
Abstract
The selective synthesis of α-functionalized ketones with two similar enolizable positions can be accomplished using allylic alcohols and iridium(III) catalysts. A formal 1,3-hydrogen shift on allylic alcohols generates catalytic iridium-enolates in a stereospecific manner, which are able to react with electrophiles to yield α-functionalized ketones as single constitutional isomers. However, the employment of nucleophiles to react with the nucleophilic catalytic enolates in this chemistry is still unknown. Herein, we report an umpolung strategy for the selective synthesis of α-alkoxy carbonyl compounds by the reaction of iridium enolates and alcohols promoted by an iodine(III) reagent. Moreover, the protocol also works in an intramolecular fashion to synthesize 3(2H)-furanones from γ-keto allylic alcohols. Experimental and computational investigations have been carried out, and mechanisms are proposed for both the inter- and intramolecular reactions, explaining the key role of the iodine(III) reagent in this umpolung approach.
Similar content being viewed by others


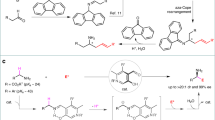
Introduction
Umpolung reactions represent a powerful approach for the introduction of functional groups into organic molecules where this would not otherwise be possible due to electronic mismatch1. This term was introduced by D. Seebach and E.J. Corey to refer the inversion of reactivity of acyl carbon atoms in their reactions with electrophiles2. Analogous methods based on switching the reactivity of amines3, imines4,5,6, or carbonyl groups7,8,9,10,11,12,13 have been developed in recent decades. This approach has played a pivotal role in the design of synthetic procedures, allowing access to target molecules that would be difficult to obtain by classical processes.
One example of a reaction where it is necessary to use an umpolung approach is the functionalization of ketones in the α position using nucleophiles14. To achieve this goal, iodine(III) compounds15,16,17,18, transition metals19, Lewis acids20,21, and other reagents22 have been used. For the synthesis of α-functionalized ketones with iodine(III) compounds, several reagents have been successfully used. These include Koser’s reagent23, (diacetoxyiodo)benzene (PIDA)24, p-iodotoluene difluoride25, and benzioxol(on)es (BX)26. When acyclic I(III) reagents are used, the nucleophile can be either an external nucleophile or a ligand of the I(III) center. However, for cyclic hypervalent iodine reagents, only examples where the nucleophile is part of the reagent have been reported, what requires an enormous synthetic effort as every reaction requires the synthesis of a new I(III) reagent27,28,29,30. In terms of the reaction substrates, dicarbonyl compounds, aromatic and cyclic ketones, and silyl enol ethers have all been used. However, no regiocontrol was achieved for ketones containing two enolizable α carbons with similar electronic/steric properties (Fig. 1a)31.

Synthesis of α-substituted carbonyl compounds. a α-Functionalization of ketones through umpolung reactions. b Isomerization/functionalization of allylic alcohols with electrophiles. c Our approach: Isomerization/umpolung functionalization of allylic alcohols
Allylic alcohols have been proven to be a very useful class of compounds as ketone synthons during the last decades32,33. Our group has reported the use of allylic alcohols as enolate equivalents for the preparation of α-functionalized carbonyl compounds as single constitutional isomers34,35,36,37,38,39,40,41. This approach relies on the generation of iridium enolates as catalytic intermediates through 1,3-hydrogen transfer, and the in situ reaction of these intermediates with a variety of heteroatomic electrophiles, including halogen and oxygen-based electrophilic species. The stoichiometric synthesis of enolates is avoided, and the reaction takes place under base-free conditions; as a result, a single substituent may be introduced at the desired α carbon. This is difficult to achieve starting from ketones with no clear electronic or steric bias (Fig. 1b). This approach has been successful, but it relies on the use of heteroatomic electrophiles. These are highly reactive species, and are less readily available than their nucleophilic counterparts. In this paper, we report an approach that inverts the polarity of the iridium enolate intermediate, allowing it to react with O-nucleophiles (Fig. 1c). In a reaction mediated by an iridium(III) complex, allylic alcohols react with nucleophiles to yield α-alkoxy ketones as single constitutional isomers. The reaction is mediated by 1-fluoro-3,3-dimethyl-1,3-dihydro-1λ3-benzo[d][1,2]iodaoxole. Furthermore, under the same conditions β-keto allylic alcohols cyclize yielding 3(2H)-furanones.
Results
Reaction development and optimization
We focused on the use of alcohols as nucleophiles; this would lead to the formation of α-alkoxy ketones. α-Alkoxy ketones are important building blocks for synthesis, and they are also present in many natural products42,43. We selected allylic alcohol 1a as a model substrate and methanol as a nucleophile. A preliminary screening of iodine(III) reagents (Fig. 2) revealed that 1-fluoro-3,3-dimethyl-1,3-dihydro-1λ3-benzo[d][1,2]iodaoxole (I) gave α-methoxy ketone 2a in a promising 43% yield as a single constitutional isomer (see Supplementary Table 1). In contrast, II–IX resulted in lower yields, ranging from 0 to 15%. I is a stable compound, and it is commercially available and easy to handle. Taking all this into account, I was chosen for further optimization studies in combination with the commercially available [Cp*IrCl2]244,45.

Iodine(III) reagents screened. Yields of 2a determined by 1H NMR spectroscopy using an internal standard (1,2,4,5-tetrachloro-3-nitrobenzene) (Supplementary Table 1)
It should be noted that the selective formation of the desired product 2 in this reaction represents an enormous challenge (Table 1). Several by-products may be formed from the allylic alcohol substrates 1, including unsubstituted ketone 3, and also enone 4. Furthermore, other nucleophiles such as water could also react, leading to the formation of α-hydroxy ketone 5. All these by-products were observed in our initial test reactions (Table 1, entry 1 and Supplementary Table 1), where common solvents previously used in related reactions were tested35,36,37,38,39,40,41. When either acetone or THF was used (Table 1, entries 1 and 2), 2a was formed in moderate yield (up to 43%) together with by-products 3–6 in yields ranging from 6 to 12%. Further testing led us to the use of 2,2,2-trifluoroethanol (TFE) (entries 3–5), which gave α-methoxy ketone 2a in 54% yield along with the same by-products in a similar ratio (Table 1, entry 4). A slightly better yield of 2a was obtained when the temperature was raised to 35 °C (57%; Table 1, entry 6). Increasing the temperature further did not prove beneficial (Table 1, entry 7). Remarkably, 2a was formed in 75% yield when the reaction mixture was diluted (from 0.2 to 0.02 M). The amounts of by-products 3 and 4 decreased, and the formation of 5 was suppressed (Table 1, entry 8). When the amount of KBF4 was increased (from 0.3 to 0.8 equiv.), 2a was formed in an excellent yield of 89% (Table 1, entry 9). The use of other additives failed to give higher yields of 2a (Table 1, entries 10 and 11). No conversion was observed when the chloride-free [Cp*Ir(H2O)3]SO4 complex was used as the catalyst. It has previously been shown that a halide ligand is essential in the isomerization of allylic alcohols45. In fact, when a chlorinated agent such as N-chlorosuccinimide is added in combination with [Cp*Ir(H2O)3]SO4, the desired product is observed (Supplementary Fig. 4). Control experiments were also carried out (Table 1, entries 13–15). In the absence of the iridium catalyst, allylic alcohol 1a was recovered in 86% yield (Table 1, entry 13), and 2a was not detected. The presence of the additive as well as TFE as cosolvent was necessary for the desired product 2a to be formed in high yield (Table 1, entry 9 vs entries 14 and 15). Saturated ketone 3 was obtained in quantitative yield when the reaction was carried out in the absence of I (Table 1, entry 16). Thus, we found that the umpolung reaction of 1a was best carried out using I in a TFE/MeOH mixture with KBF4 as additive at 35 °C catalyzed by [Cp*IrCl2]2 at a 0.02 M concentration of the allylic alcohol (Table 1, entry 9). We went on to study the substrate scope of the reaction under these optimal reaction conditions.
Reaction scope
For our studies of the substrate scope of the reaction (Fig. 3), we focused on allylic alcohols that would lead to α-methoxy ketones that are not accessible by the alternative direct α-functionalization of ketones, due to poor regioselectively. Allylic alcohols bearing terminal double bonds generally gave the corresponding α-methoxy ketones 2a–2n in moderate to good yields. Steric effects play an important role, and a more hindered allylic alcohol 1d gave 2d in only 45% yield. Aromatic allylic alcohols were found to be well tolerated, and 1e gave a 64% yield of 2e. Substrates bearing other functional groups such as an alkene, an ether, or a silyl ether (1f–1h) gave the corresponding products in excellent yields (84%, 99%, and 80%, respectively). This highlights the functional-group compatibility of the method. Remarkably, the reaction is chemoselective for the allylic alcohol functionality. Other functional groups with acidic α-methylene groups remain untouched. Allylic alcohol substrates bearing additional ketone (2i, 42% yield), nitrile (2j, 91% yield), or sulfone groups (2k, 74% yield) all gave the desired products with the reaction only taking place at the allylic alcohol. Furthermore, the reaction of chloride-containing allylic alcohol 1l proceeded in high yield (89%), and also diverse functionalized α-methoxy ketones, such as azide 2m (69%) and morpholine 2n (77%), were prepared in good yields from 1l in a one-pot two-step procedure. By simply selecting the starting allylic alcohol, constitutional isomers were prepared selectively. For instance, α-methoxy ketones 2b and 2q were selectively synthesized from allylic alcohols 1b (external double bond) and 1q (internal double bond), respectively. Allylic alcohols with internal 1,2-disubstituted double bonds (1o–1u) also reacted smoothly to give generally good yields of the corresponding products. Other alcohols such as ethanol and propanol afforded also the corresponding products, α-ethoxy ketone 6a in 40% and α-propoxy ketone 7a in 20% yield, respectively. In all examples shown in Fig. 3, the products were obtained as single constitutional isomers. Remarkably, this efficient umpolung protocol was extended to primary allylic alcohols (1v–1w) to give α-methoxy aldehydes in high yields. The method was also extended to more complex molecules derived from trans-androsterone (1x) and lythocolic acid (1y), which bear multiple functional groups such as esters and ketones, in addition to several stereocenters, and the desired products were obtained in high yields.

Scope of allylic alcohols 1. Yields by 1H NMR spectroscopy (isolated yields in parentheses). a By slow addition of the reactants. b From 2l in a one-pot two-step procedure
We also found that the reaction could be carried out in an intramolecular manner, with the oxygen of a carbonyl group acting as the nucleophile (Fig. 4). Thus, allylic alcohols 8a–8h, which have a ketone group in a 1,3-relationship with the alcohol functionality, were efficiently transformed into 3(2H)-furanones 9. This five-membered ring system is an important structural unit, and it can be found in a large number of natural products and other compounds with applications in medicine and biology46,47,48,49.

Scope of allylic alcohols 8. Yields by 1H NMR spectroscopy (isolated yields in parentheses)
Allylic alcohol 8a, with R3 = H, gave 3(2 H)-furanone 9a in 65% yield. The introduction of a substituent at R3 had a significant positive effect on the yield of the tandem reaction. Thus, alcohols 8b–8f gave the corresponding 3(2H)-furanones in excellent yields. Furthermore, allylic alcohols containing a cyclic ketone moiety 8g and 8h reacted well to give bicyclic products 9g and 9h (as a mixture of two diastereomers). The intramolecular umpolung reaction was extended to allylic alcohols 8i–8k, which contain an amide group that can act as an internal nucleophile. These substrates gave highly functionalized aminofuranones in excellent yields. Our umpolung strategy allows the construction of these highly important heterocycles from easily accessible starting materials.
Mechanism
To gain some insight into the reaction mechanism, and in particular into the role of the iodine(III) reagent I, we carried out DFT calculations at the M06/6-31G(d,p) level using the Gaussian 16 suit of programs (see the Supplementary Figs. 7 and 8, and Supplementary Data 1). In analogy with our previous work on the [Cp*Ir(III)]-catalyzed isomerization of allylic alcohols34,37,45,50 we propose that the reaction starts with the allylic alcohol undergoing a hydrogen-transfer step (1,3-hydride shift) mediated by the metal catalyst. This leads to iridium enolate species A (represented as an η3-enolate in Fig. 5)51. Our hypothesis is that the role of the additive (KBF4) is to increase the rate of the formation of iridium enolate species A. We carried out kinetic studies on the isomerisation of allylic alcohols, and found that the reaction is faster in the presence of KBF4 than in its absence (Supplementary Fig. 1). The initial reaction rate of 1a-d was found to be comparable to that of 1a, indicating that the 1,3-H shift is not rate determing (Supplementary Fig. 5).

Proposed intermolecular reaction mechanism. DFT calculations for the umpolung reaction of allylic alcohols with methanol. Values correspond to Gibbs free energies in kcal mol−1
The reaction of enolate A with I forms an enolonium intermediate B, which then reacts further with MeOH to form enolonium C52,53. From C, the final product 2 is obtained by reductive ligand coupling via TS128,54. This step has an activation energy of 16.2 kcal mol−1, which is perfectly attainable under the experimental reaction conditions. In the computational model of TS1, a molecule of TFE was introduced to activate the carbonyl group of the substrate by hydrogen bonding, and thus lower the activation barrier. When this step was modeled in the absence of TFE, the activation energy increased to 21.8 kcal mol−1 (Supplementary Fig. 8). Enolonium intermediates B′ and C′, tautomers containing an I–O bond instead of an I–C bond were also considered, but these species were excluded as they have higher energies than B and C (ΔG = 14.1 and 5.3 kcal mol−1, respectively). The energies of other isomeric forms of the enolonium intermediate (with a different arrangement of the substituents around the I(III) center) were also calculated, and these were all found to be much higher in energy (Supplementary Fig. 7). Besides, we could not locate any transition structure to form 2 starting from tautomer C′. Thus, C was the most plausible intermediate for this mechanism.
To investigate whether the iridium complex or KBF4 are involved in the reaction mechanism after enolonium B is formed, we performed control experiments in the laboratory where a pre-formed silyl enol ether was used as the starting material instead of the allylic alcohol. Neither the presence of KBF4 nor that of the iridium catalyst in the reactions from the silyl enol ether had any effect on the yield of the product (Supplementary Figs. 2 and 3). This suggests that none of them participates in the mechanism after enolonium B has been formed.
Further, reactions tested in the presence of radical scavengers, such as 2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) or 2-diphenylethylene, afforded the product (2a) in high yields (Supplementary Fig. 6). This suggests that a single-electron transfer mechanism is not operating.
We also investigated the mechanism of the cyclization reaction of type-8 allylic alcohols. An analogous enolonium species D is transformed into enolonium E by a downhill nucleophilic-addition/proton-transfer process (ΔG = −11.7 kcal mol−1, Fig. 6). The intramolecular displacement of the aryliodonium group via TS2 leads to the final (2H)-furan-3-one 9 in a very fast step, with a calculated activation energy of 8.0 kcal mol−1. Such intramolecular nucleophilic displacement reactions of aryliodonium groups have previously been suggested by Jacobsen and coworkers among others55,56.

Proposed intramolecular reaction mechanism. a DFT calculations for the umpolung reaction of γ-keto allylic alcohols. b TS for the α-methoxylation of γ-keto allylic alcohols
We also investigated why we never observed α-methoxylated products when we used 8-type substrates in our experimental work, and found out that starting from D, the competing transition state leading to the α-methoxy carbonyl compound (TS3) is higher in energy than TS2 by ca. 10 kcal mol−1 (18.2 kcal mol−1, Fig. 6).
Discussion
We have described a selective umpolung strategy which involves the reaction of an enolate species, formed from an allylic alcohol under catalytic conditions, with methanol. The catalytic enolate intermediate is formed by a 1,3-hydride shift mediated by an iridium catalyst. The two reacting species, the enolate and methanol, are both nucleophilic, so to allow them to react a polarity inversion must take place. This was achieved by carrying out the reaction in the presence of 1-fluoro-3,3-dimethyl-1,3-dihydro-1λ3-benzo[d][1,2]iodaoxole. By this approach, α-methoxy ketones were formed in high yields as single constitutional isomers under very mild conditions. Importantly, and in contrast with other methods that require the use of electrophilic sources of oxygen, a simple alcohol species can be used as a nucleophile in this reaction. The reaction also works with the carbonyl oxygen of ketones or amides acting as nucleophiles in an intramolecular fashion, yielding highly functionalized 3(2H)-furanones. Thus, this approach offers a route to these heterocyclic compounds, which are important structural units in medicinal chemistry, starting from simple and readily available substrates. The mechanisms for the inter- and intramolecular reactions were studied computationally. We found that the Cα-OMe products were formed through a reductive elimination reaction (or ligand coupling) between the C-bound I(III)-enolate and methanol, and that the TFE additive activates the substrate through hydrogen bonding promoting the C−O bond formation. We also found that the intramolecular process that leads to the formation of furanones follows a similar mechanism.
Methods
General procedure for the catalytic reaction
The corresponding allylic alcohol (0.3 mmol, 1 equiv.) was dissolved in a mixture MeOH/TFE (3:1) (15 mL). KBF4 (30 mg, 0.24 mmol, 80 mol%), 1-fluoro-3,3-dimethyl-1,3-dihydro-1-λ-3-benzo[d][1,2]iodaoxole (102 mg, 0.36 mmol, 1.2 equiv.), and [Cp*IrCl2]2 (6 mg, 0.0075 mmol, 0.025 equiv.) were added and the mixture stirred at 35 °C during 2 h. After that, H2O was added to dilute the reaction and the mixture was extracted with Et2O (3 × 5 mL). The combined organic phases were dried with MgSO4 and the solvent was evaporated under vacuum. The resulting crude was purified by column chromatography using petroleum ether/EtOAc (90:10) mixture as eluent.
DFT calculations
The calculations were carried out with the Gaussian 16 set of programs, using the M06 functional together with the 6-31G** basis sets for full structure optimization. IRC were done for both TS, where only one imaginary frequency was found and zero for the intermediate molecules. Then DEF2TZVPP in single point has been used with M06 for the energies. An implicit solvent model (IEFPCM, solvent = dimethyl formamide) was also used in all calculations (see the Supplementary Figs. 7 and 8, and Supplementary Data 1).
Data availability
The authors declare that all data supporting the findings of this report are available in the article or in their supplementary information files. This includes experimental procedures, compound characterization and full computational details. Date are also available upon request from the corresponding author.
References
Seebach, D. Methods of reactivity umpolung. Angew. Chem. Int. Ed. 18, 239–258 (1979).
Seebach, D. & Corey, E. J. Generation and synthetic applications of 2-lithio-1,3-dithianes. J. Org. Chem. 40, 231–237 (1975).
Seebach, D. & Enders, D. Umpolung of amine reactivity. Nucleophilic α‐(secondary amino)‐alkylation via metalated nitrosamines. Angew. Chem. Int. Ed. 14, 15–32 (1975).
Patra, A. et al. N-heterocyclic-carbene-catalyzed umpolung of imines. Angew. Chem. Int. Ed. 56, 2730–2734 (2017).
Liu, J., Cao, C.-G., Sun, H.-B., Zhang, X. & Niu, D. Catalytic asymmetric umpolung allylation of imines. J. Am. Chem. Soc. 138, 13103–13106 (2016).
Dickstein, J. S., Fennie, M. W., Norman, A. L., Paulose, B. J. & Kozlowski, M. C. Three component coupling of α-iminoesters via umpolung addition of organometals: synthesis of α,α-disubstituted α-amino acids. J. Am. Chem. Soc. 130, 15794–15795 (2008).
Kaiser, D., Teskey, C. J., Adler, P. & Maulide, N. Chemoselective intermolecular cross-enolate-type coupling of amides. J. Am. Chem. Soc. 139, 16040–16043 (2017).
De la Torre, A., Tona, V. & Maulide, N. Reversing polaruty: carbonyl α-aminations with nitrogen nucleophiles. Angew. Chem. Int. Ed. 56, 12416–12423 (2017).
Streuff, J. et al. Mechanism of the tiiii-catalyzed acyloin-type umpolung: a catalyst-controlled radical reaction. J. Am. Chem. Soc. 137, 14396–14405 (2015).
Leighty, M. W., Shen, B. & Johnston, J. N. Enantioselective synthesis of α-oxy amides via umpolung amide synthesis. J. Am. Chem. Soc. 134, 15233–15236 (2012).
Marion, N., Díez-González, S. & Nolan, S. P. N‐heterocyclic carbenes as organocatalysts. Angew. Chem. Int. Ed. 46, 2988–3000 (2007).
Linghu, X., Potnick, J. R. & Johnson, J. S. Metallophosphites as umpolung catalysts: the enantioselective cross silyl benzoin reaction. J. Am. Chem. Soc. 126, 3070–3071 (2004).
Burstein, C. & Glorius, F. Organocatalyzed conjugate umpolung of α,β‐unsaturated aldehydes for the synthesis of γ‐butyrolactones. Angew. Chem. Int. Ed. 43, 6205–6208 (2004).
Miyata, O., Miyoshi, T. & Ueda, M. Umpolung reactions at the α-carbon position of carbonyl compounds. Arkivoc 2, 60–81 (2013).
Dong, D.-Q., Hao, S.-H., Wang, Z.-L. & Chen, C. Hypervalent iodine: a powerful electrophile for asymmetric α-functionalization of carbonyl compounds. Org. Biomol. Chem. 12, 4278–4289 (2014).
Zhdankin, V. V. & Stang, P. J. Chemistry of polyvalent iodine. Chem. Rev. 108, 5299–5358 (2008).
Moriarty, R. M., Prakash, O., Duncan, M. P. & Vaid, R. K. Hypervalent iodine oxidation of silyl enol ethers under Lewis-acid conditions in methanol. A general route to α-methoxy ketones. J. Org. Chem. 52, 150–153 (1987).
Arava, S. et al. Enolonium species-umpoled enolates. Angew. Chem. Int. Ed. 56, 2599–2603 (2017).
Xu, Z., Chen, H., Wang, Z., Ying, A. & Zhang, L. One-pot synthesis of benzene-fused medium-ring ketones: gold catalysis-enabled enolate umpolung reactivity. J. Am. Chem. Soc. 138, 5515–5518 (2016).
Wu, Y.-K., Dunbar, C. R., McDonald, R., Ferguson, M. J. & West, F. G. Experimental and computational studies on interrupted nazarov reactions: exploration of umpolung reactivity at the α-carbon of cyclopentanones. J. Am. Chem. Soc. 136, 14903–14911 (2014).
Miyoshi, T., Miyakawa, T., Ueda, M. & Miyata, O. Nucleophilic α‐arylation and α‐alkylation of ketones by polarity inversion of N‐alkoxyenamines: entry to the umpolung reaction at the α‐carbon position of carbonyl compounds. Angew. Chem. Int. Ed. 50, 928–931 (2011).
Wei, Y., Lin, S., Xue, H., Liang, F. & Zhao, B. Halonium-initiated C–O bond formation via umpolung of α-carbon to the carbonyl: efficient access to 5-amino-3(2H)-furanones. Org. Lett. 14, 712–715 (2012).
Shneider, O. S., Pisarevsky, E., Fristrup, P. & Szpilman, A. M. Oxidative umpolung α-alkylation of ketones. Org. Lett. 17, 282–285 (2015).
Mizar, P. & Wirth, T. Flexible stereoselective functionalizations of ketones through umpolung with hypervalent iodine reagents. Angew. Chem. Int. Ed. 53, 5993–5997 (2014).
Yu, J., Tian, J. & Zhanga, C. Various α‐oxygen functionalizations of β‐dicarbonyl compounds mediated by the hypervalent iodine(III) reagent p‐iodotoluene difluoride with different oxygen‐containing nucleophiles. Adv. Synth. Catal. 352, 531–546 (2010).
Hari, D. P., Caramenti, P. & Waser, J. Cyclic hypervalent iodine reagents: enabling tools for bond disconnection via reactivity umpolung. Acc. Chem. Res. 51, 3212–3225 (2018).
Ilchenko, N. O., Hedberg, M. & Szabó, K. J. Fluorinative ring-opening of cyclopropanes by hypervalent iodine reagents. An efficient method for 1,3-oxyfluorination and 1,3-difluorination. Chem. Sci. 8, 1056–1061 (2017).
Mai, B. K., Szabó, K. J. & Himo, F. Mechanisms of Rh-catalyzed oxyfluorination and oxytrifluoromethylation of diazocarbonyl compounds with hypervalent fluoroiodine. ACS Catal. 8, 4483–4492 (2018).
Charpentier, J., Früh, N. & Togni, A. Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev. 115, 650–682 (2015).
Nagata, T., Matsubara, H., Kiyokawa, K. & Minakata, S. Catalytic activation of 1-cyano-3,3-dimethyl-3-(1H)-1,2-benziodoxole with B(C6F5)3 enabling the electrophilic cyanation of silyl enol ethers. Org. Lett. 19, 4672–4675 (2017).
Ochiai, M., Takeuchi, Y., Katayama, T., Sueda, T. & Miyamoto, K. Iodobenzene-catalyzed α-acetoxylation of ketones. In situ generation of hypervalent (diacyloxyiodo)benzenes using m-chloroperbenzoic acid. J. Am. Chem. Soc. 127, 12244–12245 (2005).
García-Álvarez, R. et al. Ruthenium (II) arene complexes with asymmetrical guanidinate ligands: synthesis, characterization, and application in the base-free catalytic isomerization of allylic alcohols. Organometallics 31, 8301–8311 (2012).
Kechaou-Perrot, M. et al. Tethered η5-oxocyclohexadienyl piano-stool ruthenium(II) complexes: a new class of catalyst? Organometallics 33, 6294–6297 (2014).
Ahlsten, N. & Martín-Matute, B. Ir-catalysed formation of C-F bonds. From allylic alcohols to α-fluoroketones. Chem. Commun. 47, 8331–8333 (2011).
Ahlsten, N., Bermejo Gómez, A. & Martín-Matute, B. Iridium-catalyzed 1,3-hydrogen shift/chlorination of allylic alcohols. Angew. Chem. Int. Ed. 52, 6273–6276 (2013).
Bermejo Gómez, A., Erbing, E., Batuecas, M., Vázquez-Romero, A. & Martín-Matute, B. Iridium-catalyzed isomerization/bromination of allylic alcohols: synthesis of α-bromocarbonyl compounds. Chem. Eur. J. 20, 10703–10709 (2014).
Vázquez-Romero, A., Bermejo Gómez, A. & Martín-Matute, B. Acid- and iridium-catalyzed tandem 1,3-transposition/3,1-hydrogen shift/chlorination of allylic alcohols. ACS Catal. 5, 708–714 (2015).
Martinez-Erro, S. et al. Base-catalyzed stereospecific isomerization of electron-deficient allylic alcohols and ethers through ion-pairing. J. Am. Chem. Soc. 138, 13408–13414 (2016).
Martinez-Erro, S., Bermejo Gómez, A., Vázquez-Romero, A., Erbing, E. & Martín-Matute, B. 2,2-Diiododimedone: a mild electrophilic iodinating agent for the selective synthesis of α-iodoketones from allylic alcohols. Chem. Commun. 53, 9842–9845 (2017).
Sanz-Marco, A., Martinez-Erro, S. & Martín-Matute, B. Selective synthesis of unsymmetrical aliphatic acyloins through oxidation of iridium enolates. Chem. Eur. J. 45, 11564–11567 (2018).
Sanz-Marco, A., Možina, S., Martinez-Erro, S., Iskra, J. & Martín-Matute, B. Synthesis of α-Iodoketones from allylic alcohols through aerobic oxidative iodination. Adv. Synth. Catal. 360, 3884–3888 (2018).
Jiang, X., Chen, W. & Hartwig, J. F. Iridiium-catalyzed diastereoselective and enantioselective allylic substitutions with acyclic α-alkoxy ketones. Angew. Chem. Int. Ed. 55, 5819–5823 (2016).
Hoyos, P., Sinisterra, J.-V., Molinari, F., Alcántara, A. R. & Domínguez de María, P. Acc. Chem. Res. 43, 288–299 (2010).
Cinar, M. E. & Schmittel, M. One-pot domino aldol reaction of indium enolates affording 6-deoxy-α-D,L-altropyranose derivatives: synthesis, mechanism, and computational results. J. Org. Chem. 80, 8175–8182 (2015).
Erbing, E. et al. General, simple, and chemoselective catalysts for the isomerization of allylic alcohols: the importance of the halide ligand. Chem. Eur. J. 22, 15659–15663 (2016).
Li, Y. & Hale, K. J. Asymmetric total synthesis and formal total synthesis of the antitumor sesquiterpenoid (+)-eremantholide A. Org. Lett. 9, 1267–1270 (2007).
Shamshina, J. L. & Snowden, T. S. Convergent synthesis of potent COX-2 inhibitor inotilone. Tetrahedron Lett. 48, 3767–3770 (2007).
Hayashi, Y. et al. Asymmetric total synthesis of pseurotin A. Org. Lett. 5, 2287–2290 (2003).
Han, Q. & Wiemer, D. F. Total synthesis of (+)-jatrophone. J. Am. Chem. Soc. 114, 7692–7697 (1992).
Bartoszewicz, A., Jeżowska, M. M., Laymand, K., Möbus, J. & Martín-Matute, B. Synthesis of β-hydroxy and β-amino ketones from allylic alcohols catalyzed by η5-(Ph5C5)Ru(CO)2Cl. Eur. J. Inorg. Chem. 2012, 1517–1530 (2012).
Krug, C. & Hartwig, J. F. Reactions of an arylrhodium complex with aldehydes, imines, ketones, and alkynones. new classes of insertion reactions. Organometallics 23, 4594–4607 (2004).
Geary, G. C., Hope, E. G., Singh, K. & Stuart, A. M. Preparation of iodonium ylides: probing the fluorination of 1,3-dicarbonyl compounds with a fluoroiodane. RSC Adv. 5, 16501–16506 (2015).
Minhas, H. K., Riley, W. & Stuart, A. M. Activation of the hypervalent fluoroiodane reagent by hydrogen bonding to hexafluoroisopropano. Org. Biomol. Chem. 16, 7170–7173 (2018).
Zhang, J., Szabó, K. J. & Himo, F. Metathesis mechanism of zinc-catalyzed fluorination of alkenes with hypervalent fluoroiodine. ACS Catal. 7, 1093–1100 (2017).
Woerly, E. M., Banik, S. M. & Jacobsen, E. N. Enantioselective, catalytic fluorolactonization reactions with a nucleophilic fluoride source. J. Am. Chem. Soc. 138, 13858–13861 (2016).
Mennie, K. M., Banik, S. M., Reichert, E. C. & Jacobsen, E. N. Catalytic diastereo- and enantioselective fluoroamination of alkenes. J. Am. Chem. Soc. 140, 4797–4802 (2018).
Acknowledgements
This project was supported by the Swedish Research Council through Vetenskapsrådet and Formas, by the Knut and Alice Wallenberg Foundation (KAW 2016.0072), and by the Göran Gustafsson Foundation. A.S.-M. thanks Universitat de València, the Generalitat Valenciana, and the European Social Fund for a post-doctoral grant. We are also grateful to the European Funding Horizon 2020-MSCA (ITN-EJD CATMEC 14/06-721223). We also thank IZO-SGI SGIker of UPV/EHU for human and technical support. Open access funding provided by Stockholm University.
Author information
Authors and Affiliations
Contributions
B.M.-M. and A.S.-M. conceived and designed the project. B.M.-M. directed the project. A.S.-M. and S.M.-E. carried out experiments and prepared the Supporting Information. E.G.-B. and M.P. carried out all the mechanistic calculations and contributed to the mechanistic understanding. They also prepared the corresponding Supporting Information. All the authors discussed the results, and all participated in writing the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Jing-Ping Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sanz-Marco, A., Martinez-Erro, S., Pauze, M. et al. An umpolung strategy to react catalytic enols with nucleophiles. Nat Commun 10, 5244 (2019). https://doi.org/10.1038/s41467-019-13175-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13175-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
