the chromosome 1A and 1D regions.
The minor impact on steady-state gene expression levels in nonsense med15 mutants in wheat parallels the lack of morphological differences across four of five growth parameters between the wild-type and mutants (Supplementary Fig. 13). In contrast, a null allele of atmed15 (nrb4-4) in Arabidopsis thaliana exhibited pleiotropic morphological defects including chlorosis, delayed growth, and sterility32. Therefore, Med15b.D exhibits a homeolog-dominant suppression of immunity that is generally compensated by homeologs in its role as a co-activator of transcription. Given the apparent degree of homeologous compensation in bread wheat, Med15 is a candidate target for gene editing as means to improve disease resistance with little morphological consequence.
Discussion
Disease resistance is a major goal of wheat breeding programs around the world. Keeping pace with pathogen evolution represents a significant challenge. Close relatives of wheat are reservoirs of genes that can be used for wheat improvement including disease resistance22. However, attempts to transfer disease resistance genes to hexaploid wheat are often unsuccessful because the resistance genes are suppressed11,12,13,14,15,16,17,18,19. Despite the many reports of suppressed resistance genes, there have been only a few reports describing the genetic basis of suppression. A molecular mechanism for suppression of disease resistance was previously described in wheat involving the suppression of powdery mildew resistance by direct interaction of NLR plant immune receptors encoded by alleles of Pm3 or Pm8 (a homeolog of Pm3)33. Interaction of homeologous genes was also implicated in the suppression of Lr2315.
Suppression of stem rust resistance by SuSr-D1 follows a different mechanism rather than that observed in the Pm3/Pm8 interactions. Med15b.D, encodes a subunit of the Mediator complex, which is a coactivator of transcription through its interaction with RNA polymerase II and general and specific transcription factors to coordinate transcription in eukaryotes34,35. The complex in Arabidopsis contains 33 subunits, and mutations within the gene affect a wide range of developmental processes as well as biotic stress responses36,37. However, there is no report of activated expression of race-specific disease resistance caused by mutations in Mediator subunits. In wheat, we identified six Med15 homologs encompassing two families, Med15a and Med15b, with three homeologs each. All copies of Med15 were expressed in CTH but only Med15b.D suppressed stem rust resistance providing an example of homeolog-specific function in wheat.
At the interface of RNA polymerase II, general transcription factors, and specific transcription factors, the Mediator complex plays a central role in coordinating transcriptional regulator proteins and core promoters38,39,40. Most of our understanding of the Mediator complex in plant immunity has come from Arabidopsis36,37, with subunits AtMED8, AtMED14, AtMED15-1, AtMED16, AtMED21, and AtMED25 having regulatory roles in plant immunity41. In addition, AtMED19a interacts with the nuclear-localized effector HaRxL44 from Hyaloperonospora arabidopsidis (an oomycete pathogen of A. thaliana), leading to degradation of MED19a in a proteasome-dependent manner42. High-throughput transcriptome experiments have been performed with several Mediator subunit mutants. Microarray-based transcriptome analysis on wild-type AtMEDIATOR15-1 (AT1G15780; NON-RECOGNITION-OF-BTH 4; NRB4) and mutants nrb4-2 (non-synonymous mutant) and nrb4-4 (gene disruption by T-DNA) found substantial differential gene expression in nrb4-4 (4416 genes), whereas nrb4-2 had no differentially expressed genes relative to wild-type32. The nrb4-4 mutation is a T-DNA insert in an intron immediately after the start codon, likely causing a significant impact on gene expression32. nrb4-4 exhibits pleiotropic morphological defects including chlorosis, delayed growth, and sterility, and impaired salicylic acid and immune signaling32. Using time course microarray-based transcriptome analysis on wild-type and atmed14-1 inoculated with an avirulent isolate of the bacterial pathogen Pseudomonas syringae pv tomato DC3000/avrRpt2, Zhang and colleagues found that atmed14-1 responded more slowly at the transcriptional level to the presence of P. syringae pv tomato DC3000/avrRpt243. The total number of differentially expressed genes was several thousand over the 0–12 hpi time-course43, unlike the substantially perturbed/abolished expression observed in med15b.D mutants. The impact of gene knockouts in Mediator complex subunits on global transcriptional reprogramming remains unclear for other subunits, as several studies have only assessed reporter genes, such as PATHOGENESIS-RELATED GENE1 (PR1)44,45,46. Similar to atmed14, atmed15, and atmed16, nonsense mutations in med15b.D led to reduced (NS2) or lost (NS1) transcriptional response to Pgt. Unlike atmed14, atmed15, and atmed16, the impaired/abolished transcriptional response at 24 hpi in med15b.D mutants was associated with immunity. While it remains unclear whether the transcriptional response is simply delayed or completely abolished relative to wild-type at other time points, the lack of expression in med15b.D-NS1 indicates that the transcriptional response at 24 hpi is not required for resistance to Pgt. All med15b.D mutants carried nonsense mutations in Med15b indicating that resistance (or suppression of transcriptome reprogramming) requires loss or truncation of the protein subunit.
The majority of differentially expressed genes in steady-state expression levels between wild-type and med15b.D mutant plants were associated with 25.7 Mb and 28.9 Mb intervals on chromosomes 1A and 1D, respectively. While genes in the chromosome 1A interval underwent a 50% reduction in expression, genes in the chromosome 1D interval showed substantial induction in expression ranging from 2 to >200 fold-change. Five NLR-encoding genes (the most prevalent cytoplasmic/nuclear immune receptors in plants) were located within the upregulated interval on chromosome 1D, and were potential candidate genes contributing to immunity to Pgt. This hypothesis will require additional genetic mapping to demonstrate that immunity to Pgt maps to the chromosome 1D interval.
The Mediator complex has a known role in fine-tuning gene-specific and pathway-specific transcriptional reprogramming47. In this study, we established a potential novel role in segmental coregulation, for which there is no known mechanism in eukaryotes that regulates genes over a >25 Mb interval. In mammals, spatial partitioning of the genome into topologically associating domains coordinates gene expression, but these regions range from 40 kb to 3 Mb48. The regulatory processes that contribute to this coregulation could involve modification of chromatin architecture such as the formation of chromatin loops or condensation of heterochromatin49. While well characterized in yeast and mammals, little is known about the role of the Mediator complex in controlling chromatin loops and heterochromatin in plants. Interestingly, the homeologous chromosome 1B interval (51 Mb) did not show any modification in gene expression, suggesting genomic features such as sequence or size could differentiate this region from the chromosome 1A and 1D intervals. Collectively, the identification of a suppressor of immunity in wheat demonstrates the complexity of interactions between its sub-genomes, and identifies a novel approach at improving agriculture through the removal of suppressors that negatively interact in wheat.
Methods
Plant materials
The wheat accession Canthatch-K (CTH-K) is a selection of Canthatch (CTH) made by E. R. Kerber (Agriculture and Agri-Food Canada, retired). CTH-K was previously used in mutagenesis experiments to generate independent mutant lines, RL5863 (NS1) and RL5864 (NS2), that carry null mutations for SuSr-D1 on chromosome arm 7DL25. Additional mutants from CTH designated W01, W02, W03, W06, W07, W10, W11, and W12 were also included in the study26. Crosses were made between Thatcher and NS1, and between Thatcher and NS2. Thatcher is the recurrent parent of CTH. The F1 plants from each cross were used to make doubled haploid (DH) populations referred to as the NS1 DH and the NS2 DH populations, respectively, for the purpose of mapping SuSr-D1. DH populations were generated using a maize pollination method50. Little Club wheat was used as susceptible check for stem rust tests. Lines CTH-K, Thatcher, NS1, NS2, CTH ditelosomic 7DL (CTH-7DL), and Williams et al.26. CTH and mutants W01, W02, W06, W07, W10, and W11 were used for chromosome isolation and sequencing experiments (Supplementary Table 3). The cytogenetic stock, CTH ditelosomic 7DS (CTH-7DS), was used in seedling stem rust tests.
Assessment of plant development
Assessment of plant developmental parameters of CTH-K, NS1, and NS2 was performed through the growth of six to seven independent plants in three different greenhouses (S52, S55, S59) in Norwich, UK from 16 April 2018 to 1 August 2018. All plants were sown on the same date and randomly assigned positions within the greenhouse. Assayed growth parameters included complete emergence of the flag leaf (Zadoks scale 47), first emergence of awns (Zadok scale 49), one-quarter spike emergence (Zadoks scale 53), total number of seeds harvested, and 1000 grain weight (Fig. 5). For 1000 grain weight, the weight was based on three randomly sampled technical measurements using 100 seeds. ANOVA was performed in R (3.5.1) aov using the factors genotype, location, and their interaction. Tests of homogeneity of variance (Levene test) and normality (Shapiro-Wilk test) were performed on individual traits. Pairwise differences were assessed using adjusted p values using Tukey Honest Significant Differences. All traits passed these tests, except for 1000 grain weight. For 1000 grain weight, the non-parametric Kruskal-Wallis test was used, with pairwise differences determined using the Wilcoxon rank sum test, adjusting p values using the Benjamini-Hochberg approach51.
Seedling and field tests for stem rust resistance
NS1, NS2, CTH-K, Thatcher, NS1 DH population, and NS2 DH population were tested for stem rust reaction at the seedling stage. Seedlings where inoculated after the first leaf was fully emerged using Pgt race QTH (C25) (AAFC-Morden isolate 1347). Classification of Pgt races is based on a previously described nomenclature system52,53. Urediniospores were suspended in light mineral oil (Bayol 55, Imperial Oil Canada, Toronto, Canada) and sprayed onto seedlings. Inoculated seedlings were incubated for 16 h in dew chambers at 100% relative humidity and then were dried slowly under light to promote infection. The seedlings were transferred to a greenhouse and grown at approximately 20 °C with 16 h of light daily including supplementary fluorescent lights. Seedlings were rated for infection type (IT) 14 days post-inoculation following the 0–4 scale previously described10. ITs 0–2 were classified as resistant and ITs 3–4 were classified as susceptible. Seedlings of CTH-K, CTH-7DL, CTH-7DS, NS1, and NS2 were tested with Pgt races QTHJC (C25; isolate 1541), TPLKC (C33; isolate 1427), TPMKC (C53; isolate 1373), QCCSC (C56; isolate 901), RTHJF (C57; isolate 1561), SPMMC (C74; isolate 972), RKQQC (C35; isolate 1312), MCCFC (C17; isolate 1541), QFCSC (isolate 06ND76C), QTHJC (isolate 75ND717C), TTKSK (isolate 04KEN156/04), TTKST (isolate 06KEN19v3), TTTSK (isolate 07KEN24-4), TRTTF (isolate 06YEM34-1), TPMKC, 21-2,5 (49) and 34-1,2,3,5,6,7 (313) as described above52,54,55,56. CTH-K, NS1, and NS2 were tested in field trials in 2009, 2010, 2011, 2012, 2016, and CTH-W, W01 at Njoro, Kenya that were inoculated with Ug99 races of Pgt following previously described procedures57. Field plots were rated for stem rust severity and infection response using the modified Cobb scale58.
Chromosome isolation and DNA amplification
Chromosome 7D from CTH, Thatcher, NS1, NS2, and five CTH mutants (W01, W02, W06, W07, and W10)26 and chromosome arm 7DL from CTH-ditelosomic 7DL (CTH-DT7DL), were isolated by flow cytometric sorting (Supplementary Figs. 3, 4; Supplementary Table 3)27. Suspensions of intact mitotic metaphase chromosomes were prepared from synchronized root tips of young seedlings according to Vrána et al.59. Prior to chromosome analysis by flow cytometry, fluorescence in situ hybridization in suspension (FISHIS) was used to label GAA microsatellites by FITC following the protocol of Giorgi et al.60, with modifications. Briefly, chromosomal DNA was denatured by adding 10 M NaOH to the solution to reach pH 12.8–13.3. Following incubation at room temperature for 15 min, pH of the solution was changed to 8.5–9.1 using 1 M Tris-HCl (pH 7.5) and the sample was incubated on ice for 1 min. Then, (GAA)7-FITC probe was added to the suspension to final concentration 4.6 ng/μL and the sample was incubated in darkness at room temperature for 1 h. After FISHIS, chromosomal DNA was stained by fluorochrome DAPI (4′,6-diamidino-2-phenylindole) at 2 μg/mL final concentration and the suspension was analyzed by FACSAria SORP II flow sorter (BD Biosciences, San Jose, USA) at rates of 1000–2000 particles/s.
A total of 1000 particles from the population putatively representing 7D or 7DL were sorted onto a microscope slide and chromosome identity was checked under a fluorescence microscope based on the GAA labeling pattern. Once a target population was confirmed, three independent samples of 1000 chromosomes were sorted onto microscope slides to estimate the extent of contamination by other chromosomes using FISH with probes for Afa-family repeat and (GAA)7 microsatellites (Supplementary Table 3)61. Three batches of 33,000 and 66,000 copies of chromosome 7D and the 7DL telosome, respectively, were then sorted from each sample into PCR tubes containing 40 µL sterile deionized water. The number of sorted chromosomes was determined so that an equivalent of approximately 50 ng DNA was obtained per sample. To produce DNA for sequencing, chromosomal DNA was purified and amplified by Illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare, Piscataway, USA) according to Šimková et al.62. Each sample of sorted chromosomes was amplified separately and three amplification products from the same chromosome were pooled to reduce possible amplification bias. DNA amounts thus obtained are listed in Supplementary Table 3.
Chromosomal DNA sequencing and assembly
PCR-free libraries for Illumina sequencing were prepared by Novogene (Hong Kong, China) using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs, Ipswich, USA) following the manufacturer’s protocols and index codes were added to attribute sequences to each sample. Sequencing was performed for chromosome 7D isolated from CTH-K, NS1, NS2, Williams et al.26 wild-type (CTH-W) and mutants W01, W02, W06, W07, W10, Thatcher, and isolated chromosome arm 7DL from CTH-DT7DL. Samples were sequenced on an Illumina HiSeqX (Illumina, San Diego, USA) to generate 150 bp paired-end reads with two samples per lane except for CTH-K and CTH-W with 250 bp paired-end reads which were each run singly in a lane (HiSeq2500). Data quality from paired end reads was assessed with FastQC and reads were removed using Trimmomatic (v0.36) with parameters set at ILLUMINACLIP:TruSeq3-PE.fa:2:30:10, LEADING:5, TRAILING:5, SLIDINGWINDOW:4:15, and MINLEN:15063. These parameters remove all reads with the adapter sequence, ambiguous bases, or a substantial reduction in read quality. The de novo assembly of chromosome 7DL was performed using Edena (v3.131028) with default parameters, use of paired end relationships, and a minimum overlap of 100 bp64,65.
Identification of genes and SNVs in the SuSr-D1 interval
The assembly of chromosome arm 7DL from CTH-DT7DL was masked for repetitive sequence using RepeatMasker (version open-4.0.5; repeat library version 20140131)66. A self-alignment of reads from wild-type CTH, NS1, and NS2 chromosome 7DL was performed using bwa aln with default parameters (version 0.7.10-r789). Paired end reads were merged using bwa sampe. Samtools (version 1.9) was used to select only paired end reads, the removal of duplicate sequences, and generate pileup files for SNV identification. The VarScan (version 2.3.8) pileup2snp command was used to identify polymorphisms relative to the reference sequence. Alignments for mutants NS1 and NS2 were performed using identical parameters67. Python scripts mutant_flow_sorting_analysis.py and analyze_HQ_SNPs.py were used to identify SNVs that differed between wild-type and mutants NS1 and NS2. In wild-type and mutant, a requirement of at least ten reads was used for genotype calls. CTH mutants NS1 and NS2 were generated using EMS mutagenesis. EMS induces alkylation of guanine (G), which results in conversion of G to A (adenine) due to pairing of O6-ethylguanine with thymine (T). After replication, equal numbers of mutations should be observed as conversions of cytosine (C) to T. For an initial screen of SNVs, two parameter sets were selected: the strict set using parameters of 95% mutant and 5% wild-type allele frequency and the relaxed set using 70% mutant and 20% wild-type allele frequencies (Supplementary Table 5).
Tophat (version 2.0.9) was used to perform spliced alignment of RNAseq reads derived from CTH, NS1, and NS268. Default parameters were used with the exception of maximum intron length of 20,000. In total, 27,664,945 reads were used. Approximately 4.0% of reads aligned to the chromosome 7D assembly. Aligned reads were used to generate gene models using cufflinks (version 2.1.1) with default parameters69. Transdecoder.LongOrfs (version 2.0.1) was used to identify open reading frames (ORFs) in protein encoding genes70. BLASTn (version 2.2.26) was used to align contigs onto the reference wheat genome (IWGSC RefSeq v1.0) using default parameters with the exception of no filtering of the query sequence. The Python script link_position_expression.py was generated to export all ORFs from gene models, independently integrating SNVs in NS1 and NS2. ORFs were translated and mutations that generated non-synonymous changes were identified. All mutations shared between NS1 and NS2 relative to the reference were removed, under the assumption they were due to contamination or were shared mutations relative to CTH-K. Selection of SNVs for analysis on NS1 and NS2 DH populations was prioritized based on contigs with SNVs in leaf expressed genes that led to non-synonymous changes that were evenly spread across chromosome arm 7DL. The complete analytical pipeline and custom scripts are available on a GitHub repository (https://github.com/matthewmoscou/Canthatch).
Genetic mapping of SuSr-D1
The populations segregating for wildtype and mutant SuSr-D1 were fixed for the suppressed Sr genes. SuSr-D1 mutant lines25 NS1 and NS2 were crossed with Thatcher, the recurrent parent of CTH, and the hybrids were used to generate doubled-haploid (DH) populations. The Thatcher × NS1 (N = 159) and Thatcher × NS2 (N = 129) DH populations segregated for reaction to stem rust race QTHJC, and both populations fitted 1:1 single gene ratios (p = 0.48 and p = 0.06, respectively). A total of 35 Kompetitive Allele Specific PCR (KASP) markers (LGC, Teddington, UK) based on EMS-induced SNVs28 were assayed on the two DH populations to localize SuSr-D1 to a 1.3 cM genetic interval flanked by markers kwh239 and kwh281 (Fig. 2a, b and Supplementary Tables 5, 6)29. Linkage maps of chromosome arm 7DL were constructed for both DH populations using MapDisto71. Genetic distances were calculated using the Kosambi mapping function72. The physical positions of the SNV markers were determined by locating flanking sequences on the wheat reference genome assembly (IWGSC RefSeq v1.0) using BLAST. To compare the map positions of the cross-specific SNV markers and SuSr-D1 with commonly used markers, markers from the wheat 90k iSelect consensus map73 on chromosome arm 7DL were also located on the IWGSC RefSeq v1.0. Flanking markers were anchored on the International Wheat Genome Sequencing Consortium reference sequence of Chinese Spring (IWGSC RefSeq v.1.0) for chromosome 7D30. A minimum physical interval of 1079 kb in Chinese Spring wheat corresponded to the region in CTH carrying SuSr-D130 (Fig. 2c, Supplementary Table 7). De novo assembled contigs from CTH, NS1, and NS2 were aligned to the SuSr-D1 physical region. Chinese Spring and CTH share an isogenic haplotype for the interval, facilitating the identification of SNVs based on the Chinese Spring reference annotation. The region carrying SuSr-D1 contained 17 high confidence and 10 low confidence genes (Fig. 2c and Supplementary Table 4). Only one gene carried mutations in both NS1 and NS2 generating nonsense mutations in Med15b.D, a member of the Mediator complex (Fig. 2d)37. SNVs responsible for nonsense mutations in Med15b.D cosegregated with the phenotype in both DH populations (Fig. 2b). De novo assembly of flow-sorted chromosomes from eight mutants identified two mutants with large chromosome deletions and six mutants with nonsense mutations in Med15b.D (Fig. 2d and Supplementary Figs. 7, 8)74. The nonsense mutations in the encoded protein ranged from changes at amino acid positions 33 (W06) to 820 (W10 and W11) (Supplementary Figs. 7, 9). A common mutation in W10 and W11 suggested they were sib lines from the mutagenesis experiment26 as both lines possessed common SNVs across many contigs. Taken together, independent mutations confirmed that inactivation or deletion of Med15b.D was responsible for the activation of suppressed stem rust resistance.
Identification of SNVs in additional CTH mutants
De novo assemblies for each of the wild-type accessions, CTH-W and CTH-K, were created from their quality-controlled, paired-end sequencing reads using CLC Genomics Workbench (v9) with a length fraction of 0.95, a similarity fraction of 0.98 and the remaining parameters as default. Assembly FASTA files consisted of 767,884 contigs spanning approximately 841 Mbp and 749,802 contigs spanning approximately 806 Mbp for CTH-W and CTH-K, respectively. Assemblies were masked for repeat sequences using RepeatMasker (v4.0.6)66 and the custom Triticeae, non-redundant repeat library trep-db_nr_Rel-16.fasta (release 16) downloaded from the TRansposable Elements Platform (TREP) (http://botserv2.uzh.ch/kelldata/trep-db/index.html). Alignment to wild-type assemblies of quality-controlled reads from wild-types CTH-K and CTH-W, and mutants W01, W02, W06, W07, W10, was performed using bwa aln followed by bwa sampe to map paired-end reads using default parameters (bwa v0.7.15). Samtools (v0.1.19) was used for subsequent processing of alignments to select only paired-end reads, remove duplicates and generate pileup files for SNV calling. SNV identification was performed using an in-house pipeline. The program Noisefinder.pyc was used on alignments for the wild-types (CTH-W, CTH-K) to detect regions with high density noise possibly due to misalignments and presence of allelic variants. SNVs were logged in all wild-types and mutants using the program SNPlogger.pyc, with noisy regions detected by Noisefinder.pyc being masked. Subsequent discovery of candidate sequences was performed using SNPtracker.pyc to shortlist sequences containing polymorphisms coinciding across multiple mutants. A threshold of 80% mutant and 20% wild-type allele frequency was used and variant positions detected in the wild-types were masked from the analysis. In both wild-type assemblies, a contig of approximately 16 kb was identified for which all five mutants presented independent SNVs that were canonical for EMS mutagenesis (C->T/G->A) (Fig. 2 and Supplementary Fig. 7). Custom programs used in this analysis are available on GitHub (https://github.com/TC-Hewitt/MuTrigo).
Protein sequence and domain analyses
Amino acid frequency was assessed using a sliding window of 50 amino acids to identify regions that were rich for specific amino acids using QKutilities_protein_analysis.py (https://github.com/matthewmoscou/QKutilities). Motif and structure-based domain analysis was performed using InterProScan75,76 and Phyre277. Coiled coils predictions were made using Coils78, Marcoil79, and Pcoils80.
Phylogenetic and molecular evolution analyses
Multiple data sources were used to identify homologs of Med15. Rice (O. sativa) and B. distachyon Med15 homologs were identified using the Department of Energy-Joint Genome Institute Phytozome database (https://phytozome.jgi.doe.gov), barley (H. vulgare) homologs from the 2017 genome sequence81, and bread wheat (T. aestivum) from the reference genome sequence (IWGSC RefSeq v1.0). We accessed publicly available leaf RNAseq data for eight grass species (Supplementary Table 13)82,83, performed de novo transcriptome assembly using Trinity84 (version r20140717) using the parameters “--min_kmer_cov 2 --normalize_max_read_cov 20 –trimmomatic”, and identified Med15 homologs using BLAST. PRANK (v.140603) and RAxML (v8.2.9) were used for codon-based alignment and phylogenetic tree construction, respectively85,86,87. Default parameters were used for PRANK and RAxML parameters include the GTRCAT model using rice Med15 as an outgroup. A total of 2000 bootstraps were performed. In addition, pairwise comparisons were made between DNA and protein alignments (Supplementary Table 14). Molecular evolutionary analyses were performed with PAML (v4.8)88. A reduced phylogenetic tree based on a requirement of 90% coverage was used for estimating ω (dN/dS), as several sequences lacked sufficient coverage of Med15 due to truncated transcript assemblies (Supplementary Fig. 12).
RNAseq
Two RNAseq experiments were performed. The first experiment included non-inoculated seedlings of CTH, NS1, and NS2, and the second experiment included mock-inoculated and Pgt-inoculated seedlings of CTH, NS1, and NS2. For the non-inoculated seedlings, first and second leaf tissue was harvested at 10 days after sowing of CTH, NS1, and NS2 grown in the greenhouse. For the second experiment, mock (mineral oil application) and Pgt isolate QTH inoculation was performed after the first leaf was fully elongated. Tissue was collected from CTH, NS1, and NS2 at 0 hpi and 24 hpi. Both experiments were performed using three independent biological replicates. Tissue was flash frozen in liquid nitrogen and stored at −80 °C. Tissues were homogenized into fine powder in liquid nitrogen-chilled pestle and mortars. RNA was extracted, purified, and assessed for quality as described previously89. RNA libraries were constructed using TruSeq RNA Library Prep Kit v2 (Illumina). Barcoded libraries were sequenced using a 150 bp paired-end reads. All library preparation and sequencing was performed at Novogene. Initial quality was assessed using FastQC and reads were trimmed using Trimmomatic (v0.36) with parameters ILLUMINACLIP:TruSeq3-PE.fa:2:30:10, LEADING:5, TRAILING:5, SLIDINGWINDOW:4:15, and MINLEN:36. RNAseq data quality was assessed with FastQC. Read mapping for expression analysis was performed using Kallisto (v0.43.1) and BBmap (v37.77). Default parameters were used for kallisto, whereas subgenome-specific read mapping with BBmap was carried out using parameters set to require 100% identity (perfectmode = t), unambiguous mapping (ambiguous = toss), and proper paired reads only (samtools view –f 2). Differential expression (DE) analysis was performed DEseq2 (Bioconductor v3.2) using default parameters90. Multiple hypothesis testing was controlled using a false discovery rate of 5%. To identify putative homeologs in wheat, clustering of high and low confidence proteins from the IWGSC RefSeq 1.0 was performed using CD-HIT, with a requirement of 90% or greater identity30.
Arabidopsis microarray reanalysis
Data from Canet et al.32 was obtained from EBI ArrayExpress experiment E-MEXP-3602. This data set includes transcriptome analysis of A. thaliana wild-type (Col-0) and a nonsynonymous EMS mutant nrb4-2 (3-week-old plants), and T-DNA mutant nrb4-4 (5-week-old plants) in replicated experiments (three replicates). Raw CEL files were normalized using R/BioConductor rma package91. Pairwise comparisons using log2 transformed expression data between wild-type and mutants were performed using R t-tests with multiple hypothesis testing controlled using q-value92 at false discovery rate of 20%.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All high-throughput sequencing data described in this manuscript have been deposited in the NCBI BioProject “PRJNA401266 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA401266]” and European Nucleotide Archive (ENA) “PRJEB23265 [https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJEB23265]”. Illumina sequencing data for flow-sorted chromosomes are deposited in sequential NCBI SRR accessions SRR6001708 to SRR6001719 and ENA ERS1988338 to ERS1988347, de novo assemblies of flow-sorted chromosomes for CTH-K (NCBI “NTGG00000000 [https://www.ncbi.nlm.nih.gov/nuccore/NTGG00000000.1/]”, ENA “ERS1988348 [https://www.ebi.ac.uk/ena/data/view/GCA_900235935.1]”), CTH-W (ENA “ERS1988349 [https://www.ebi.ac.uk/ena/data/view/GCA_900235945.1]”), NS1 (NCBI “NTGH00000000 [https://www.ncbi.nlm.nih.gov/nuccore/NTGH00000000.1/]”), and NS2 (NCBI “NTGI00000000 [https://www.ncbi.nlm.nih.gov/nuccore/NTGI00000000.1/]”). RNAseq sequencing data are deposited in NCBI SRR accessions SRR6003621 to SRR6003628 and SRR10426855 to SRR10426890. The source data underlying Figs. 1, 2b, 3b, 4, and 5, and Supplementary Figs. 1, 4, 7, 10–13, and Supplementary Table 9 have been deposited in a “Figshare project [https://figshare.com/projects/Stem_rust_resistance_in_wheat_is_suppressed_by_a_subunit_of_the_Mediator_complex/28056]”.
Code availability
Bioinformatic analyses and scripts can be found on Github https://github.com/matthewmoscou/Canthatch, https://github.com/matthewmoscou/QKutilities, and https://github.com/TC-Hewitt/MuTrigo.
References
Shewry, P. R. Wheat. J. Exp. Biol. 60, 1537–1553 (2009).
Bilgic, H., Hakki, E. E., Pandey, A., Khan, M. K. & Akkaya, M. S. Ancient DNA from 8400 year-old Çatalhöyük wheat: implications for the origin of Neolithic agriculture. PLoS One 11, e0151974 (2016).
Huang, S. et al. Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc. Natl Acad. Sci. USA 99, 8133–8138 (2002).
Marcussen, T. et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 345, 1250092 (2014).
Chalupska, D. et al. Acc homoeoloci and the evolution of wheat genomes. Proc. Natl Acad. Sci. USA 105, 9691–9696 (2008).
Peturson, B. Wheat rust epidemics in western Canada in 1953, 1954 and 1955. Can. J. Plant Sci. 38, 16–28 (1958).
Olivera, P. et al. Phenotypic and genotypic characterization of race TKTTF of Puccinia graminis f. sp. tritici that caused a wheat stem rust epidemic in Southern Ethiopia in 2013-14. Phytopathology 105, 917–928 (2015).
Olivera, P. D. et al. Races of Puccinia graminis f. sp. tritici with combined virulence to Sr13 and Sr9e in a field stem rust screening nursery in Ethiopia. Plant Dis. 96, 623–628 (2012).
Pretorius, Z. A., Singh, R. P., Wagoire, W. W. & Payne, T. S. Detection of virulence to wheat stem rust resistance gene Sr31 in Puccinia graminis f. sp. tritici in Uganda. Plant Dis. 84, 203 (2000).
Stakman E. C., Stewart D. M. & Loegering W. Q. Identification of Physiologic Races of Puccinia graminis var. tritici. (USDA Agricultural Research Service, Washington, 1962).
Bai, D. & Knott, D. R. Suppression of rust resistance in bread wheat (Triticum aestivum L.) by D-genome chromosomes. Genome 35, 276–282 (1992).
Liu, W. et al. Development and characterization of a compensating wheat-Thinopyrum intermedium Robertsonian translocation with Sr44 resistance to stem rust (Ug99). Theor. Appl. Genet. 126, 1167–1177 (2013).
The, T. T. & Baker, E. P. Basic studies relating to the transference of genetic characters from Triticum monococcum L. to hexaploid wheat. Aust. J. Biol. Sci. 28, 189–199 (1975).
Innes, R. L. & Kerber, E. R. Resistance to wheat leaf rust and stem rust in Triticum tauschii and inheritance in hexaploid wheat of resistance transferred from T. tauschii. Genome 37, 813–822 (1994).
Nelson, J. C., Singh, R. P., Autrique, J. E. & Sorrells, M. E. Mapping genes conferring and suppressing leaf rust resistance in wheat. Crop Sci. 37, 1928–1935 (1997).
Kema, G. H. J., Lange, W. & van Silfhout, C. H. Differential suppression of stripe rust resistance in synthetic wheat hexaploids derived from Triticum turgidum subsp. dicoccoides and Aegilops squarrosa. Phytopathology 85, 508–512 (1995).
Hanušová, R., Hsam, S. L. K., Bartoš, P. & Zeller, F. J. Suppression of powdery mildew resistance gene Pm8 in Triticum aestivum L. (common wheat) cultivars carrying wheat-rye translocations T1BL.1RS. Heredity 77, 383–387 (1996).
Ren, S. X., McIntosh, R. A. & Lu, Z. J. Genetic suppression of the cereal rye-derived gene Pm8 in wheat. Euphytica 93, 353–360 (1997).
Assefa, S. & Fehrmann, H. Resistance to wheat leaf rust in Aegilops tauschii Coss. and inheritance of resistance in hexaploid wheat. Genet. Resour. Crop. Evol. 47, 135–140 (2000).
Assefa, S. & Fehrmann, H. Evaluation of Aegilops tauschii Coss. for resistance to wheat stem rust and inheritance of resistance genes in hexaploid wheat. Genet. Resour. Crop Evol. 51, 663–669 (2004).
Chen, W., Liu, T. & Gao, L. Suppression of stripe rust and leaf rust resistances in interspecific crosses of wheat. Euphytica 192, 339–346 (2013).
Wulff, B. B. & Moscou, M. J. Strategies for transferring resistance into wheat: from wide crosses to GM cassettes. Front. Plant Sci. 5, 692 (2014).
Kerber, E. R. & Green, G. J. Suppression of stem rust resistance in the hexaploid wheat cv. Canthatch by chromosome 7DL. Can. J. Bot. 58, 1347–1350 (1980).
Kerber, E. R. Wheat: reconstitution of the tetraploid component (AABB) of hexaploids. Science 143, 253–255 (1964).
Kerber, E. R. Stem-rust resistance in ‘Canthatch’ hexaploid wheat induced by a nonsuppressor mutation on chromosome 7DL. Genome 34, 935–939 (1991).
Williams, N. D., Miller, J. D. & Klindworth, D. L. Induced mutations of a genetic suppressor of resistance to wheat stem rust. Crop Sci. 32, 612–616 (1992).
The International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788 (2014).
Sánchez-Martín, J. et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 17, 221 (2016).
Allen, A. M. et al. Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol. J. 9, 1086–1099 (2011).
International Wheat Genome Sequencing C. et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191 (2018).
Mathur, S., Vyas, S., Kapoor, S. & Tyagi, A. K. The Mediator complex in plants: structure, phylogeny, and expression profiling of representative genes in a dicot (Arabidopsis) and a monocot (rice) during reproduction and abiotic stress. Plant Physiol. 157, 1609–1627 (2011).
Canet, J. V., Dobón, A. & Tornero, P. Non-recognition-of-BTH4, an Arabidopsis mediator subunit homolog, is necessary for development and response to salicylic acid. Plant Cell 24, 4220–4235 (2012).
Hurni, S. et al. The powdery mildew resistance gene Pm8 derived from rye is suppressed by its wheat ortholog. Plant J. 79, 904–913 (2014).
Flanagan, P. M. et al. Resolution of factors required for the initiation of transcription by yeast RNA polymerase II. J. Biol. Chem. 265, 11105–11107 (1990).
Kim, Y. J., Bjorklund, S., Li, Y., Sayre, M. H. & Kornberg, R. D. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell 77, 599–608 (1994).
Backstrom, S., Elfving, N., Nilsson, R., Wingsle, G. & Bjorklund, S. Purification of a plant mediator from Arabidopsis thaliana identifies PFT1 as the Med25 subunit. Mol. Cell 26, 717–729 (2007).
Bourbon, H. M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 36, 3993–4008 (2008).
Bjorklund, S. & Gustafsson, C. M. The yeast mediator complex and its regulation. Trends Biochem. Sci. 30, 240–244 (2005).
Conaway, R. C., Sato, S., Tomomori-Sato, C., Yao, T. & Conaway, J. W. The mammalian mediator complex and its role in transcriptional regulation. Trends Biochem. Sci. 30, 250–255 (2005).
Kornberg, R. D. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 30, 235–239 (2005).
An, C. & Mou, Z. The function of the mediator complex in plant immunity. Plant Signal. Behav. 8, e23182 (2013).
Caillaud, M. C. et al. A downy mildew effector attenuates salicylic acid-triggered immunity in Arabidopsis by interacting with the host mediator complex. PLoS Biol. 11, e1001732 (2013).
Zhang, X., Yao, J., Zhang, Y., Sun, Y. & Mou, Z. The Arabidopsis Mediator complex subunits MED14/SWP and MED16/SFR6/IEN1 differentially regulate defense gene expression in plant immune responses. Plant J. 75, 484–497 (2013).
Wang, C., Du, X. & Mou, Z. The mediator complex subunits MED14, MED15, and MED16 are involved in defense signaling crosstalk in Arabidopsis. Front. Plant Sci. 7, 1947 (2016).
Wathugala, D. L. et al. The mediator subunit SFR6/MED16 controls defence gene expression mediated by salicylic acid and jasmonate responsive pathways. New Phytol. 195, 217–230 (2012).
Zhang, X., Wang, C., Zhang, Y., Sun, Y. & Mou, Z. The Arabidopsis mediator complex subunit16 positively regulates salicylate-mediated systemic acquired resistance and jasmonate/ethylene-induced defense pathways. Plant Cell 24, 4294–4309 (2012).
Balamotis, M. A. et al. Complexity in transcription control at the activation domain-mediator interface. Sci. Signal. 2, ra20 (2009).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Allen, B. L. & Taatjes, D. J. The mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 16, 155–166 (2015).
Thomas, J., Chen, Q. & Howes, N. Chromosome doubling of haploids of common wheat with caffeine. Genome 40, 552–558 (1997).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Jin, Y. Detection of virulence to resistance gene Sr24 within race TTKS of Puccinia graminis f. sp. tritici. Plant Dis. 92, 923–926 (2008).
Roelfs, A. P. & Martens, J. W. An internaltional system of nomenclature of Puccinia graminis f. sp. tritici. Phytopathology 78, 526–533 (1988).
Jin, Y. et al. Characterization of seedling infection types and adult plant infection responses of monogenic Sr gene lines to race TTKS of Puccinia graminis f. sp. tritici. Plant Dis. 91, 1096–1099 (2007).
McIntosh, R. A., Wellings, C. R. & Park, R. F. Wheat Rusts—An Atlas of Resistance Genes. (CSIRO Publications, 1995).
Newcomb, M. et al. Kenyan isolates of Puccinia graminis f. sp. tritici from 2008 to 2014: Virulence to SrTmp in the Ug99 race group and implications for breeding programs. Phytopathology 106, 729–736 (2016).
Hiebert, C. W. et al. Genetics and mapping of seedling resistance to Ug99 stem rust in Canadian wheat cultivars ‘Peace’ and ‘AC Cadillac’. Theor. Appl. Genet. 122, 143–149 (2011).
Peterson, R. F., Campbell, A. B. & Hannah, A. E. A diagrammatic scale for estimating rust intensity on leaves and stems of cereals. Can. J. Res. 26, 496–500 (1948).
Vrána, J. et al. Flow sorting of mitotic chromosomes in common wheat (Triticum aestivum L.). Genetics 156, 2033–2041 (2000).
Giorgi, D. et al. FISHIS: fluorescence in situ hybridization in suspension and chromosome flow sorting made easy. PLoS One 8, e57994 (2013).
Kubaláková, M. et al. Analysis and sorting of rye (Secale cereale L.) chromosomes using flow cytometry. Genome 46, 893–905 (2003).
Šimková, H. et al. Coupling amplified DNA from flow-sorted chromosomes to high-density SNP mapping in barley. BMC Genomics 9, 294 (2008).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Hernandez, D., Francois, P., Farinelli, L., Osteras, M. & Schrenzel, J. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 18, 802–809 (2008).
Hernandez, D. et al. De novo finished 2.8 Mbp Staphylococcus aureus genome assembly from 100 bp short and long range paired-end reads. Bioinformatics 30, 40–49 (2014).
Smit A. F. A., Hubley R. & Green P. RepeatMasker Open-4.0. http://repeatmasker.org (2013–2015).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Lorieux, M. MapDisto: fast and efficient computation of genetic linkage maps. Mol. Breed. 30, 1231–1235 (2012).
Kosambi, D. D. The estimation of map distances from recombination values. Ann. Eugen. 12, 172–175 (1943).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 12, 787–796 (2014).
Akhunov, E. D. et al. Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genomics 11, 702 (2010).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Lupas, A., Van Dyke, M. & Stock, J. Predicting coiled coils from protein sequences. Science 252, 1162–1164 (1991).
Delorenzi, M. & Speed, T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625 (2002).
Gruber, M., Soding, J. & Lupas, A. N. REPPER-repeats and their periodicities in fibrous proteins. Nucleic Acids Res. 33, W239–W243 (2005).
Mascher, M. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017).
Grønvold, L., Schubert, M., Sandve, S. R., Fjellheim, S. & Hvidsten, T. R. Comparative transcriptomics provides insight into the evolution of cold response in Pooideae. bioRxiv (2017).
Liu, J., Zhou, Y., Luo, C., Xiang, Y. & An, L. De novo transcriptome sequencing of desert herbaceous Achnatherum splendens (Achnatherum) seedlings and identification of salt tolerance genes. Genes 7, E12 (2016).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170 (2014).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Meharg, C. et al. Trait-directed de novo population transcriptome dissects genetic regulation of a balanced polymorphism in phosphorus nutrition/arsenate tolerance in a wild grass, Holcus lanatus. New Phytol. 201, 144–154 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Dawson, A. M. et al. Isolation and fine mapping of Rps6: an intermediate host resistance gene in barley to wheat stripe rust. Theor. Appl. Genet. 129, 831–843 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Huber, W. et al. Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods 12, 115–121 (2015).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445 (2003).
Parker, D. et al. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol. Cell Biol. 16, 694–703 (1996).
Acknowledgements
This work was supported in part by funds provided through Agriculture and Agri-Food Canada, Western Grains Research Foundation, Biotechnology and Biological Sciences Research Council (BB/P012574/1, BB/P016855/1), National Science Foundation (NSF BREAD program IOS-0965429), CSIRO, Gatsby Foundation, and the ERDF project “Plants as a tool for sustainable global development” (No. CZ.02.1.01/0.0/0.0/16_019/0000827). We thank Peter Dodds and Jeff Ellis for critical review of the manuscript prior to submission. We acknowledge the technical expertize of M. Popovic, G. Mardli, D. Edwards, T. Malasiuk, T. Zegeye, Z. Dubská, J. Weiserová, R. Šperková, W. Schnippenkoetter, and S. Chandramohan. We thank D. Klindworth and S. Xu for providing seed of mutant lines developed at the USDA (Fargo, North Dakota).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: C.H., M.M., W.S., J.D., L.H., B.W. and E.L. Performed transcript profiling: I.H.P. and N.U. Performed disease phenotyping: C.H., T.F., V.P., P.Z., M.R., Y.J., R.M., S.B., E.L. Performed chromosome flow sorting: J.V., M.K., J.D. Performed bioinformatic analyses: M.M., T.H., B.S. Designed software used in analysis: M.M., T.H., and B.S. Analyzed the data: C.H., M.M., W.S., T.H., B.S., V.P., P.Z., P.G., J.Z., R.M., S.B. and E.L. Developed mapping populations: C.H., V.P., and E.L. Contributed reagents/materials/analysis tools: P.Z. and R.M. Wrote the paper: C.H., M.M., W.S., J.D., T.H. and E.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hiebert, C.W., Moscou, M.J., Hewitt, T. et al. Stem rust resistance in wheat is suppressed by a subunit of the mediator complex. Nat Commun 11, 1123 (2020). https://doi.org/10.1038/s41467-020-14937-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-14937-2
This article is cited by
-
Wheat Pm55 alleles exhibit distinct interactions with an inhibitor to cause different powdery mildew resistance
Nature Communications (2024)
-
Genetic dissection and identification of stripe rust resistance genes in the wheat cultivar Lanhangxuan 121, a cultivar selected from a space mutation population
Molecular Breeding (2024)
-
Global transcriptome analysis of allopolyploidization reveals large-scale repression of the D-subgenome in synthetic hexaploid wheat
Communications Biology (2023)
-
Identification of a suppressor for the wheat stripe rust resistance gene Yr81 in Chinese wheat landrace Dahongpao
Theoretical and Applied Genetics (2023)
-
FHB resistance conferred by Fhb1 is under inhibitory regulation of two genetic loci in wheat (Triticum aestivum L.)
Theoretical and Applied Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
