
Abstract
Oceanic emissions represent a highly uncertain term in the natural atmospheric methane (CH4) budget, due to the sparse sampling of dissolved CH4 in the marine environment. Here we overcome this limitation by training machine-learning models to map the surface distribution of methane disequilibrium (∆CH4). Our approach yields a global diffusive CH4 flux of 2–6TgCH4yr−1 from the ocean to the atmosphere, after propagating uncertainties in ∆CH4 and gas transfer velocity. Combined with constraints on bubble-driven ebullitive fluxes, we place total oceanic CH4 emissions between 6–12TgCH4yr−1, narrowing the range adopted by recent atmospheric budgets (5–25TgCH4yr−1) by a factor of three. The global flux is dominated by shallow near-shore environments, where CH4 released from the seafloor can escape to the atmosphere before oxidation. In the open ocean, our models reveal a significant relationship between ∆CH4 and primary production that is consistent with hypothesized pathways of in situ methane production during organic matter cycling.
Similar content being viewed by others


Introduction
Methane (CH4) is a potent greenhouse gas with a 100-year global warming potential that is ~23 times that of carbon dioxide1. Its atmospheric mixing ratio has increased more than two-fold since the preindustrial, contributing ~20% of the radiative climate forcing for all greenhouse gases2. Future anthropogenic impacts on the atmospheric CH4 budget are not restricted to direct emissions (e.g. during agriculture and energy production), but will also include climate-driven perturbation of the natural CH4 cycle3. This motivates recent efforts to place strong baseline constraints on natural CH4 sources and understand their environmental sensitivity4.
The global ocean is a highly uncertain term in the atmospheric CH4 budget, emitting 5–25 Tg of CH4 per year (hereafter Tg yr−1) or 1–13% of all natural emissions4. The dominant source of this methane is traditionally thought to be the sea floor, where it is produced biologically in anoxic sediments5 or released from geological reservoirs at hydrocarbon seeps6 and degrading methane hydrate deposits7. Methane is emitted to the atmosphere by two processes: diffusive gas transfer and ebullition (i.e. bubbling) across the air–sea interface8. Ebullitive emissions are only significant in regions that combine very shallow water columns with aggressive rates of CH4 bubbling through the seafloor9. Elsewhere, efficient dissolution of CH4 from rising bubbles produces supersaturated waters that drive a diffusive flux to the atmosphere10, although this pathway is limited by rapid oxidation of dissolved CH4 during its transport through the water column11. More recently, novel methanogenesis pathways have been identified that may produce CH4 in situ in the surface ocean mixed layer, providing a more direct conduit to atmosphere12,13,14.
Globally, both diffusive and ebullitive CH4 emissions remain uncertain due to sparse data constraints and the crude extrapolation methods used to upscale their rates4, limiting our understanding of the ocean’s leverage over atmospheric CH4. In this study, we provide a new robust estimate for the global diffusive flux and combine it with upper and lower bounds on ebullition rates, thus narrowing the uncertainty range for the total oceanic methane source.
Results
Global distribution of methane disequilibrium
Diffusive air–sea gas fluxes can be estimated from their ocean–atmosphere disequilibrium (denoted ∆) using gas transfer theory15. Previous attempts to constrain marine diffusive CH4 emissions have extrapolated from limited cruise track data, estimating a global flux between 0.2 and 18 Tg yr−1 to the atmosphere16,17,18,19. We improved upon this approach using machine-learning models to map methane disequilibrium (∆CH4) at the global scale, before computing the air–sea flux.
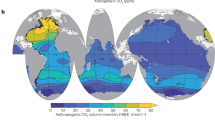
Our work is underpinned by a large compilation of shipboard CH4 concentration measurements collected between 1980 and 201620,21, which we combined with atmospheric pCH4 from a global monitoring network to determine ∆CH4 (see the “Methods” section). Data from the surface mixed layer was then assembled into a monthly climatology at 0.25° horizontal resolution (Fig. 1a, see the “Methods” section). This ∆CH4 climatology shows that open ocean waters (>2000 m deep) are most weakly supersaturated (0.02–0.2 nM, IQ range), reaching undersaturation in some polar regions (Fig. 1a, b). Surface supersaturation increases sharply towards coastlines, typically ranging between 0.08 and 0.7 nM across continental slopes (200–2000 m), 0.1–2 nM on the outer shelf (50–200 m), and 0.7–20 nM in near-shore environments (0–50 m). In these very shallow waters, ∆CH4 can occasionally reach many hundreds of nM (~5% above 100 nM, maximum of ~1500 nM). Our climatology contains 8725 gridded data points that are well distributed between marine environments, with ~65% coming from the open ocean and ~10% each from the slope, outer shelf and near-shore regions (Fig. 1b). Normalizing by their areas, this means that data density increases towards coastal waters that are critical regions of elevated flux (Fig. 1b)22.

Global ∆CH4 climatology. a Annual-mean ∆CH4, computed after binning all data into 0.25 × 0.25 monthly climatology. Data points are drawn larger than the grid cells for clarity. b Probability distributions of observed ∆CH4, grouped into four bathymetric regions (see also Supplementary Fig. 2). Boxes span the interquartile range, with black line at median. Black diamonds are mean values, and whiskers span the 5–95th percentiles. Number of datapoints (n) and data density per 109 m2 (N) after binning are listed
Our database is still too sparse for traditional gap-filling approaches applied to oceanographic data (e.g. ref. 23), especially given the sharp spatial gradients in ∆CH4. We therefore employed two different machine-learning methods that have previously been applied to map sparse marine data24,25,26: artificial neural networks (ANN) and random regression forests (RRF). These methods build nonlinear statistical models for ∆CH4 based on its relationship to physical and biogeochemical predictor variables, whose distributions are well known and are plausibly linked to ∆CH4 (see the “Methods” section), allowing global extrapolation of ∆CH4 in the mixed layer (Fig. 2a, b). Both ANN and RRF models are trained using randomly selected subsets of the data, and are designed to maximize the prediction of residual validation data while minimizing overfitting (Supplementary Fig. 1). Repeating the training process generates a large ensemble of maps that are used for error propagation (see the “Methods” section).

Machine-learning mapping of ∆CH4. a Annual mean ∆CH4 averaged across an ensemble of 100,000 individual maps generated by the artificial neural network (ANN) method. b Same as a but from random regression forest (RRF) method. c Taylor diagram summarizing the fit of a subset of 100 randomly selected ANN and RRF models to observed ∆CH4, after transformation (see the “Methods” section). Correlation coefficient (R) is shown on the outer angular axis, centered root-mean-squared difference is given by radial distance from REF point, and standard deviation (s.d.) normalized by observed s.d. is the radial distance from the origin (points on the 1.0 line have the same s.d. as observations). ANN and RRF dramatically outperform linear regression and multiple linear regression models by all three metrics
The machine-learning methods accurately capture the observed magnitude, variance, and spatial patterns of ∆CH4 both regionally (Supplementary Figs. 2 and 3) and globally (Fig. 2c; R2 = 0.7–0.8 for log-transformed data, see the “Methods” section). They dramatically outperform traditional linear regression (R2 = 0–0.15) and multiple linear regression (R2 = 0.2) models developed from the same predictor variables, according to multiple metrics of model skill (Fig. 2c).
Diffusive ocean–atmosphere methane flux
Having mapped ∆CH4 across the global ocean, we computed the diffusive sea–air CH4 flux at daily resolution using a wind-dependent gas transfer velocity (k) and accounting for sea ice cover, which acts as a barrier to gas exchange27 (see the “Methods” section). A Monte Carlo method was used to propagate uncertainties in ∆CH4, gas transfer velocity, and ice coverage into our calculation (see the “Methods” section), generating an ensemble of 200,000 different flux estimates (100,000 each for ANN and RRF methods).
The spatial pattern of air–sea flux predicted by these model ensembles is qualitatively similar to the ∆CH4 distribution, with highest fluxes in shallow shelf regions that often exceed rates of 10 mmol m−2 yr−1 (Fig. 3, Supplementary Table 1). Only in outer shelf environments of the Arctic Ocean is there a strong mismatch between the magnitude of ∆CH4 and flux, due to ice coverage over most of the year. The open ocean is mostly a weak source of CH4 (generally 0–0.5 mmol m−2 yr−1), with the exception of the Southern Ocean, which takes up ~0.04 mmol m−2 yr−1 on average south of 45°S. The North Atlantic Ocean polewards of 45°N is either a weak sink (ANN method) or weak source (RRF method) of CH4, marking the only region where the two mapping methods systematically disagree (Figs. 2 and 3), likely due to data scarcity (Fig. 1).

Diffusive ocean–atmosphere CH4 flux. a Annual diffusive CH4 emissions, averaged across an ensemble 100,000 individual calculations using the artificial neural network mapping method. b Same as a, but using the random regression forest mapping method
Integrating the fluxes regionally across near-shore, shelf, slope, and open ocean regions reveals a highly disproportionate contribution of shallow waters to oceanic methane emissions (Fig. 4a). The near-shore environment contributes the largest but most uncertain diffusive flux of the four, despite accounting for only ~3% of the ocean area. Emissions in these environments sum to 2.1 ± 1.6 and 2.0 ± 1.45 Tg yr−1 (mean ± s.d.) according to the ANN and RRF methods, respectively, with a likely range (defined here as 10–90th percentile range) between 0.8 and 3.8 Tg yr−1 when ensembles from both mapping methods are combined (Fig. 3a). The open ocean is the second largest emitter (likely range 0.6–1.4 Tg yr−1) because its vast area (~85% of ocean) compensates for low flux rates (Fig. 3a), followed by outer shelf (likely range 0.3–1.0 Tg yr−1) and continental slope (likely range 0.2–0.6 Tg yr−1) environments. Integrated globally, we find an ocean–atmosphere CH4 flux of 4.3 ± 2.2 or 3.9 ± 1.8 Tg yr−1 (mean ± s.d.) in the ANN and RRF ensembles, respectively, with a likely range between 2.2 and 6.3 Tg yr−1 combining all estimates (Fig. 4b).

Regional and global diffusive CH4 emissions. a Violin plot for annual diffusive CH4 emissions integrated across four bathymetric regions, computed using Monte Carlo method to propagate uncertainty in ∆CH4 and gas transfer velocity. Violin thickness corresponds to probability density, with think black lines at 25th and 75th percentiles, thick line at median, and diamond at mean value. Light gray shading for each region spans the 10–90th percentiles for all estimates, combining artificial network (ANN) and random regression forest (RRF) ensembles. b Probability density functions for globally integrated CH4 emissions from ANN and RRF methods. Diamonds and light gray shading as defined in a
Sensitivity tests revealed that the global flux is relatively insensitive to increasing the model grid resolution, the choice of biological predictor variables, and the propagation of potential measurement errors (Supplementary Figs. 1 and 4)28. We found that the largest contributor to the range of flux estimates is uncertainty in the ∆CH4 distribution introduced by our mapping methods, although uncertainty in the gas transfer velocity also makes a significant contribution (Supplementary Figs. 5 and 6).
Our new global estimate of 2.2–6.3 Tg yr−1 is larger than previous estimates based on basin-scale cruises (0.2–3 Tg yr−1)16,17,18, which may have undersampled strongly supersaturated coastal waters, but significantly smaller than estimated from a compilation of shelf data (11–18 Tg yr−1)19, which likely extrapolated high ∆CH4 too broadly18. In the Arctic Ocean—a region where methane emissions are highly sensitive to future climate warming29—we find annual diffusive CH4 emissions of ~0.5 Tg yr−1 (Supplementary Table 1). This is substantially lower than a previous estimate from the East Siberian Arctic Shelf30 (3.3 Tg yr−1), despite the fact that our statistical mapping methods skillfully reproduce the ∆CH4 distribution in this region (Supplementary Fig. 3). This implies that total Arctic CH4 emissions have previously been overestimated, consistent with more recent oceanic and atmospheric observations in this region31,32,33.
Ebullitive and total oceanic methane emissions
Direct constraints on methane ebullition across the air–sea interface are extremely rare34, meaning that our statistical mapping methods cannot be applied to scale-up this process. Instead, we attempt to place upper and lower bounds on the global ebullitive emission rate by combining previous estimates of ebullition at the seafloor with bubble model calculations to predict the transfer efficiency of CH4 from the seafloor to the atmosphere.
Extrapolation of rate measurements from active seafloor seeps across areas of likely seepage suggests that global CH4 ebullition from continental shelf sediments (0–200 m) likely falls between 18 and 48 Tg yr−1 9,35, with a most likely rate of ~35 Tg yr−1 8,36. Due to its rapid diffusion from bubbles, the fraction of this CH4 that reaches the atmosphere is governed by the release depth and size-dependent rise velocity of bubbles, and is estimated here using a numerical bubble model that has been validated against observations10 (see the “Methods” section). Recent observations from high-resolution imaging37 show that the vast majority of bubbles escaping seafloor sediments (~99% by volume) are between 2 and 8 mm in diameter (Supplementary Fig. 7). Even the largest of these bubbles lose > 99% of their initial CH4 when rising through a 100 m water column (Fig. 5a), suggesting that seeps beyond the continental shelf7 transfer negligible CH4 to the atmosphere and can be omitted from our global estimate, which is further supported by recent isotopic constraints38.

Ebullitive and total CH4 emissions. a Modeled transfer efficiency of CH4 in bubbles from the seafloor to surface ocean, for 2 and 8 mm diameter bubbles, and integrated across a characteristic bubble size spectrum (Supplementary Fig. 7). Diamond and circle points represent the mean transfer efficiency for bubbles released uniformly between 0–100 and 0–200 m, respectively, and gray shading marks the range of 11–17% bounded by these cases. b Probability density functions for total oceanic CH4 emissions, combining the distribution for diffusive fluxes (Fig. 4b) with two uniform probability distributions for ebullitive emissions that are obtained by applying 11–17% transfer efficiency to seafloor ebullition rates of 35 and 18–48 Tg yr−1. Dark and light gray shading mark the likely range (10–90th percentiles) for the two estimates
Integrated across a representative bubble size spectrum with a volume-weighted mean diameter of ~4 mm (Supplementary Fig. 7)37, CH4 transfer to the atmosphere decreases rapidly as a function of release depth, even in water columns tens of meters deep (Fig. 5a). The distribution of seeps across the continental shelf is therefore an important determinant of ebullitive emissions, but remains poorly constrained9. Based on a compilation of shelf seep locations35, we consider two limiting scenarios (see the “Methods” section): one in which seeps are uniformly distributed between 0 and 200 m and another in which seeps are confined to waters shallower than 100 m, in which 11% and 17% of the ebullitive CH4 flux is transferred to the atmosphere, respectively (see the “Methods” section).
Applying a transfer efficiency range of 11–17% to seafloor ebullition rates of 35 or 18–48 Tg yr−1, we estimate global ebullitive emissions of 4–6 or 2–8 Tg yr−1 respectively, which overlap a previous estimate of 0.5–12 Tg yr−1 based on simpler bubble transfer assumptions39,40. Combined with our probability distributions for diffusive fluxes (Fig. 4b), this implies that the global ocean likely emits 7–11 or 6–12 Tg yr−1 of CH4 to the atmosphere (10–90th percentile range, Fig. 5b, see the “Methods” section), depending on the degree of uncertainty in seafloor ebullition rates. Even the broader estimate of 6–12 Tg yr−1 constrains oceanic emissions towards the lower end of the range incorporated in previous atmospheric budgets (5–25 Tg yr−1) 4. The previous range incorporates assumptions and extrapolations that have not been updated in many years41, and can be replaced by our new robust estimate in future appraisals. In part, this will help close the gap between bottom-up estimates of natural CH4 emissions, and the lower rates implied by top-down atmospheric constraints4.
Discussion
While our machine-learning models cannot directly constrain the origins of CH4 in the surface ocean, the large-scale distribution of ∆CH4 they infer may provide useful insights into production mechanisms. We employed a correlation analysis (see the “Methods” section) to determine which of our set of physical and biogeochemical predictor variables most closely approximates the ensemble-mean distribution of ΔCH4 mapped by our machine learning models (Supplementary Table 2 and Fig. 6).

Controls on surface ocean ∆CH4. a Joint probability distribution for mapped ∆CH4 and seafloor depth (zsf) in coastal ocean regions (<2000 m depth). Color scale represents the frequency of gridcells with a given combination of log10(depth) and log10(∆CH4), after averaging together all 200,000 machine-learning maps. Black line is the best fit for the mapped data (∆CH4 = 67zsf−0.7, R2 = 0.55). b Scatter plot of observed ∆CH4 versus depth. Gray points show raw data; black circles with errorbars show mean ± s.d. ∆CH4 within depth bins. Red line is best fit to the binned data (CH4 = 69zsf−0.8, R2 =0.94). c, d Same as a and b, but for the relationship of mapped c and observed d ∆CH4 to net primary production (NPP) in open ocean (>2000 m depth) environments. In c, black line is the best fit for mapped data (∆CH4 = (0.5NPP − 62)/103, R2 = 0.30), and symbols represent large-scale averages (Supplementary Fig. S8). In d, black circles show mean ± s.d. ∆CH4 within NPP bins, and red line is best fit to the binned data (∆CH4 = (0.3NPP + 14)/103, R2 = 0.91)
In coastal ocean regions (<2000 m) where ΔCH4 spans orders of magnitude, log10(ΔCH4) correlates strongly with seafloor depth (zsf, R2 = 0.37), whereas other predictor variables can explain at most ~10% of its spatial variance (Supplementary Table 2). The correlation is further strengthened against log10(zsf) (R2 = 0.55), indicating that the first-order pattern identified by our machine-learning models is a decline in ΔCH4 away from coastlines following a power-law relationship: ΔCH4 = 67zsf−0.7. A similar relationship can be derived directly from the raw dataset used to train our models (Fig. 6b), and the same qualitative pattern is apparent in observations across the shelf at individual locations22. The strong dependence of ΔCH4 on depth reflects the important role of the seafloor as a CH4 source to the surface ocean in coastal regions, supplied by rising gas bubbles that dissolve within meters of the seafloor (Fig. 5a), or by diffusion from anoxic sediments followed by transport to the surface. In the latter case, bathymetry controls both the rain rate of organic carbon that fuels anaerobic metabolism in sediments8, and the mixing timescale between bottom waters and the surface. The lack of strong relationships with other predictor variables suggests that the environmental controls of seafloor CH4 sources are complex and vary significantly between regions.
Beyond the continental slope (>2000 m), the more subtle open-ocean gradients in ∆CH4 no longer resemble bathymetry (R2 = 2 × 10−5), and the almost ubiquitous CH4 supersaturation implies in situ production in the water column rather than transfer from the sediments8. Without such a source, rapid CH4 oxidation in the marine environment should leave surface waters undersaturated, driving ingassing from the atmosphere. We only find this condition in the Southern Ocean (Fig. 2), where extensive upwelling supplies CH4-depleted deep water to the surface, and in the central Arctic Ocean, where ice cover mostly prevents air–sea exchange (Supplementary Fig. 4). The predictor variable that most closely approximates ensemble-mean ∆CH4 in the open ocean is net primary production (NPP), as determined from a carbon-based satellite algorithm42. The two are positively correlated and NPP explains ~30% of the variance in ∆CH4, and ~95% of its large-scale latitudinal pattern, which is highest in the tropics and lowest in polar oceans, with subtropical and subpolar regions falling between (Fig. 6c). A similar although somewhat weaker correlation to NPP emerges from our raw ∆CH4 database (Fig. 6d), demonstrating that this relationship is not generated artificially during the mapping procedure.
Methane production has been reported during growth of coccolithophores13 and other ubiquitous members of the prymnesiophyte class of marine phytoplankton43, which may contribute in part to the correlation we find between ∆CH4 and NPP. However, a number of alternative pathways have been proposed for methanogenesis in surface ocean waters, which could give rise to the relationship indirectly. CH4 may be released from sinking organic aggregates that harbor anoxic microzones suitable for methanogensis44, but this should result in a stronger relationship of ∆CH4 to particulate organic carbon (POC) flux than to NPP, which is not borne out in our analysis (R2 = 0.14, Supplementary Table 2). Similarly, CH4 may be produced in the anoxic digestive tracts of zooplankton and egested to the watercolumn at potentially significant rates14. Because zooplankton biomass and productivity scales with NPP45, this mechanism is broadly consistent with the surface distribution of ∆CH4.
In addition, two aerobic pathways have been identified for methanogenesis during the microbial cycling of dissolved organic matter (DOM) compounds, which are ultimately a product of phytoplankton growth (i.e. NPP). First, microbial transformations of dimethylsulfide (DMS) are thought to yield CH4 (ref. 46), but we find only a weak correlation between DMS and ∆CH4 (Supplementary Table 2), suggesting this is not an important pathway at the global scale. Second, CH4 is produced by the degradation of methylphosphonate12 (MPn)—an important constituent of the surface DOM inventory47—especially under phosphate (PO4) limited conditions. We find that a multiple linear regression model combining a positive relationship to NPP and a negative relationship to [PO4] explains surface ∆CH4 significantly better than NPP alone (∆CH4 = 5 × 10−3 NPP–0.1[PO4]–0.03, R2 = 0.35). This relationship is consistent with timeseries evidence for coincident variations in ∆CH4 and [PO4] in the North Pacific Ocean while NPP remained constant48, and supports an important role for MPn cycling as a CH4 source.
Ultimately, a combination of pathways may control the open ocean surface ∆CH4 distribution and contribute to its correlation with NPP. Methanogenesis by phytoplankton and in zooplankton guts may dominate in productive ocean regions, with MPn becoming the dominant pathway in oligotrophic regions, where PO4 stress acts as the driving variable by selecting for phosphonate decomposing metabolisms49. Additionally, we cannot definitively conclude that the NPP vs. CH4 relationship arises mechanistically from methanogenesis, and not from spatial variations in CH4 oxidation or the physical CH4 supply, which may also be correlated with NPP.
This work has narrowed the uncertainty range of total oceanic CH4 emissions to 6–12 Tg yr−1, providing a robust baseline to assess anthropogenic perturbations against, and contributing towards an improved accounting of the natural atmospheric methane budget. The majority of the remaining uncertainty in our estimate is attributed to shallow near-shore environments, where ∆CH4 and diffusive emissions vary most among our model ensembles (Fig. 4a), and where relatively unconstrained ebullitive fluxes are concentrated (Fig. 5a). To further refine our estimate, future observational efforts should focus on these shallow environments and sample with the resolution to capture sharp coastal gradients in ∆CH4 (ref. 22), while employing new imaging technologies37 to further constrain bubble dynamics and ebullition. Understanding and resolving interlaboratory discrepancies in [CH4] measurements28 should also be prioritized, so that consistent data may be synthesized across multiple sources.
By contrast, open ocean CH4 emissions are relatively well constrained (Fig. 4a) and are driven by ∆CH4 variations that appear systematically linked to organic matter cycling (Fig. 6). Our work supports previous hypotheses for CH4 release during phytoplankton growth, zooplankton egestion, and MPn degradation, and we encourage future work to distinguish and quantify the contributions of these process. The global relationship between ∆CH4 and NPP reported here also potentially provides a simple approach to represent open ocean emissions in coupled ocean–atmosphere models, and tentatively predict future perturbations in this source as ocean warming and stratification impact marine productivity50.
Methods
CH4 concentration database
We compiled a large database of CH4 concentration measurements from the ocean mixed layer, to form the basis of a ∆CH4 climatology that was used train machine-learning models. The majority of [CH4] data were taken from the MarinE MethanE and NiTrous Oxide (MEMENTO) Database, which has compiled published trace gas measurements from research cruises dating back to 1970 (ref. 20,21). The full dataset and references for individual data contributions can be found at https://memento.geomar.de. We downloaded the version of MEMENTO available as of June 2018, and retained only data that was collected within the mixed layer depth, as determined by interpolation from the MIMOC global mixed layer climatology51. We rejected data points that were not accompanied by temperature data, which is required to compute CH4 solubility. Data points with missing salinity data were accepted, due to its weaker effect on solubility, and salinity was filled by interpolating from the MIMOC salinity climatology51. We also rejected data collected outside the time interval 1980–2016, when atmospheric pCH4 could not be determined (see below).
We combined this subset of the MEMENTO database with other recent published [CH4] measurements from the surface ocean to expand data coverage in critical regions, mostly polar oceans and marginal seas30,32,52,53,54,55,56,57. Again, data collected below the climatological mixed layer was rejected, and missing salinity data was filled from MIMOC. Only the data from ref. 30 was accepted without accompanying temperature data, which was filled by interpolation from MIMOC. This data has previously been used to infer very large CH4 emissions from Arctic shelves and was included in our database to test this inference.
Mixed layer ∆CH4 climatology
Each mixed layer [CH4] measurement in our database was converted to CH4 disequilibrium (∆CH4) using:
In Eq. (1), \(S_{{\mathrm{CH}}_4}\) is the solubility of methane computed from temperature and salinity at each data point58, and \(p_{{\mathrm{CH}}_4}^{{\mathrm{moist}}}\) is the partial pressure of CH4 in moist air. \(p_{{\mathrm{CH}}_4}^{{\mathrm{moist}}}\) was determined by first interpolating dry-air \(p_{{\mathrm{CH}}_4}\) to the location of each ocean data point from atmospheric measurements taken in the same year and month, using ordinary kriging. Atmospheric data was taken from the NOAA Global Monitoring Division archive, which has collected flask samples from a global network of monitoring stations since 1980 (https://www.esrl.noaa.gov/gmd/ccgg/). Dry \(p_{{\mathrm{CH}}_4}\) was then converted to \(p_{{\mathrm{CH}}_4}^{{\mathrm{moist}}}\) following ref. 59.
Finally, our complete ∆CH4 database of ~120,000 observations was compiled into a monthly climatology. For each month, all data collected during that month (regardless of year) was binned onto a 0.25° × 0.25° latitude/longitude grid, and the average value for each grid cell was calculated. This step was necessary to minimize the impact of a few high-resolution cruise tracks, which contribute orders of magnitude more datapoints than others. We note that by combining data from the years 1985–2016 into a single monthly climatology, we have made the implicit assumption that ∆CH4 remains relatively constant over time, even as atmospheric \(p_{{\mathrm{CH}_{4}}}\) has increased by ~10% from ~1650 to ~1850 ppb. This assumption is supported by observations from open ocean waters in the Atlantic18,60 and Pacific17 oceans, where ∆CH4, and therefore air–sea flux, remained constant over interannual to decadal timescales while [CH4] increased in track with \(p_{{\mathrm{{CH}}_{4}}}.\) It is consistent with the view that ∆CH4 is controlled by internal sources and sinks of CH4 that maintain a disequilibrium between the ocean and atmosphere, regardless of the atmospheric mixing ratio8,18.
Machine-learning mapping
Our monthly ∆CH4 climatology was used to train an ensemble of ANN and RRF models to generate continuous, mapped climatologies. These are both machine-learning methods that exploit pattern similarities between ∆CH4 and other physical, chemical, and biological properties (termed predictor variables) whose climatological distributions are well known, to generate skillful predictive models for ∆CH4. Employing the two independent mapping methods and taking an ensemble approach allows us to propagate uncertainties introduced by the mapping process into our flux estimates.
Predictor data used in our models include: seafloor depth taken from the ETOPO2 high-resolution bathymetry (https://rda.ucar.edu/datasets/ds759.3/, available at 0.033° resolution); surface temperature and salinity from the MIMOC climatology (0.5° resolution)51; a net primary production (NPP) climatology constructed from data collected between 2002 and 2016 by a carbon-based remote-sensing algorithm (http://www.science.oregonstate.edu/ocean.productivity/, 0.25° resolution); POC export flux at the base of the euphotic zone, estimated by combining our NPP climatology with the export ratio algorithm of ref. 61 phosphate ([PO4]) in the surface ocean, taken from the World Ocean Atlas 2013 (WOA13) climatology23 (0.25° resolution); oxygen ([O2]) in shallow subsurface waters (50 m below mixed layer, or at seafloor depth if seafloor is within 50 m of mixed layer) from the WOA13 climatology; sediment gas hydrate inventory, taken from the global model of ref. 62 (1° resolution). All predictor data were interpolated from their original grids to the same 0.25° × 0.25° as the ∆CH4 climatology. We note that while we have chosen the most up-to-date global data products for use in our work, each is likely subject to its own uncertainties, and some have been subjected to their own gap-filling procedures.
Each ANN and RRF ensemble member was trained using a random subset of 70% of the dataset, leaving 30% of the data for validation. Before training, ∆CH4 was transformed using an inverse hyperbolic sine (IHS) transform, which is similar to a log transform except it is defined at negative ∆CH4. Because ∆CH4 spans more than four orders of magnitude, this transform prevents a few data points with very high ∆CH4 from dominating the training process. While the transformation is not necessary for the RRF method, it was undertaken for operational consistency between our two approaches.
Our ANN model structure is similar to that used in ref. 25, with a single hidden layer of 20 neurons (sigmoid response functions), fully connected to a single-node output layer (linear response function), and is trained using a Bayesian regularization method. The individual regression trees comprising our RRF ensemble are structured with a maximum of 100 decision splits and trained using a standard CART algorithm. The complexity of these models is chosen to maximize predictive skill while minimizing overfitting. More complex models (i.e. more neurons in the ANN or more decision splits in RRF trees) achieves a better fit to the full dataset, because the majority of that data is used in training the model. However, when the fit to validation data does not improve in tandem, it suggests the model is overfitting the training data, rather than improving its predictive power. We therefore experimented with different levels of complexity (Supplementary Fig. 1a, b), and chose the level at which the fit to validation data began to plateau.
An ensemble of 100,000 ANN and RRF models was trained for error propagation (see below). All ensemble members were able to reproduce the IHS-transformed validation data with R > 0.75, and closely matched the variance of the data (Fig. 2c) and its probability distribution in different environments (Supplementary Fig. 2). After training, each ensemble member was used to generate a 0.25° × 0.25° monthly mapped ∆CH4 climatology by applying the model to gridded climatologies of the predictor data.
Diffusive CH4 fluxes and error propagation
To estimate diffusive CH4 fluxes (Fdiff) across the air–sea interface, we applied a standard gas transfer model to our ∆CH4 climatologies:
Here, fice is the fractional sea ice cover of a grid cell, εice is the efficiency with which ice cover blocks gas exchange (1 means no exchange through ice), and k is the gas transfer velocity. A number of different empirical algorithms have been proposed relating k to wind speed at 10 m above the air–sea interface, and diverge by >20% at characteristic ocean wind speeds between 5 and 10 m s−1 (ref. 63). Additionally, a number of wind speed and ice coverage climatologies have been assembled from different methodologies, which all agree in their large-scale patterns but can differ at smaller scales.
To propagate these sources of uncertainty into our flux calculation, we used a Monte Carlo procedure in which each ∆CH4 climatology was combined in Eq. (2) with random selections between five different wind climatologies, three different sea ice climatologies, and four different empirical algorithms for k (refS. 15,64,65,66). We note that the most recent and perhaps best constrained of these algorithms15 yields k values close to the average of all four. Daily wind climatologies were obtained from the cross-calibrated multi-platform (CCMP) product67 (http://www.remss.com/measurements/ccmp/) that combines satellite and buoy data with model predictions, the QuickScat product (http://www.remss.com/missions/qscat/) from satellite scatterometry, the WindSat product (http://www.remss.com/missions/windsat/) from satellite radiometry, the ECMWF ERA-Interim product from model reanalysis (https://www.ecmwf.int/en/forecasts/datasets/reanalysis-datasets/era-interim) and the NCEP product (https://www.esrl.noaa.gov/psd/data/gridded/data.ncep.reanalysis.html) from model reanalysis. Monthly sea ice climatologies were obtained from the ECMWF ERA-Interim and NCEP reanalysis products (links above), and the HadISST product that combines in situ and satellite observations (https://catalogue.ceda.ac.uk/uuid/facafa2ae494597166217a9121a62d3c).
Flux calculations were conducted at daily resolution to limit the impact of temporal smoothing of windspeeds, given that the relationship between k and windspeed is nonlinear. Windspeed and fice climatologies were first interpolated to our 0.25° × 0.25° grid, and then monthly ∆CH4 and fice were interpolated to each day of the year before applying Eq. (2). While most estimates of air–sea gas exchange assume that ice coverage completely blocks gas exchange (εice = 1), we allow gas transfer across sea ice to occur up to 10% as fast as in ice-free water, based on radon measurements in Arctic Ocean27. Each Monte Carlo iteration therefore randomly selected from the range 0.9 < εice < 1 for application in Eq. (2).
Sensitivity tests
To inform our selection of grid resolution, we applied the full procedure outlined above using grids ranging from 2° to 0.125° in resolution (Supplementary Fig. 1). In each case, ∆CH4 data were binned into a climatology at the specified resolution, predictor variables were interpolated to the specified resolution, and an ensemble of 200 ∆CH4 and flux estimates were generated (100 each from ANN and RRF). The total global flux decreased as the grid resolution was improved, because coarser grids spread high coastal ∆CH4 values over larger areas. This trend plateaued between 0.5° and 0.25° resolution, so we selected a 0.25° grid (~25 × 25 km near equator) for our full model ensemble, to balance accuracy and computational efficiency.
To test whether selecting different biological predictor variables would impact our results, we conducted a sensitivity test in which NPP was replaced by the high-resolution MODIS chlorophyll-a (Chl) climatology (https://oceancolor.gsfc.nasa.gov/, 4 km resolution) and a new suite of 200 flux estimates was generated. The global fluxes predicted by this ensemble were not significantly different from those using NPP as the biological predictor variable. Furthermore, improving the grid resolution beyond 0.25° again had no impact on the global flux, suggesting this plateau is not dependent on predictor resolution.
We tested whether potential errors in our [CH4] database would greatly impact our results, because recent work has revealed interlaboratory discrepancies in [CH4] measurements28. Prior to generating our ∆CH4 climatology and applying our mapping methods, a synthetic database was generated by randomly selecting a [CH4] value for each datapoint in the range (1 − R.E.)[CH4]obs to (1 + R.E.)[CH4]obs, where [CH4]obs is the reported value. Measurements from individual laboratories can diverge up to 25% from the interlaboratory mean in strongly supersaturated waters and up to 50% in weakly supersaturated waters28. We therefore conducted tests with R.E. = 0.25 and R.E. = 0.5, and generated an ensemble of 200 flux estimates in each case (Supplementary Fig. 4). We find that propagating potential measurement errors does not change the ensemble-mean global diffusive flux (~4 Tg yr−1 in each case), but expands the likely range to 1.8–6.4 or 1.5–6.9 Tg yr−1 (R.E. = 0.25, 0.5 respectively).
Finally, we attempted to compare the degree of uncertainty introduced to our flux calculations by the ∆CH4 distribution and by gas transfer velocity (Supplementary Fig. 6). First, each permutation of windspeed climatologies, fice climatologies, and algorithms for k (60 permutations) was used to calculate (1 − εicefice)k, and each was applied to the same ∆CH4 climatology (average from our full ensemble). Second, the same (1 − εicefice)k climatology (average across the 60 permutations) was applied to 60 different ∆CH4 maps generated by the ANN and RRF methods. The variance across these two ensembles can be used to compare the uncertainty introduced by gas transfer velocity (first ensemble) versus ∆CH4 (second ensemble).
Ebullitive and total CH4 fluxes
We attempted to place broad bounds on ebullitive CH4 emissions from the ocean. The globally integrated ebullitive flux to the atmosphere (ΣFeb) can be estimated from:
In Eq. (3), ΣFsf is the globally integrated ebullitive flux from the seafloor to the water column, εtr denotes the transfer efficiency of the CH4 through the water column and to the atmosphere, and \(\overline {\varepsilon _{{\mathrm{tr}}}}\) represents the flux-weighted global average of εtr. We take two previous literature values of ΣFsf : the most likely flux of 35 Tg yr−1 from ref. 36 and the full range of 18–48 Tg yr−1 based on a compilation of seepage rates by ref. 9. We note that these ΣFsf estimates apply only to shelf regions between 0 and 200 m, but because εtr approaches 0 in waters beyond the shelf10,38, this is sufficient to estimate ΣFeb (flux to atmosphere).
We estimated εtr using output from a model of rising gas bubbles, which simulates the diffusive loss of CH4 to predict the fraction that reaches the surface as a function of bubble size and release depth10. Because environmental conditions have a relatively small impact on CH4 transfer in this model, we use model output generated previously under idealized conditions that is recommended for application in most marine environments10. First, we integrated this output across a characteristic volume-weighted bubble size distribution to determine εtr as a function of release depth (Fig. 5a). This size distribution is generated by combining the individual distributions from four seep sites observed recently using high-resolution imaging37. While we note that these observations are from deeper seeps than the shelf seeps we are interested in, the bubble sizes reported are consistent with older, less well-resolved observations from shelf seeps and shallow lake9,68.
To determine \(\overline {\varepsilon _{{\mathrm{tr}}}}\) we must know the depth distribution of the seafloor ebullitive flux (ΣFsf). While relatively few individual seep locations have been charted, these are widely distributed across continental shelves at depths between 0 and 200 m (ref. 35). However, some of the world’s most active seep sites are situated in waters shallower than 100 m (e.g. Santa Monica Channel, ~60 m; Norwegian North Sea, 60–80 m). Based on these observations, we use two limiting scenarios to bracket \(\overline {\varepsilon _{{\mathrm{tr}}}}\). First, to derive a lower limit, we assume that ΣFsf is uniformly distributed between 0 and 200 m, and average the depth-dependent εtr across this interval, weighted by the ocean area with each depth, yielding \(\overline {\varepsilon _{{\mathrm{tr}}}}\) = 11%. To derive an upper limit, we assume that ΣFsf is confined to regions between 0 and 100 m depth, and repeat the calculation to yield \(\overline {\varepsilon _{{\mathrm{tr}}}}\) = 17%. This range of \(\overline {\varepsilon _{{\mathrm{tr}}}}\) (11–17%) was combined with the two estimates of ΣFsf (35 and 18–48 Tg yr−1) in Eq. (3) to specify likely ranges for ΣFeb, and we assumed uniform probability within these ranges. Finally, total oceanic CH4 emissions were estimated by combining these uniform probability distributions for ΣFeb with the probability distributions derived previously for diffusive fluxes (Fig. 5b).
Analysis of ∆CH4 distribution
To evaluate which physical or biogeochemical properties drive the global distribution of methane disequilibrium in our machine-learning models, we correlated annual-mean mapped ∆CH4 (averaged across all 200,000 climatologies, Supplementary Fig. 8) against each predictor variable in turn. A climatology of DMS69 was also correlated against ∆CH4 to test hypothesized production during DMS cycling46. This analysis was conducted separately for coastal oceans (<2000 m depth) and the open ocean (>2000 m depth), given that different drivers are likely dominant in these environments8.
To compare the large-scale open-ocean patterns of NPP and ∆CH4 across latitude, both variables were averaged across polar, subpolar, subtropical, and tropical regions. In the Southern, Atlantic, and Pacific and Arctic Oceans, these regions were defined as in ref. 70, and the Indian Ocean was split into tropics and subtropics along 15°S (Supplementary Fig. 8).
Data availability
All datasets used in this work are described in the Methods section, and links are provided to the online repositories where they can be obtained. Gridded climatologies of methane disequilibrium and air-sea methane fluxes generated by this study are available at https://figshare.com/articles/ocean_ch4_nc/9034451.
Code availability
This work makes extensive use of functions from MATLAB’s Machine Learning Toolbox (https://www.mathworks.com/solutions/machine-learning.html). Custom codes for applying those functions are available upon request from the corresponding author.
References
Hartmann, D. L., Klein Tank, A. M. G., Rusticucci, M. & Alexander, L. V. in Climate Change 2013 – The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (ed. Change Intergovernmental Panel on Climate) 159–254 (Cambridge University Press, 2014).
Etminan, M., Myhre, G., Highwood, E. J. & Shine, K. P. Radiative forcing of carbon dioxide, methane, and nitrous oxide: a significant revision of the methane radiative forcing. Geophys. Res. Lett. 43, 12614–12623 (2016).
Hamdan, L. J. & Wickland, K. P. Methane emissions from oceans, coasts, and freshwater habitats: new perspectives and feedbacks on climate. Limnol. Oceanogr. 61, S3–S12 (2016).
Saunois, M. et al. The global methane budget: 2000–2012. Earth Syst. Sci. Data Discuss. 1–79, https://doi.org/10.5194/essd-2016-25 (2016).
Barnes, R. O. & Goldberg, E. D. Methane production and consumption in anoxic marine sediments. Geology 4, 297–300 (1976).
Du, M. et al. High resolution measurements of methane and carbon dioxide in surface waters over a natural seep reveal dynamics of dissolved phase air–sea flux. Environ. Sci. Technol. 48, 10165–10173 (2014).
Skarke, A., Ruppel, C., Kodis, M., Brothers, D. & Lobecker, E. Widespread methane leakage from the sea floor on the northern US Atlantic margin. Nat. Geosci. 7, 657 (2014).
Reeburgh, W. Oceanic methane biogeochemistry. Am. Chem. Soc. 107, 486–513 (2007).
Hornafius, J. S., Quigley, D. & Luyendyk, B. P. The world's most spectacular marine hydrocarbon seeps (Coal Oil Point, Santa Barbara Channel, California): quantification of emissions. J. Geophys. Res.-Oceans 104, 20703–20711 (1999).
McGinnis, D. F., Greinert, J., Artemov, Y., Beaubien, S. E. & Wüest, A. Fate of rising methane bubbles in stratified waters: How much methane reaches the atmosphere? J. Geophys. Res. 111, https://doi.org/10.1029/2005jc003183 (2006).
Leonte, M. et al. Rapid rates of aerobic methane oxidation at the feather edge of gas hydrate stability in the waters of Hudson Canyon, US Atlantic Margin. Geochim. Cosmochim. Acta 204, 375–387 (2017).
Karl, D. M. et al. Aerobic production of methane in the sea. Nat. Geosci. 1, 473–478, https://doi.org/10.1038/ngeo234 (2008).
Lenhart, K. et al. Evidence for methane production by the marine algae Emiliania huxleyi. Biogeosciences 13, 3163–3174 (2016).
Schmale, O. et al. The contribution of zooplankton to methane supersaturation in the oxygenated upper waters of the central Baltic Sea. Limnol. Oceanogr. 63, 412–430 (2018).
Wanninkhof, R. Relationship between wind speed and gas exchange over the ocean revisited. Limnol. Oceanogr. 12, 351–362 (2014).
Conrad, R. & Seiler, W. Methane and hydrogen in seawater (Atlantic Ocean). Deep Sea Res. Part A 35, 1903–1917 (1988).
Bates, T. S., Kelly, K. C., Johnson, J. E. & Gammon, R. H. A reevaluation of the open ocean source of methane to the atmosphere. J. Geophys. Res.-Atmos. 101, 6953–6961 (1996).
Rhee, T. S., Kettle, A. J. & Andreae, M. O. Methane and nitrous oxide emissions from the ocean: a reassessment using basin-wide observations in the Atlantic. J. Geophys. Res.-Atmos. 114, https://doi.org/10.1029/2008JD011662 (2009).
Bange, H. W., Bartell, U. H., Rapsomanikis, S. & Andreae, M. O. Methane in the Baltic and North Seas and a reassessment of the marine emissions of methane. Glob. Biogeochem. Cycles 8, 465–480 (1994).
Kock, A. & Bange, H. W. Counting the ocean’s greenhouse gas emissions. EOS 96, https://doi.org/10.1029/2015EO023665 (2015).
Bange, H. W. et al. MEMENTO: a proposal to develop a database of marine nitrous oxide and methane measurements. Environ. Chem. 6, 195–197 (2009).
Borges, A. V., Champenois, W., Gypens, N., Delille, B. & Harlay, J. Massive marine methane emissions from near-shore shallow coastal areas. Sci. Rep.-Uk. 6, https://doi.org/ARTN 2790810.1038/srep27908 (2016).
Garcia, H. E. et al. World Ocean Atlas 2013, Vol. 4: Dissolved Inorganic Nutrients (phosphate, nitrate, silicate). S. Levitus, Ed., A. Mishonov Technical Ed.; NOAA Atlas NESDIS 76, 25 pp. (2013).
Landschutzer, P. et al. The reinvigoration of the Southern Ocean carbon sink. Science 349, 1221–1224 (2015).
Roshan, S. & DeVries, T. Efficient dissolved organic carbon production and export in the oligotrophic ocean. Nat. Commun. 8, https://doi.org/ARTN 203610.1038/s41467-017-02227-3 (2017).
Sherwen, T. et al. A machine learning based global sea-surface iodide distribution. Earth Syst. Sci. Data Discuss. 2019, 1–40 (2019).
Rutgers van der Loeff, M. M., Cassar, N., Nicolaus, M., Rabe, B. & Stimac, I. The influence of sea ice cover on air–sea gas exchange estimated with radon-222 profiles. J. Geophys. Res.-Oceans 119, 2735–2751 (2014).
Wilson, S. T. et al. An intercomparison of oceanic methane and nitrous oxide measurements. Biogeosciences 15, 5891–5907 (2018).
Biastoch, A. et al. Rising Arctic Ocean temperatures cause gas hydrate destabilization and ocean acidification. Geophys. Res. Lett. 38, https://doi.org/10.1029/2011gl047222 (2011).
Shakhova, N. et al. Extensive methane venting to the atmosphere from sediments of the East Siberian Arctic Shelf. Science 327, 1246–1250 (2010).
Berchet, A. et al. Atmospheric constraints on the methane emissions from the East Siberian Shelf. Atmos. Chem. Phys. 16, 4147–4157 (2016).
Thornton, B. F., Geibel, M. C., Crill, P. M., Humborg, C. & Morth, C. M. Methane fluxes from the sea to the atmosphere across the Siberian shelf seas. Geophys. Res. Lett. 43, 5869–5877 (2016).
Warwick, N. J. et al. Using δ13C-CH4 and δD-CH4 to constrain Arctic methane emissions. Atmos. Chem. Phys. 16, 14891–14908 (2016).
Shakhova, N. et al. Ebullition and storm-induced methane release from the East Siberian Arctic Shelf. Nat. Geosci. 7, 64, https://doi.org/10.1038/ngeo2007 (2013).
Hovland, M., Judd, A. G. & Burke, R. A. The global flux of methane from shallow submarine sediments. Chemosphere 26, 559–578 (1993).
Kvenvolden, K. A. & Rogers, B. W. Gaia’s breath—global methane exhalations. Mar. Pet. Geol. 22, 579–590 (2005).
Wang, B., Socolofsky, S. A., Breier, J. A. & Seewald, J. S. Observations of bubbles in natural seep flares at MC 118 and GC 600 using in situ quantitative imaging. J. Geophys. Res.-Oceans 121, 2203–2230 (2016).
Leonte, M. et al. Using carbon isotope fractionation to constrain the extent of methane dissolution into the water column surrounding a natural hydrocarbon gas seep in the Northern Gulf of Mexico. Geochem. Geophys. Geosyst. 19, 4459–4475 (2018).
Judd, A. et al. Contributions to atmospheric methane by natural seepages on the UK continental shelf. Mar. Geol. 137, 165–189 (1997).
Judd, A. G. in Atmospheric Methane: Its Role in the Global Environment (ed. Mohammad Aslam Khan Khalil) 280–303 (Springer, Berlin, Heidelberg, 2000).
Ruppel, C. D. & Kessler, J. D. The interaction of climate change and methane hydrates. Rev. Geophys. 55, 126–168 (2017).
Behrenfeld, M. J., Boss, E., Siegel, D. A. & Shea, D. M. Carbon‐based ocean productivity and phytoplankton physiology from space. Glob. Biogeochem. Cycles 19, 3GB1006 (2005).
Klintzsch, T. et al. Methane production by three widespread marine phytoplankton species: release rates, precursor compounds, and relevance for the environment. Biogeosci. Discuss. 2019, 1–25 (2019).
Sasakawa, M. et al. Carbon isotopic characterization for the origin of excess methane in subsurface seawater. J. Geophys. Res. 113, https://doi.org/10.1029/2007jc004217 (2008).
Strömberg, K. H. P., Smyth, T. J., Allen, J. I., Pitois, S. & O'Brien, T. D. Estimation of global zooplankton biomass from satellite ocean colour. J. Mar. Syst. 78, 18–27 (2009).
Florez-Leiva, L., Damm, E. & Farías, L. Methane production induced by dimethylsulfide in surface water of an upwelling ecosystem. Prog. Oceanogr. 112–113, 38–48 (2013).
Repeta, D. J. et al. Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nature Geoscience, 9, https://doi.org/10.1038/NGEO2837 (2016).
Wilson, S. T., Ferrón, S. & Karl, D. M. Interannual variability of methane and nitrous oxide in the North Pacific Subtropical Gyre. Geophys. Res. Lett. 44, 9885–9892 (2017).
Sosa, O. A., Repeta, D. J., DeLong, E. F., Ashkezari, M. D. & Karl, D. M. Phosphate-limited ocean regions select for bacterial populations enriched in the carbon–phosphorus lyase pathway for phosphonate degradation. Environ. Microbiol. 21, 2402–2414 (2019).
Bopp, L. et al. Multiple stressors of ocean ecosystems in the 21st century: projections with CMIP5 models. Biogeosciences 10, 6225–6245 (2013).
Schmidtko, S., Johnson, G. C. & Lyman, J. M. MIMOC: a global monthly isopycnal upper-ocean climatology with mixed layers. J. Geophys. Res.-Oceans 118, 1658–1672 (2013).
Geprägs, P. et al. Carbon cycling fed by methane seepage at the shallow Cumberland Bay, South Georgia, sub-Antarctic. Geochem. Geophys. Geosyst. 17, 1401–1418 (2016).
Mau, S. et al. Seasonal methane accumulation and release from a gas emission site in the central North Sea. Biogeosciences 12, 5261–5276 (2015).
Mau, S. et al. Widespread methane seepage along the continental margin off Svalbard— from Bjørnøya to Kongsfjorden. Sci. Rep.-UK 7, 42997 (2017).
Steinle, L. et al. Effects of low oxygen concentrations on aerobic methane oxidation in seasonally hypoxic coastal waters. Biogeosciences 14, 1631–1645 (2017).
Kudo, K. et al. Spatial distribution of dissolved methane and its source in the western Arctic Ocean. J. Oceanogr. 74, 305–317 (2018).
Yoshikawa, C. et al. Methane sources and sinks in the subtropical South Pacific along 17°S as traced by stable isotope ratios. Chem. Geol. 382, 24–31 (2014).
Wiesenburg, D. A. & Guinasso, N. L. Jr Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and sea water. J. Chem. Eng. Data 24, 356–360 (1979).
Weiss, R. F. & Price, B. A. Nitrous oxide solubility in water and seawater. Mar. Chem. 8, 347–359 (1980).
Dlugokencky, E. J., Masarie, K. A., Lang, P. M. & Tans, P. P. Continuing decline in the growth rate of the atmospheric methane burden. Nature 393, 447–450 (1998).
Dunne, J. P., Armstrong, Ra, Gnnadesikan, A. & Sarmiento, J. L. Empirical and mechanistic models for the particle export ratio. Glob. Biogeochem. Cycles 19, 1–16 (2005).
Kretschmer, K., Biastoch, A., Rüpke, L. & Burwicz, E. Modeling the fate of methane hydrates under global warming. 610–625, https://doi.org/10.1002/2014GB005011 (2015).
Sarmiento, J. L. Ocean Biogeochemical Dynamics (Princeton University Press, 2013).
Wanninkhof, R. Relationship between wind-speed and gas-exchange over the ocean. J. Geophys. Res.-Oceans 97, 7373–7382 (1992).
Nightingale, P. D. et al. In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers. Glob. Biogeochem. Cycles 14, 373–387 (2000).
Liss, P. S. & Merlivat, L. in The Role of Air–Sea Exchange in Geochemical Cycling (ed. Patrick Buat-Ménard) 113–127 (Springer, Netherlands, 1986).
Atlas, R. et al. A cross-calibrated, multiplatform ocean surface wind velocity product for meteorological and oceanographic applications. Bull. Am. Meteorol. Soc. 92, 157–174 (2011).
Ostrovsky, I., Mcginnis, D. F., Lapidus, L. & Eckert, Quantifying gas ebullition with echosounder: the role of methane transport by bubbles in a medium-sized lake. Limnology and Oceanography: Methods 6, 105–118 (2008).
Lana, A. et al. An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Glob. Biogeochem. Cycles 25, https://doi.org/10.1029/2010gb003850 (2011).
Weber, T., Cram, J. A., Leung, S. W., DeVries, T. & Deutsch, C. Deep ocean nutrients imply large latitudinal variation in particle transfer efficiency. Proc. Natl Acad Sci USA 113, 201604414 (2016).
Acknowledgements
T.W. and N.A.W. were supported by NASA-IDS grant NNX17AK11G. A.K. was supported by the Baltic Earth joint project BONUS INTEGRAL (FKZ03F0773B). We thank all data collectors that contributed to the MEMENTO archive, and D. McGinnis for providing bubble model output. We also acknowledge the Ocean Carbon and Biogeochemistry Project Office for facilitating a workshop at Lake Arrowhead in October 2018, which facilitated interactions among the ocean methane community and greatly improved this work.
Author information
Authors and Affiliations
Contributions
T.W. designed the study. A.K. and N.A.W. assembled the methane supersaturation dataset. N.A.W. and T.W. developed the statistical mapping method and generated methane emissions estimates. T.W. wrote the paper with input from N.A.W. and A.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Alberto Borges, David Karl and other, anonymous, reviewer(s) for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weber, T., Wiseman, N.A. & Kock, A. Global ocean methane emissions dominated by shallow coastal waters. Nat Commun 10, 4584 (2019). https://doi.org/10.1038/s41467-019-12541-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-12541-7
This article is cited by
-
Coastal sink outpaces open ocean
Nature Climate Change (2024)
-
Methane emissions decreased in fossil fuel exploitation and sustainably increased in microbial source sectors during 1990–2020
Communications Earth & Environment (2024)
-
Effects of pockmark activity on iron cycling and mineral composition in continental shelf sediments (southern Baltic Sea)
Biogeochemistry (2024)
-
Methane emissions offset atmospheric carbon dioxide uptake in coastal macroalgae, mixed vegetation and sediment ecosystems
Nature Communications (2023)
-
Gravity complexes as a focus of seafloor fluid seepage: the Rio Grande Cone, SE Brazil
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
