
Abstract
Supported gold catalysts play a crucial role in the chemical industry; however, their poor on-stream stability because of the sintering of the gold nanoparticles restricts their practical application. The strong metal-support interaction (SMSI), an important concept in heterogeneous catalysis, may be applied to construct the structure of catalysts and, hence, improve their reactivity and stability. Here we report an ultrastable Au nanocatalyst after calcination at 800 °C, in which Au nanoparticles are encapsulated by a permeable TiOx thin layer induced by melamine under oxidative atmosphere. Owning to the formed TiOx overlayer, the resulting Au catalyst is resistant to sintering and exhibits excellent activity and stability for catalytic CO oxidation. Furthermore, this special strategy can be extended to colloidal Au nanoparticles supported on TiO2 and commercial gold catalyst denoted as RR2Ti, providing a universal way to engineer and develop highly stable supported Au catalysts with tunable activity.
Similar content being viewed by others

Introduction
Supported catalysts, dispersed on high specific area, are one of the most important heterogeneous catalysts1,2. The primary role of the support is ascribed to increase the dispersion of metal and stabilize the active sites. And the interaction between metal and support named carried effects are also recognized to play a vital role in tuning the activity, selectivity and stability of the catalysts2,3,4,5,6,7,8. However, much less is known about how meal-support interaction will affect the activity and selectivity of metal-oxide supported catalysts9,10.
Gold catalysts have been developed for several industrial processes because of their unique catalytic activity11,12,13,14,15,16,17. However, poor on-stream stability resulting from Au nanoparticles (NPs) sintering restricts their practical application18,19. Up to now, some strategies have been proposed to increase the stability of Au NPs in high-temperature reactions, including fixing the Au NPs inside porous channels, coating the Au NPs with thin shells of carbon or porous oxide and strengthening the meal-support interaction. However, there are still some issues remain to be resolved although great progresses have been achieved20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35. For example, Au NPs inside the porous channels are tending to sinter because the pores are poorly controlled20,21. As for coating strategy, which may cover the active sites and/or lead diffusion limitations, therefore will reduce the activity although it can stabilize the Au NPs in a degree22,36. Comparatively, encapsulating Au NPs with reducible oxide overlayer is valid, achieved by constructing an oxide barrier on the surface of Au NPs, which is known as strong metal-support interaction (SMSI)27,28,29,30. Generally, strong metal-support interaction (SMSI; now denoted as classical SMSI) was first described by Tauster et al. in the late 1970s to explain the dramatically suppressed CO and H2 adsorption on titania-supported platinum group metals after high-temperature reduction treatment37. From then on, SMSI has been investigated comprehensively and an agreement has been reached that SMSI emanates from the encapsulation of metals by supports38,39. Up to now, SMSI has been exploited to enhance catalytic performance of the catalysts by modifying the electronic and geometric factors of the metal NPs27,40,41,42,43,44,45,46,47,48.
For long ages it has been well-recognized that Au cannot manifest SMSI, which was ascribed to its low work function and surface energy and its low ability to dissociate H2 to active the support in the previous investigation49,50,51,52. Under this assumption, a few successes have been achieved that an oxide overlayer can be formed on gold supported on ZnO, hydroxyapatite and phosphate supports in oxygen atmosphere, contrary to the condition required for classical SMSI27,53,54. Later, we demonstrated the existence of classical SMSI between Au and TiO2 and the encapsulation of Au by TiOx overlayer was also observed28. Recently, Xiao et al. constructed a TiOx overlayer on Au NPs via wet-chemical strategy in a relative mild condition without reduction pretreatment30. More excitingly, Christopher et al. proposed that the adsorbates HCOx on TiO2- and Nb2O5-supported Rh catalysts could induce the encapsulation of Rh NPs by supports in CO2–H2 environment at temperatures of 150–300 °C, denoted as adsorbate-mediated SMSI (A-SASI)46.
Herein, we demonstrate that Au NPs can be encapsulated by a permeable TiOx overlayer under oxidative atmosphere induced by melamine, the reverse of condition required for classical SMSI between Au and TiO228. The key to construct the overlayer is the application of melamine and pretreatment in nitrogen gas, followed by calcination at 800 °C in air atmosphere, which strengthen the interaction between Au and TiO2. Owning to the formation of the overlayer, the resultant catalyst is sintering-resistant with high activity after calcination at 800 °C as well as excellent durability in a simulated practical testing, implying the potential for practical application. More importantly, this special strategy can be extended to colloidal Au NPs supported on TiO2 and commercial gold catalyst denoted as RR2Ti, which enables to rationally devise and develop highly stable supported Au catalysts with controllable activity.
Results
Physical and chemical nature of the encapsulation layer
The catalysts were prepared with deposition-precipitation (DP) method and followed by modification with melamine and pretreatment at 600 °C in N2 atmosphere and further calcination at 800 °C in air. High-resolution transmission electron microscopy (HRTEM) (Fig. 1) showed the morphology change of Au NPs during synthesis. For the as-prepared Au/TiO2, the average particle size of gold was about 3.5 nm after pretreatment at 250 °C in air (Fig. 1a and Supplementary Fig. 1a). After modification with melamine (denoted as Au/TiO2@M), the particle size increased to 3.6 nm. However, no melamine was observed on the surface of Au NPs, which was different from the previous work (Fig. 1b and Supplementary Fig. 1b)23. When calcination at 600 °C in N2 atmosphere for 3 h (denoted as Au/TiO2@M-N), the change in the interface was observed, suggesting the variation of the interaction between Au NPs and support (Fig. 1c). Meanwhile, the Au NPs size increased from 3.6 to 6.7 nm (Supplementary Fig. 1c). Surprisingly, after being subjected to calcination at 800 °C in air (denoted as Au/TiO2@M-N-800) it was found that Au NPs were covered with an overlayer, which should be from TiO2 as melamine would decomposed completely in this condition (Fig. 1d and Supplementary Fig. 2). Meanwhile, the Au NPs size was only about 7.5 nm, indicating a robust sintering-resistant Au nanocatalyst (Supplementary Fig. 1d). It is worth noting that the smaller Au NPs can also be encapsulated similarly to that of the larger ones (Supplementary Fig. 3). However, it is difficult to observe the morphology and encapsulation of gold when the Au NPs size is less than 2 nm. And it was calculated that more than 90% of Au NPs was encapsulated based on the particle size distribution. Comparatively, for the Au/TiO2 catalyst without melamine modification after pretreatment at 800 °C (denoted as Au/TiO2-800), the catalyst sintered seriously with a particle size about 32.6 nm and no cover layer was observed (Supplementary Fig. 4).

The above results were further examined by X-ray diffraction (XRD) patterns. As pictured in Supplementary Fig. 5, there was no diffraction peaks ascribed to Au NPs for Au/TiO2 and Au/TiO2@M, suggesting that Au NPs were dispersed uniformly, in agreement with TEM results. While after calcination, diffraction peaks of Au (111) at 38.2° and Au (200) at 44° were observed for Au/TiO2@M-N, Au/TiO2@M-N-800 and Au/TiO2-800, especially for the last two. Furthermore, the crystal structure of TiO2 was also analyzed (Supplementary Table 1). And anatase was identified as the primary phase for Au/TiO2, Au/TiO2@M and Au/TiO2@M-N. After calcination at 800 °C, anatase transformed to rutile completely in Au/TiO2-800. However, for Au/TiO2@M-N-800 sample 12% anatase still existed, implying the phase conversion from anatase to rutile was delayed in a degree. The inhibition may be ascribed to the enhanced interaction between Au and support, in line with the recent reports23.
To identify the chemical composition of the cover layer in Au/TiO2@M-N-800, the sample was probed by electron energy loss spectroscopy (EELS). Figure 2b and Supplementary Fig. 6b showed the obvious Ti L-edge signals in the amorphous overlayer, demonstrating the encapsulation by Ti-containing coating. While no C or N signal was observed in the overlayer region, proving the composition of overlayer was pure TiOx (Supplementary Figs. 7, 8). (Note: The signals of C and N in EELS are located at 280 and 400 eV, respectively). On the other hand, N 1s XPS spectra in Au/TiO2@M definitely demonstrated that melamine was adsorbed on the Au/TiO2 (Supplementary Fig. 9). And a decrease of N 1s XPS intensity in Au/TiO2@M-N may be ascribed to the carbonization of melamine and no N 1s XPS signal was observed in Au/TiO2@M-N-800 which demonstrated melamine had decomposed completely. The EELS results also revealed that Ti species on the overlayer existed as Ti3+ oxidation state (region II) while those on the support were Ti4+ oxidation state (region III), respectively, in consistent with the previous reports (Fig. 2c and Supplementary Fig. 6c)28,46,55. However, no reduced Ti3+ species were observed in the Ti 2p XPS spectra (Supplementary Fig. 10), in contrast to the case of classical SMSI, may be due to the smaller number of TiOx species51,56. The EELS analysis of Au/TiO2@M-N-800 catalyst definitely demonstrated the existence and the composition of the overlayer on Au NPs. To the best of our knowledge, this phenomenon has never been reported before as it was well-recognized that Au NPs can only be encapsulated by TiOx in reductive condition at high temperature (classical SMSI), inconsistent with the condition in this work28,37,38,39.

Changes of Au structure during treatments
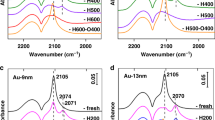
To determine if there is adsorption change and/or electron transfer between gold and support, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements of CO adsorption were examined. As shown in Fig. 3, two bands were detected on Au/TiO2 at 2174 and 2104 cm−1, respectively. The former was ascribed to gaseous CO as the peak decreased rapidly and disappeared totally under purge with He (Supplementary Fig. 11a, b). The latter was due to CO adsorbed on metallic Au NPs57. When modification with melamine, CO absorption band red-shifted to 2098 cm−1 assigned to Auδ− and accompanied by a decrease in intensity. The red-shift of CO adsorption may be due to the electron transfer from melamine to Au and therefore the adsorption intensity decreased, as it was difficult for CO to adsorb on Auδ− and there was no change in the structure morphology of Au NPs28. However, after pretreatment at 600 °C in N2 flow, 2098 cm−1 band was blue-shifted to 2106 cm−1 and accompanied by a decrease in intensity. The blue-shift of CO adsorption band may be attributed to electron withdrawing from Au NPs to melamine due to the carbonization of melamine. Furthermore, the decrease of the intensity may be ascribed to the particle size increase. After being subjected to calcination at 800 °C, 2106 cm−1 adsorption band blue-shifted to 2116 cm−1, suggesting much more positive charge on the surface Au species, which may be attributed to the electron transfer from Au NPs to TiO2 support as melamine had decomposed completely at this high temperature. It is noteworthy that 2116 cm−1 is not the band of gas-phase CO as it still existed under purge with He while gaseous CO band (2174 cm−1) decreased rapidly after 30 s and disappeared completely within 2 min (Supplementary Fig. 11c, d), suggesting that this band is the peak of CO adsorption on Auδ+ as CO-Auδ+ is quite stable53,58. The intensity decrease can be ascribed to the loss of CO adsorption sites resulting from the encapsulation of Au NPs by TiOx instead of sintering of the Au NPs as the mean size of Au NPs only increased from 6.7 to 7.5 nm, in good consistent with high-resolution transmission electron microscopy (HRTEM) results. Comparatively, the adsorption band at 2090 cm−1 and loss CO adsorption were observed for Au/TiO2−800, which was ascribed to the serious sintering of Au NPs leading to the decrease of adsorption sites, consistent with HRTEM results. The red-shift was due to the decrease of the positively charged Au species, in accordance with previous report27. While no obvious changes in the binding energy of Au 4f were observed, suggesting that melamine treatment and subsequent calcinations did not affect the oxidation state of the surface Au significantly (Supplementary Fig. 12).

In situ DRIFT spectra of CO adsorption on Au/TiO2, Au/TiO2@M, Au/TiO2@M-N, Au/TiO2@M-N-800, and Au/TiO2-800.
To further investigate the interaction between Au NPs and TiO2, Au LIII-edge XAFS spectra were collected on a series of samples to follow the coordination environment and valence state of the gold. As displayed in Fig. 4a, we showed the normalized Au LIII-edge XANES spectra recorded on a series of Au/TiO2 samples. The white line intensity was much stronger for Au/TiO2 and Au/TiO2@M than other samples, which was ascribed to the presence Au3+59. Moreover, the white line intensity of Au/TiO2@M-N-800 was stronger than Au/TiO2@M-N and Au/TiO2-800, indicating much more positive charge on the gold. In the Fourier transforms of k3-weighted EXAFS oscillations (Fig. 4b), a two-shells fitting was performed. In the first shell, the coordination number of Au-Au1 increased from 5.9 to 8.4 and the average bond length of Au-Au1 increased from 2.82 to 2.85 Å when the pretreatment temperature increased from 250 to 800 °C. While for Au/TiO2-800 the coordination number and the average bond length of Au-Au1 were 9.8 and 2.86 Å, respectively, which was much closer to that of Au foil that suggested the d orbital of Au was almost entirely occupied, in good accordance with the XANES results. Moreover, it was found that including additional Au–O shells would lower the quality of fitting results, which may be ascribed to the growth of the Au NPs size. Furthermore, a two-shell analysis of the narrow window region data of Au/TiO2@M-N-800 using Au–Au and Au–Ti scattering parameters gave a good fitting result. The best fitting was obtained with Au-Au1 bond distance of 2.85 Å (N = 8.4) and Au–Ti bond distance of 2.79 Å (N = 5.6). The expected Au–Ti distance of 2.79 Å was similar to that of AuTi alloy phase60. The fitting results were summarized in Supplementary Table 2 and the k-space curves were depicted in Supplementary Fig. 13. And the previous studies on Pt/TiO2 and Rh/TiO2 catalysts in the SMSI state provided direct evidence for Pt–Ti bond and Rh–Ti bond with a distance of 2.76 and 2.56 Å, respectively61,62. Therefore, there was no doubt that the encapsulation state leaded to a structural reorganization of the TiO2 support in the neighborhood of the Au NPs. And this reorganization of the support only generated where there was Au NPs and encapsulation occurred. Otherwise, no reorganization would occur.

a Normalized XANES spectra at the Au LIII-edge of Au foil, Au/TiO2, Au/TiO2@M, Au/TiO2@M-N, Au/TiO2@M-N-800, Au/TiO2-800. b Fourier transform of k3-weighted EXAFS spectra of Au foil, Au/TiO2, Au/TiO2@M, Au/TiO2@M-N, Au/TiO2@M-N-800, and Au/TiO2-800.
Catalytic performance in a series of reactions
CO oxidation was performed as a typical probe reaction due to its great importance in fundamental study, especially its size-dependent behavior63. As shown in Fig. 5a, Au/TiO2 revealed the highest activity with a T50 (the temperature 50% conversion is acquired) of −2.1 °C. After calcination at 800 °C, Au/TiO2@M-N-800 exhibited a much higher activity compared with Au/TiO2-800, explicitly displaying the strong effect of particle size on catalytic performance. According to the active curve, the T50 was determined to be 32 °C for Au/TiO2@M-N-800. While for Au/TiO2-800 the activity was negligible below 220 °C with a T50 of 245 °C. On the basis of TEM and DRIFTS characterization results, the poor activity of Au/TiO2-800 was attributed to the serious sintering of Au NPs. However, for Au/TiO2@M-N-800 it was due to the encapsulation of Au by TiOx overlayer leading to the partial decrease of accessible active sites. To investigate the kinetic study of the above catalysts, the specific reaction rates and turn over frequency (TOF) were determined at 25 °C. For Au/TiO2 catalyst, the reaction rate and TOF were 0.536 molCO h−1 gAu−1 and 0.12 s−1, respectively, in good accordance with the above results. While for Au/TiO2@M-N-800, the reaction rate was induced by a 3-fold decrease from 0.536 to 0.186 molCO h−1 gAu−1. As for Au/TiO2-800, the sintering of Au NPs decreased the reaction rate by about 90-fold from 0.536 to 0.006 molCO h−1 gAu−1.

a CO oxidation curves of Au/TiO2, Au/TiO2@M-N-800 and Au/TiO2-800 with a feed gas comprising 1 vol% CO/ 1 vol% O2/ 98 vol% He at 33.3 mL min−1. b Conversion of CO from 0 to 800 °C with 1st-10th cycles on Au/TiO2@M-N-800 catalyst for ten ignition-extinction cycles with a feed gas comprising 1 vol% CO/1 vol% O2/98 vol% He at 33.3 mL min−1. c Long-term simulated CO emission control reaction at 400 °C on the Au/TiO2@M-N-800 with space velocity of 220 L h−1 gcat−1. Reaction gas composition: 1.6 vol% CO, 1 vol% O2, 0.01 vol% propene, 0.00852 vol% toluene, 10 vol% water and balanced with He. d HRTEM image of Au/TiO2@M-N-800 after simulated CO emission control reaction at 400 °C for 10 days. The scale bar corresponds to 5 nm.
Furthermore, the activity of the Au/TiO2@M-N-800 was also compared with that of Au/TiO2 catalysts in classical SMSI state before and after calcination. It was showed that the T50 of Au/TiO2-H500 and Au/TiO2-H500-O400 were −19.5 and 16.5 °C and the corresponding particle size were about 3.2 and 4.2 nm, respectively (Supplementary Figs. 14–16). HRTEM results showed that Au NPs were encapsulated with a TiOx overlayer in Au/TiO2-H500, which retreated after further calcination under oxidation condition, consistent with our previous report28. It should be noted that the TiOx overlayer in classical SMSI was much thinner than the overlayer induced by melamine in this work. After calcination at 800 °C in air for 3 h, the T50 of Au/TiO2-H500–800 and Au/TiO2-H500-O400–800 were 191 and 168 °C and the particle size were 28.6 and 27.2 nm, respectively, in which no overlayer was observed and Au NPs sintered seriously (Supplementary Figs. 17, 18). Therefore, the overlayer formed in the reduction condition retreated under oxidation condition (classical SMSI) may account for the sintering of Au in Au/TiO2-H500–800, leading to invalid effect on the catalytic and stability property of the underlying metal. And the T50 of Au/TiO2@M-N-800 was much lower than that of Au/TiO2-H500–800, exhibiting the excellent sintering-resistant ability and superiority of Au/TiO2@M-N-800 compared with Au/TiO2-H500–800. For comparison, the specific rate and/or TOF of the above the catalysts and some reported catalysts are listed in Supplementary Table 3. To the best of our knowledge, it is the first time to report that Au NPs can be encapsulated by titania under oxidative atmosphere at high temperature, in contrast with condition required for classical SMSI. Although the resultant Au/TiO2@M-N-800 does not outperform the state-of-the-art, it is still comparable to the Au/TiO2-HAP-800 and Au/TiO2-SiO2-800 catalysts, which are the most excellent sintering-resistance catalyst up to now.
The reaction stability of Au/TiO2@M-N-800 catalyst was tested with CO oxidation at 400 °C with a high space velocity of 500 L h−1 gcat−1. As shown in Supplementary Fig. 19, the conversion increased from 79.1 to 90.1% during 100 h test instead of deactivation, demonstrating the excellent sintering-resistance and stability. HRTEM images of the used Au/TiO2@M-N-800 catalyst proved that the overlayer was still covering Au NPs, explaining the distinguished stability (Supplementary Fig. 20c, d). And the increase of the activity during the reaction may be due to much more active sites were accessible, as the overlayer on the used catalyst was not so dense compared with the fresh catalyst (Supplementary Fig. 20e, f). The recycle performance of Au/TiO2@M-N-800 was also examined by successive 10 ignition-extinction cycles up to 800 °C. Figure 5b displayed that the catalyst exhibited a slight decrease of the activity after 10th cycles. HRTEM images of the used Au/TiO2@M-N-800 catalyst demonstrated that TiOx overlayer still existed on the Au NPs which afforded the sinter-resistant Au nanocatalyst (Supplementary Fig. 21). And it has been acknowledged that water would expedite the sintering of Au NPs. Therefore, water-gas shift (WGS) reaction was performed on Au/TiO2@M-N-800 catalyst at 500 °C with space velocity of 18 L h−1 gcat−1. As expected, the conversion almost kept at 45 % during the 90 h test, further certifying the excellent stability of Au/TiO2@M-N-800 catalyst (Supplementary Fig. 22). TEM image of the used Au/TiO2@M-N-800 catalyst exhibited that the particle size of Au NPs remained unchanged. HRTEM images demonstrated that the Au NPs were still encapsulated by TiOx overlayer after reaction, leading to ultrastable Au NPs (Supplementary Fig. 23). To further evaluate the stability of Au/TiO2@M-N-800 in presence of water and other gases, simulated CO emission control was performed at 400 °C with the feed mixture gas containing 1.6 vol% CO, 1 vol% O2, 0.01 vol% propene, 0.00852 vol% toluene, 10 vol% water and balanced with He. Figure 5c showed that CO conversion of Au/TiO2@M-N-800 increased from 90.3 % to 92.5 % during the test for 10 days, exhibiting an excellent durability. TEM image of the used Au/TiO2@M-N-800 catalyst showed that the mean size of Au NPs was 7.3 nm, almost similar with the fresh catalyst (Supplementary Fig. 24a, b). HRTEM images (Fig. 5d and Supplementary Fig. 24c) revealed the still existence of the TiOx overlayer on the Au NPs after reaction for 10 days, which meant that water and other gases in the reaction almost had no influence on the TiOx overlayer. And the increase of the activity during the reaction may be due to much more accessible active sites (Supplementary Fig. 24d–f). However, for the catalyst without the encapsulation, the reactivity decreased continuously because of the aggregation of Au NPs27,30. Therefore, the encapsulation of Au by TiOx cover layer plays a crucial role in influencing the catalytic performances.
Relating encapsulation induced by melamine to SMSI behavior
In above, we have demonstrated the encapsulation of Au NPs by TiOx overlayer under oxidative condition induced by melamine while high temperature reduction pretreatment is prerequisite for the encapsulation of Au NPs by TiOx in classical SMSI. Furthermore, this kind of encapsulation may not belong to classical SMSI as the mean size of the Au NPs increases from 3.5 to 7.5 nm while the mean diameter of Au NPs remains almost unchanged in classical SMSI. Therefore, the formation mechanisms of these two kinds of overlayer are different. The formation process of classical SMSI overlayer and melamine-induced TiOx overlayer can be illustrated by Fig. 6. To investigate whether the overlayer will recede, Au/TiO2@M-N-800 sample was further calcined in air atmosphere at 400 °C, 500 °C and 600 °C for 3 h, respectively. HRTEM results showed that Au NPs were still encapsulated by TiOx overlayer, which was different from classical SMSI in which TiOx overlayer will retreat under further heating in oxidation condition (Supplementary Figs. 25–27)28,37,38,55. Moreover, the encapsulation TiOx overlayer induced by melamine is porous and permeable to reactant and very stable under oxidative reaction condition, which may minimize the reactants diffusion on Au surface and strongly enhance the sintering-resistance and catalytic performance of the Au.

a Bare Au nanoparticles on TiO2. b Au/TiO2 catalyst that forms an impermeable SMSI TiOx overlayer after treatment with 10 vol% H2/He at 500 °C. c SMSI TiOx overlayer retreats when exposed to oxidation condition at 400 °C which is almost similar with that in a. d Melamine-modified catalyst that forms a permeable TiOx overlayer after treatment with N2 at high temperature (600 °C) followed by treatment with air at 800 °C. e Stable melamine-induced TiOx overlayer under air condition modifies the Au NPs catalytic bahavior.
As shown in Fig. 1c and Supplementary Fig. 28, encapsulation did not occur when calcination in N2 flow at 600 °C. And no cover layer was observed even calcination in N2 at 800 °C (Supplementary Fig. 29), which indicated that encapsulation could not occur only calcination in N2 follow. As displayed in Supplementary Figs. 30–31, when Au/TiO2@M sample was heated directly in air at 600 or 800 °C without pretreatment in N2 atmosphere no overlayer was observed, suggesting the indispensable of pretreatment in N2. Supplementary Figs. 32–33 showed that overlayer also could not be formed when calcination in air at 500 or 600 °C after pretreatment in N2, which meant higher anneal temperature was requisite. For Au/TiO2 catalyst without modification with melamine and pretreatment under similar condition with that of Au/TiO2@M-N-800 (denoted as Au/TiO2-N-800), no cover layer was observed instead of the sintering of Au NPs, indicating the significant role of melamine in the formation of TiOx overlayer (Supplementary Fig. 34). To identify whether the encapsulation can be extended to other related materials, two catalysts (denoted as Au/anatase@M-N-800 and Au/rutile@M-N-800) were prepared with the same method as Au/TiO2@M-N-800. As shown in Supplementary Figs. 35–36, a TiOx overlayer was observed on both catalysts, evidencing a common encapsulation phenomenon between Au and theses two titania supports. Moreover, similar encapsulations can also be obtained on colloidal Au NPs supported on TiO2 (denoted as C–Au/TiO2@M-N-800) and commercial gold catalyst RR2Ti (denoted as RR2Ti@M-N-800), suggesting the universality of this strategy and providing a widespread way which enables to rationally design sintering-resistant gold catalysts via TiOx overlayer encapsulation strategy (Supplementary Figs. 37–38).
It is worth to summarize the mechanism and the effect of adsorbate melamine on the formation of the TiOx overlayer and stabilization of Au NPs. The HRTEM and EELS results verify the role of melamine in mediating TiOx overlayer formation. On the basis of identification Ti3+ in the cover layer by EELS, we hypothesize the plausible mechanism is that electron is transferred from Au NPs to TiO2 under the effect of melamine at high annealing temperature under oxidative condition, leading to the reduction of Ti4+ to Ti3+ and partial positive charge on gold, in consistent with DRIFTS results. Meanwhile, EXAFS fitting results corroborate the presence of Au–Ti bond in the encapsulation state, which may lead to the migration of titania onto Au NPs thermodynamically favorable46,56, similar with classical SMSI in which Pt–Ti and Rh–Ti bond are observed after high temperature reduction pretreatment. However, the specific effect of melamine in electron transfer is still unclear and much more work is deserved to be done. And further research is also required to clarify the real driving force for this kind of encapsulation.
Discussion
In summary, we have presented an ultrastable titania-supported Au nanocatalyst in which Au NPs were encapsulated with a permeable TiOx overlayer mediated by melamine under oxidative atmosphere. The indispensable pretreatment condition is anneal in N2 flow and further calcination at 800 °C under air condition, in contrast with classical SMSI which occurs under reductive condition. And the overlayer is stable even further calcination under oxidative condition, which is contrary to classical SMSI. Owing to the formation of permeable and stable TiOx layer, the designed catalyst is sintering-resistant with high catalytic activity and excellent durability. In addition, this methodology can be extended to colloidal Au catalyst and even commercial RR2Ti catalyst. More importantly, this work paves a new avenue for designing sintering-resistant Au nanocatalysts with high activity via TiOx overlayer encapsulation strategy.
Methods
Synthesis of Au/TiO2
As a typical run, Au/TiO2 was synthesized with the DP method. A 40 mL of HAuCl4·4H2O aqueous solution (6.3 mmol L−1) was adjusted to PH 9.0 with 0.1 M NaOH. Then, 1 g of TiO2 (Degussa P25) was added to the above solution and the PH was kept to 9.0 for 1 h by adding NaOH. The slurry was heated to 65 °C and magnetically stirred for 1 h. After centrifugation and washed with ultrapure water until free of chloride ions, the resultant solid was dried at 60 °C overnight and calcined at 250 °C for 2 h in air. The actual content of Au was 3.84 wt % determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES).
Synthesis of C–Au/TiO2
C–Au/TiO2 was synthesized by colloidal deposition method. 2.6 mL of 0.045 M HAuCl4·4H2O solution and 3.33 mL of 0.5 wt% PVA (Mw 10,000; 80% hydrolyzed) solution were added to 100 mL ultrapure water. After stirring for 30 min, 6.6 mL NaBH4 (0.1 mol L−1) solution was rapidly added into the above solution and a dark-brown solution was produced, indicating the formation of gold colloid. 0.5 g of TiO2 (Degussa P25) was added under vigorous stirring. After stirred for 6 h, the solid was washed with ultrapure water and dried at 60 °C overnight, followed by heating at 300 °C for 2 h in 10 vol% O2/He to remove PVA. The resulting sample was denoted as C-Au/TiO2.
Synthesis of Au/TiO2-H500
The Au/TiO2-H500 was synthesized by reducing Au/TiO2 under 10 vol% H2/He at 500 °C for 1 h following a previously reported procedure28.
Synthesis of Au/TiO2-H500-800
The Au/TiO2-H500-800 was synthesized by directly calcining Au/TiO2-H500 at 800 °C in air for 3 h at a heating rate of 2 °C min−1.
Synthesis of Au/TiO2-H500-O400
The Au/TiO2-H500-O400 was synthesized by reoxidizing Au/TiO2-H500 under 10 vol% O2/He flow at 400 °C for 1 h following a previously reported procedure28.
Synthesis of Au/TiO2-H500-O400–800
The Au/TiO2-H500-O400–800 was synthesized by directly calcining Au/TiO2-H500-O400 at 800 °C in air for 3 h at a heating rate of 2 °C min−1.
Synthesis of Au/TiO2@M-N-800
Typically, 0.15 g as-synthesized Au/TiO2 was added to the melamine-containing (1.67 mg mL−1) solution (30 mL) and the slurry was stirred at 65 °C for 24 h. After centrifugation and washed with ultrapure water, the product was dried at 60 °C overnight. The obtained sample was denoted as Au/TiO2@M and then annealed at 600 °C in N2 flow for 3 h at a heating rate of 5 °C min−1. The resulting sample was denoted as Au/TiO2@M-N. Then the Au/TiO2@M-N-800 was synthesized by calcining Au/TiO2@M-N at 800 °C in air for 3 h at a heating rate of 2 °C min−1.
Synthesis of Au/TiO2@M-T
The Au/TiO2@M-T series were synthesized by directly calcining Au/TiO2@M in air at different temperature for 3 h at a heating rate of 2 °C min−1, where T (600, 800) represents the calcining temperature (°C).
Synthesis of Au/TiO2@M-N800
The Au/TiO2@M-N800 was synthesized by calcining Au/TiO2@M in N2 at 800 °C for 3 h at a heating rate of 5 °C min−1.
Synthesis of Au/TiO2@M-N-T
The Au/TiO2@M-N-T series were synthesized by calcining Au/TiO2@M-N in air at different temperature for 3 h at a heating rate of 2 °C min−1, where T (500, 600) represents the calcining temperature (°C).
Synthesis of Au/TiO2@M-N-800-T
The Au/TiO2@M-N-800-T series were synthesized by calcining Au/TiO2@M-N-800 in air at different temperature for 3 h at a heating rate of 2 °C min−1, where T (400, 500, 600) represents the calcining temperature (°C).
Synthesis of Au/TiO2-800
The Au/TiO2-800 was synthesized by directly calcining Au/TiO2 at 800 °C in air for 3 h at a heating rate of 2 °C min−1.
Synthesis of Au/TiO2-N-800
The Au/TiO2-N-800 was synthesized by calcining Au/TiO2 in N2 at 600 °C for 3 h at a heating rate of 5 °C min−1, followed by calcining at 800 °C in air for 3 h at a heating rate of 2 °C min−1.
Synthesis of Au/anatase@M-N-800
The synthesis procedure of Au/anatase@M-N-800 is similar to that of Au/TiO2@M-N-800 except anatase was sued instead of TiO2.
Synthesis of Au/rutile@M-N-800
The synthesis procedure of Au/rutile@M-N-800 is similar to that of Au/rutile@M-N-800 except rutile was sued instead of TiO2.
Synthesis of C-Au/TiO2@M-N-800
The synthesis procedure of C-Au/TiO2@M-N-800 is similar to that of Au/TiO2@M-N-800 except C-Au/TiO2 was sued instead of Au/TiO2.
Synthesis of RR2Ti@M-N-800
The commercial gold catalyst RR2Ti was supplied by Haruta Gold Inc. The synthesis procedure of RR2Ti@M-N-800 is similar to that of Au/TiO2@M-N-800 except RR2Ti was sued instead of Au/TiO2.
Characterization
XRD patterns were collected on a PANalytical X’Pert Pro diffractometer with Cu Kα radiation (λ = 0.15432 nm). The gold content was determined by inductively coupled plasma-optical emission spectroscopy analysis (Perklin–Elmer 2100 DV). TEM and HRTEM were performed on JEM-2100 electron microscopy (JEOL) with an acceleration voltage of 200 kV. EELS analysis was carried out on an FEI Tecnai G2 F20 microscope equipped with Gatan Imaging Filter (GIF) system operated at 200 kV. The chemical compositions of the covering layer of Au NPs were characterized by directly putting electron beam at the Au NPs in a STEM mode. In situ DRIFTS spectra were recorded on Bruker Equinox 70 spectrometer equipped with MCT detector and operated at a resolution of 4 cm−1. Before CO adsorption, the samples were pretreated with He at 120 °C for 1 h and then cooled to room temperature. The background spectrum was collected in following He and then the gas (3 vol% CO/He) was introduced into the reaction cell at a total flow rate of 33.3 mL min−1. The spectra were recorded until the peak intensity was steady. The blank test was carried out following the exactly same procedure just without catalyst. X-ray photoelectron spectroscopy measurements (XPS) were collected on Thermo Fisher Scientific ESCALAB 250 spectrometer with monochromatic Al Kα (1486.6 eV) X-ray source. The binding energy (BE) value of C 1 s at 284.6 eV was used as the reference. X-ray adsorption fine structure (XAFS) measurements at Au LIII-edge in fluorescence mode were performed at Beamline 1W1B in Beijing Synchrotron Radiation Facility (BSRF). The electron storage ring was operated at ~200 mA and ~2.2 GeV with a top-up injection mode. A double-crystal Si (111) monochromator was used to scan X-ray energy for X-ray adsorption near edge (XANES) spectra and extended X-ray fine structure (EXAFS) spectra, respectively. The Au foil was used as reference for X-ray energy calibration. Data processing and analysis were performed by Demeter software package. The crystallographic structure of Au, Au2O3, AuTi were used as models for EXAFS fitting.
Catalytic activity measurement
CO oxidation was carried out in a fixed-bed reactor under atmospheric pressure. The gaseous mixture of 1 vol% CO + 1 vol% O2 and He balance (33.3 mL min−1) was passed through the catalysts. The unreacted reactants and products were analyzed online by gas chromatograph equipped with a TCD detector. WGS reaction was performed in the same reactor as was used for CO reaction and a mixture of 2 vol% CO + 10 vol% H2O and He balance was used with a space velocity of 18 L h−1 gcat−1. The simulated CO emission control test was carried out in the same reactor and the feed gas was 1.6 vol% CO + 1 vol% O2 + 0.01 vol% propene + 0.0087 vol% toluene + 10 vol% water and He balance with a space velocity of 220 L h−1 gcat−1. Specific reaction rates and TOF were evaluated by changing the weight of catalysts from 100 mg to 2 mg to guarantee that CO conversion was below 15%. For each run, the CO conversion was averaged at the steady state and the TOF was calculated according to equation: TOF = rCO*MAu/D, where rCO is the specific reaction rates.
Data availability
All data are available within the article and its Supplementary Information file are available from the authors upon reasonable request.
References
Bell, A. T. The impact of nanoscience on heterogeneous catalysis. Science 299, 1688–1691 (2003).
Xia, Y. N. Nanoparticles for catalysis. Acc. Chem. Res. 46, 1671–1672 (2013).
Zheng, N. & Stucky, G. D. A general synthetic strategy for oxide-supported metal nanoparticle catalysts. J. Am. Chem. Soc. 128, 14278–14280 (2006).
Schauermann, S., Nilius, N., Shaikhutdinov, S. & Freund, H.-J. Nanoparticles for heterogeneous catalysis: new mechanistic insights. Acc. Chem. Res. 46, 1673–1681 (2013).
Zhang, S. et al. High catalytic activity and chemoselectivity of sub-nanometric Pd clusters on porous nanorods of CeO2 for hydrogenation of nitroarenes. J. Am. Chem. Soc. 138, 2629–2637 (2016).
Noh, H. et al. An exceptionally stable metal-organic-framework supported molybdenum(VI) oxide catalyst for cyclohexene epoxidation. J. Am. Chem. Soc. 138, 14720–14726 (2016).
Gu, D. et al. Gold on different manganese oxides: ultra-low-temperature CO oxidation over colloidal gold supported on bulk-MnO2 nanomaterials. J. Am. Chem. Soc. 138, 9572–9580 (2016).
Zhang, J. et al. A Pd@zeolite catalyst for nitroarene hydrogenation with high product selectivity by sterically controlled adsorption in the zeolite micropores. Angew. Chem., Int. Ed. 56, 9747–9751 (2017).
Behrens, M. et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 336, 893–897 (2012).
Graciani, J. et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 345, 546–550 (2014).
Haruta, M., Kobayashi, T., Sano, H. & Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 16, 405–408 (1987).
Haruta, M. Gold rush. Nature 437, 1098–1099 (2005).
Corma, A. & Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 313, 332–334 (2006).
Chen, M. S. & Goodman, D. W. The structure of catalytically active gold on titania. Science 306, 252–255 (2004).
Hutchings, G. A golden future. Nat. Chem. 1, 584 (2009).
K. Hashmi, A. S. Homogeneous catalysis by gold. Gold. Bull. 37, 51–65 (2004).
Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 36, 153–166 (1997).
Wolf, A. & Schüth, F. A systematic study of the synthesis conditions for the preparation of highly active gold catalysts. Appl. Catal., A. 226, 1–13 (2002).
Zanella, R. & Louis, C. Influence of the conditions of thermal treatments and of storage on the size of the gold particles in Au/TiO2 samples. Catal. Today 107-108, 768–777 (2005).
Laursen, A. B. et al. Substrate size-selective catalysis with zeolite-encapsulated gold nanoparticles. Angew. Chem., Int. Ed. 49, 3504–3507 (2010).
Wang, S. et al. Aggregation-free gold nanoparticles in ordered mesoporous carbons: toward highly active and stable heterogeneous catalysts. J. Am. Chem. Soc. 135, 11849–11860 (2013).
Arnal, P. M., Comotti, M. & Schuth, F. High-temperature-stable catalysts by hollow sphere encapsulation. Angew. Chem., Int. Ed. 45, 8224–8227 (2006).
Zhan, W. et al. A sacrificial coating strategy toward enhancement of metal-support interaction for ultrastable Au nanocatalysts. J. Am. Chem. Soc. 138, 16130–16139 (2016).
Zhan, W. et al. Surfactant-assisted stabilization of Au colloids on solids for heterogeneous catalysis. Angew. Chem., Int. Ed. 56, 4494–4498 (2017).
Zhang, J. et al. Sinter-resistant metal nanoparticle catalysts achieved by immobilization within zeolite crystals via seed-directed growth. Nat. Catal. 1, 540–546 (2018).
Wang, C. et al. Precisely applying TiO2 overcoat on supported Au catalysts using atomic layer deposition for understanding the reaction mechanism and improved activity in CO oxidation. J. Phys. Chem. C 120, 478–486 (2015).
Tang, H. et al. Ultrastable hydroxyapatite/titanium-dioxide-supported gold nanocatalyst with strong metal-support interaction for carbon monoxide oxidation. Angew. Chem., Int. Ed. 55, 10606–10611 (2016).
Tang, H. et al. Classical strong metal–support interactions between gold nanoparticles and titanium dioxide. Sci. Adv. 3, e1700231 (2017).
Wang, L. et al. Strong metal–support interactions achieved by hydroxide-to-oxide support transformation for preparation of sinter-resistant gold nanoparticle catalysts. ACS Catal. 7, 7461–7465 (2017).
Zhang, J. et al. Wet-chemistry strong metal-support interactions in titania-supported Au catalysts. J. Am. Chem. Soc. 141, 2975–2983 (2019).
Dick, K., Dhanasekaran, T., Zhang, Z. & Meisel, D. Size-dependent melting of silica-encapsulated gold nanoparticles. J. Am. Chem. Soc. 124, 2312–2317 (2002).
Li, W. Z. et al. Stable platinum nanoparticles on specific MgAl2O4 spinel facets at high temperatures in oxidizing atmospheres. Nat. Commun. 4, 2481 (2013).
Yang, X. et al. Taming the stability of Pd active phases through a compartmentalizing strategy toward nanostructured catalyst supports. Nat. Commun. 10, 1611 (2019).
Lu, J. et al. Coking- and sintering-resistant palladium catalysts achieved through atomic layer deposition. Science 335, 1205–1208 (2012).
Aydin, C., Lu, J., Browning, N. D. & Gates, B. C. A “smart” catalyst: sinter-resistant supported iridium clusters visualized with electron microscopy. Angew. Chem., Int. Ed. 51, 5929–5934 (2012).
Cao, A., Lu, R. & Veser, G. Stabilizing metal nanoparticles for heterogeneous catalysis. Phys. Chem. Chem. Phys. 12, 13499–13510 (2010).
Tauster, S. J., Fung, S. C. & Garten, R. L. Strong metal-support interactions. Group 8 noble metals supported on TiO2. J. Am. Chem. Soc. 100, 170–175 (1978).
Tauster, S. J., Fung, S. C., Baker, R. T. K. & Horsley, J. A. Strong interactions in supported-metal catalysts. Science 211, 1121–1125 (1981).
Tauster, S. J. Strong metal-support interactions. Acc. Chem. Res. 20, 389–394 (1987).
Belton, D. N., Sun, Y.-M. & White, J. M. Encapsulation and electronic effects in a thin-film model of a Rhodium-Titania strong metal-support interaction catalysts. J. Am. Chem. Soc. 106, 3059–3060 (1984).
Bracy, J. D. & Burch, R. Enhanced activity of Pd/TiO2 catalysts for the CO/H2 reaction in the absence of strong metal-support interactions (SMSI). J. Catal. 86, 384–391 (1984).
Belzunegui, J. P., Sanz, J. & Rojo, J. M. Contribution of physical blocking and electronic effect to establishment of strong metal-support interaction in Rh/TiO2 catalysts. J. Am. Chem. Soc. 114, 6749–6754 (1992).
Fan, L. & Fujimoto, K. Promotive SMSI effect for hydrogenation of carbon dioxide to methanol on a Pd/CeO2 catalyst. J. Catal. 150, 217–220 (1994).
Weerachawanasak, P. et al. Effect of strong metal–support interaction on the catalytic performance of Pd/TiO2 in the liquid-phase semihydrogenation of phenylacetylene. J. Catal. 262, 199–205 (2009).
Li, S. et al. Tuning the selectivity of catalytic carbon dioxide hydrogenation over Iridium/Cerium oxide catalysts with a strong metal-support interaction. Angew. Chem., Int. Ed. 56, 10761–10765 (2017).
Matsubu, J. C. et al. Adsorbate-mediated strong metal-support interactions in oxide-supported Rh catalysts. Nat. Chem. 9, 120–127 (2017).
Dong, J. H., Fu, Q., Jiang, Z., Mei, B. B. & Bao, X. H. Carbide supported Au catalysts for water gas shift reactions: a new territory for the strong metal-support interaction efect. J. Am. Chem. Soc. 140, 13808–13816 (2018).
Hernandez Mejia, C., van Deelen, T. W. & de Jong, K. P. Activity enhancement of cobalt catalysts by tuning metal-support interactions. Nat. Commun. 9, 4459 (2018).
Gao, Y., Liang, Y. & Chambers, S. A. Thermal stability and the role of oxygen vacancy defects in strong metal support interaction-Pt on Nb-doped TiO2(100). Surf. Sci. 365, 638–648 (1996).
Meyer, R., Lemire, C., Shaikhutdinov, S. K. & H.-J., F. Surface chemistry of catalysis by gold. Gold. Bull. 37, 72–124 (2004).
Fu, Q., Wagner, T., Olliges, S. & Carstanjen, H.-D. Metal-oxide interfacial reactions: encapsulation of Pd on TiO2 (110). J. Phys. Chem. B. 109, 944–951 (2005).
Goodman, D. W. “Catalytically active Au on titania”: yet another example of a strong metal support interaction (SMSI)? Catal. Lett. 99, 1–4 (2005).
Liu, X. et al. Strong metal-support interactions between gold nanoparticles and ZnO nanorods in CO oxidation. J. Am. Chem. Soc. 134, 10251–10258 (2012).
Tang, H. et al. Strong metal−support interactions between gold nanoparticles and nonoxides. J. Am. Chem. Soc. 138, 56–59 (2016).
Zhang, S. et al. Dynamical observation and detailed description of catalysts under strong metal-support interaction. Nano Lett. 16, 4528–4534 (2016).
Pesty, F., Steinriick, H.-P. & Madey, T. E. Thermal stability of Pt films on TiO2(110): evidence for encapsulation. Surf. Sci. 339, 82–95 (1995).
Green, I. X., Tang, W., Neurock, M. & Yates, J. T. Jr Spectroscopic observation of dual catalytic sites during oxidation of CO on a Au/TiO2 catalyst. Science 333, 736–739 (2011).
Mihaylov, M., Knözinger, H., Hadjiivanov, K. & Gates, B. C. Characterization of the oxidation states of supported gold species by IR spectroscopy of adsorbed CO. Chem. Ing. Tech. 79, 795–806 (2007).
Abdel-Mageed, A. M., Kučerová, G., Bansmann, J. & Behm, R. J. Active Au species during the low-temperature water gas shift reaction on Au/CeO2: a time-resolved operando XAS and DRIFTS study. ACS Catal. 7, 6471–6484 (2017).
Svanidze, E. et al. High hardness in the biocompatible intermetallic compound β-Ti3Au. Sci. Adv. 2, e1600319 (2016).
Beard, B. C. & Ross, P. N. Pt-Ti alloy formation from high-temperature reduction of a Titania-impregnated Pt catalyst: implications for strong metal-support interaction. J. Phys. Chem. 90, 6811–6817 (1986).
Sakellson, S., McMillan, M. & Haller, G. L. EXAFS evidence for direct metal-metal bonding in reduced Rh/TiO2 catalysts. J. Phys. Chem. 90, 1733–1736 (1986).
Freund, H. J., Meijer, G., Scheffler, M., Schlogl, R. & Wolf, M. CO oxidation as a prototypical reaction for heterogeneous processes. Angew. Chem., Int. Ed. 50, 10064–10094 (2011).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21476232, 21607029), the International Partnership Program of Chinese Academy of Sciences (No.121421KYSB20170020) and the State Key Laboratory of Catalysis in Dalian Institute of Chemical Physics (N-16-07).
Author information
Authors and Affiliations
Contributions
S.F.L. performed all the experimental work, including material synthesis, catalyst characterization and test. W.X. and L.R.Z. carried out the XAFS experiment. Y.M.N. and B.S.Z. performed EELS experiment. S.F.L. and W.L. performed the electron-microscopy characterization. L.L. carried out CO-DRIFT experiment. All the authors discussed the result and commented on the manuscript. S.F.L. and J.H.W. wrote and revised the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks James H. Carter and Liang Wang for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, S., Xu, W., Niu, Y. et al. Ultrastable Au nanoparticles on titania through an encapsulation strategy under oxidative atmosphere. Nat Commun 10, 5790 (2019). https://doi.org/10.1038/s41467-019-13755-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13755-5
This article is cited by
-
Applying heteroatom co-doped carbon nanotube for manifesting high performance in the electrochemical reduction of aqueous nitrogen oxide by gold nanoparticles
Nano Research (2024)
-
Stable Au nanoparticles confined in boron nitride shells for optimizing oxidative desulfurization
Nano Research (2023)
-
Enhanced catalytic activity and thermal stability by highly dispersed Pd-based nanocatalysts embedded in ZrO2 hollow spheres
Frontiers of Materials Science (2023)
-
Direct environmental TEM observation of silicon diffusion-induced strong metal-silica interaction for boosting CO2 hydrogenation
Nano Research (2023)
-
One-Step Solvothermal Synthesis of Ni Nanoparticle Catalysts Embedded in ZrO2 Porous Spheres to Suppress Carbon Deposition in Low-Temperature Dry Reforming of Methane
Nanoscale Research Letters (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
