
Abstract
While cobalt-based catalysts have been used in industrial Fischer-Tropsch synthesis for decades, little is known about how the dynamics of the Co-Co2C phase transformation drive their performance. Here we report on the occurrence of hysteresis effects in the Fischer-Tropsch reaction over potassium promoted Co/MnOx catalyst. Both the reaction rate and the selectivity to chain-lengthened paraffins and terminally functionalized products (aldehydes, alcohols, olefins) show bistability when varying the hydrogen/carbon monoxide partial pressures back and forth from overall reducing to carbidizing conditions. While the carbon monoxide conversion and the selectivity to functionalized products follow clockwise hysteresis, the selectivity to paraffins shows counter-clockwise behavior. In situ X-ray diffraction demonstrates the activity/selectivity bistability to be driven by a Co-Co2C phase transformation. The conclusions are supported by High Resolution Transmission Electron Microscopy which identifies the Co-Co2C transformation, Mn5O8 layered topologies at low H2/CO partial pressure ratios, and MnO at high such ratios.
Similar content being viewed by others
Introduction
Catalytic reactions occur far from equilibrium. Consequently, multiple steady states may be observed along with kinetic hysteresis effects. Both clockwise and counter-clockwise dynamic hysteresis may occur upon variation of critical reaction parameters. The reason for the occurrence of hysteretic response behavior can frequently be related to competitive and asymmetric reactant adsorption or, more generally, to inhibition effects following, for example, concentration-dependent catalyst phase transitions. While considerable foundational knowledge has been acquired over the past three decades on kinetic non-linearities of seemingly simple catalytic surface reactions like the CO oxidation or NOx reduction1, much less information is presently available for complex reaction networks with varying product selectivity. An excellent example is the CO hydrogenation according to Fischer–Tropsch (F–T), which may lead to the production of chain-lengthened hydrocarbons and terminally functionalized oxygenates over Co-based and Fe-based catalysts2,3.
Historically, one of the earliest observations of a F–T reaction-induced chemical phase transition was made for Fe-based catalysts4, which were found to transform into carbide structures through pre-carbidization of iron oxide with CO or syngas5 or, more recently, during the ongoing F–T reaction using Fe(0)6,7,8 as activated precursor. By contrast, bulk cobalt carbides were commonly not observed, and therefore not considered as being relevant for the construction of the catalytically active phase9,10,11,12,13,14,15,16. Moreover, bulk cobalt carbides were actually considered thermodynamically metastable since they decomposed under inert atmosphere and in the presence of H2 at elevated temperatures17,18. Mohandas et al.15 and Claeys et al.19 observed that, under F–T reaction conditions, as-prepared Co2C can be converted into metallic cobalt at temperatures above 220 °C. Nevertheless, the formation of Co2C was occasionally observed through exposure of reduced cobalt to pure CO and found to be strongly dependent on the choice of the support material and time-on-stream in syngas16,20. More recently, Paterson et al. reported a nice demonstration of Ostwald’s Stufenregel (“rule of stages”) when transforming Co3O4 spinel stepwise into hexagonal Co2C by exposure to CO21. Particular attention received on Co2C formation during F–T synthesis over Co-based catalysts promoted by Mn and/or alkali22,23,24,25,26,27. Such catalysts demonstrated a remarkable capacity in producing terminally functionalized products, such as n-aldehydes22, n-alcohols25,28,29, or olefins23,24,26,27. The promoting effect of Mn on the formation of Co2C during the F–T reaction was recently investigated by Ding et al.25. The authors suggested that Mn facilitates the dissociation and disproportionation of CO and suppresses H2 adsorption on the catalyst surface, thereby creating a relatively C-rich and H-lean surface chemical environment. Similar arguments were previously advanced by Johnson et al.30,31 to explain the high selectivity of C5+ products and olefins, at the expense of methane. Overall, however, little is known about how the kinetics of the Co–Co2C phase transformation drive both reaction rate and selectivity.
Here, we show kinetic hysteresis in the F–T reaction and identify compositional changes of the catalyst to drive this hysteresis in the absence of thermokinetic effects. Using a potassium-promoted Co/MnOx catalyst with Co4Mn1K0.1 metal atomic composition, which was previously shown to produce more than 60 wt% n-aldehydes under H2-lean conditions of the F–T reaction22, we will demonstrate the occurrence of rate and selectivity hysteresis while cycling the syngas composition from high PH2/PCO to low and back to high such ratio. Two steady states with distinct rate-selectivity signatures are produced this way for a unique point in the parameter space; only the direction of the PH2/PCO variation matters when approaching such points within the bistability regime. This history dependence of the system will be shown to be exclusively dependent on and driven by the kinetic Co–Co2C phase transition.
Results
Catalytic results
We first turn to our catalytic results, which are shown in Fig. 1. Obviously, both the CO conversion and the selectivity depend on whether the PH2/PCO ratio is increased or decreased. Each data point in Fig. 1 represents quasi-steady-state behavior after ~12 h time-on-stream. We also note that three samples of the same batch of Co4Mn1K0.1 catalyst are being investigated while varying the PH2/PCO ratio. The results of these measurements fit into the same Fig. 1 without difficulties. With regard to the catalytic activity, the CO conversion (panel a) obviously shows clockwise hysteresis, i.e., a low reactivity state (LRS) is followed when decreasing the PH2/PCO ratio from initially high (30/1) to low and, vice versa, a high-reactivity state (HRS) is followed when increasing this ratio back to high again. In particular, while the CO conversion is up to 20%, at the most (at PH2/PCO = 30), and less than 10% for 5 ≤ PH2/PCO ≤ 10, on the LRS branch, it increases significantly to 75% at PH2/PCO = 9 and up to 90% at PH2/PCO = 30 on the HRS branch. The bifurcation between LRS and HRS seems to occur at PH2/PCO ~3. The hysteresis loop finally closes by running the catalyst under pure H2, i.e., removing CO from the reaction mixture at 220 °C and 40 bar (indicated by a green dashed line in Fig. 1). A similar hysteresis distinguishing the low reactivity from the high-reactivity branch is obtained when plotting the specific reaction rate rather than conversion (Supplementary Fig. 1). The slight misfit between “start” and “end” in both conversion and rate hysteresis (as well as the selectivity hysteresis discussed below) is probably associated with irreversible changes of the Co metallic phase which is mainly fcc besides less hcp when starting and rather equally distributed between hcp and fcc when finishing the experiments (see below for XRD and HRTEM results). We note that the transition from H2/CO = 30 to pure H2 (rather than systematically establishing higher such ratios) has technical reasons related to the back pressure control at very low CO gas pressures.

Kinetic hysteresis in the catalytic CO hydrogenation. a CO conversion; b–e selectivities of aldehydes, alcohols, paraffins and olefins, respectively (ex CO2); f CO2 selectivity. The same batch of catalyst is tested independently three times (circle), (square), and (triangle) over 0.5 g Co4Mn1K0.1 at 220 °C, 40 bar, and GHSV = 3600 h−1. During each run, the PH2/PCO ratio is varied systematically as follows: (circle) 30/1 → 7/1 → 3/1 → 1/1 → 7/1 → 30/1 → 30/1 (after H2 treatment for 8 h: green dashed line), (square) 7/1 → 5/1 → 1.5/1 → 5/1 → 7/1 → 9/1, (triangle) 0.5/1 → 1.5/1 → 3/1 → 5/1. Each data point reflects quasi-steady-state conditions after 12 h time-on-stream
While CO conversion follows clockwise hysteresis, both clockwise and counter-clockwise hysteresis are observed for selectivity. Most notable hysteresis loops occur for paraffins and alcohols. Starting with essentially 100% selectivity (of which 70% are allotted to methane, see Supplementary Fig. 2a for methane selectivity hysteresis, at PH2/PCO = 30/1), the yield in paraffins drops significantly at PH2/PCO ≤ 5/1. Under partial pressure conditions typically applied in F–T synthesis (1 ≤ PH2/PCO ≤ 3), paraffin selectivities are between 30 and ~40%. For under-stoichiometric reaction conditions, PH2/PCO = 0.5, only 20% of paraffins are detected. When increasing the PH2/PCO ratios from low to high, paraffin selectivities set out on their way back, but remain considerably lower than those of the forward run. A (nearly) closed hysteresis loop is obtained when subjecting the catalyst to pure H2 conditions. Thus, compared with the clockwise hysteresis of the CO conversion, an overall counter-clockwise behavior is seen for the paraffin selectivity. Interestingly, terminal alcohols and total oxygenates (see Supplementary Fig. 2b for total oxygenates selectivity hysteresis) run counter paraffins, i.e., show clockwise hysteresis: while alcohol selectivities are low for initially high PH2/PCO ratios, they are significantly higher when increasing these ratios from low to high. A maximum alcohol production of ~45% is found for PH2/PCO = 5. Larger such ratios cause a gradual decline of alcohols.
Turning to aldehydes and olefins, we note that both products can be easily hydrogenated into alcohols and paraffins, respectively, under hydrogen-rich conditions. Therefore, the hysteresis loops for aldehydes and olefins are much less distinct and absent for PH2/PCO ≥ 5. Remarkably, while neither aldehydes nor olefins are ever produced for PH2/PCO > 5, overall 60% of aldehydes and 15% of olefins (ex CO2) are obtained at PH2/PCO = 0.5. Under these conditions, methane formation is low (~5%), see Supplementary Fig. 2a. More detailed product distributions along the LRS and HRS branches under the same reaction conditions can be found in Supplementary Fig. 3.
Finally, CO2 formation also shows significant clockwise hysteresis. For very high partial pressure ratios, PH2/PCO > 9, no CO2 at all is formed. The highest CO2 selectivites occur at low such ratios, but remain below ~13% all the time. The observed hysteresis suggests CO2 formation to involve similar surface precursors as hydrocarbons and oxygenates rather than originating from the water–gas shift reaction, CO + H2O ↔ CO2 + H2.
Hysteresis effects as reported in this study raise the question for the intrinsic kinetics by which the steady states of the reaction are approached. We therefore measured the time dependence of changes in activity and product selectivity after switching the partial pressure ratios. One such measurement, carried out for a switch in PH2/PCO from 5/1 to 1.5/1, is provided in Supplementary Fig. 4. The data clearly show a nonlinear response behavior consisting in moderate changes of the product selectivity during the first 5 h time-on-stream (TOS) followed by a rapid drop of paraffins and raise of olefins (from < 5 to 40%) and oxygenates (from < 5 to 20%) within a short period of time. For 6 ≤ TOS ≤ 12, oxygenates further increase to reach a selectivity of ~50% at the expense of both olefins and paraffins; close-to-steady state behavior for all product classes is obtained after ~12 h.
Product distributions in Fig. 1 have also been evaluated in terms of chain lengthening probabilities. Anderson–Schulz–Flory (ASF) plots have been constructed and are compiled in Fig. 2. Accordingly, a linear ASF behavior with unique chain lengthening probability α = 0.5 is obtained for C4+ product distributions on both the LRS and HRS branch. However, significant deviations from the linear ASF behavior are seen for C1–C3 on the low rate branch. These deviations seem to be general; they also occur at pressure ratios other than PH2/PCO = 5 and are probably associated with variations in the composition of the “most abundant surface (reaction) intermediate (“masi” or “mari”).

ASF distribution for low and high-reactivity hysteresis branches. Obtained at a H2/CO partial pressure ratio of 5/1 and otherwise identical reaction conditions as in Fig. 1. Wn stands for the mass fraction of all products containing n carbon atoms: Wn = Wn, paraffins + Wn, olefins + Wn, alcohols + Wn, aldehydes
Structural characterization
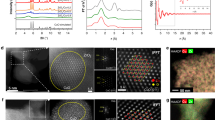
To demonstrate that changes in the structure and composition of the catalyst correlate with the occurrence of kinetic hysteresis effects as described above, we have performed X-ray diffraction (XRD) experiments. To ensure this correlation also applies under in-operando (atmospheric) conditions, some of the XRD experiments have been made while following the catalytic production on-line. The general trend of hysteretic reaction behaviors has been reproduced this way. In Fig. 3, we focus on in situ XRD experiments to systematically follow the bulk Co composition for varying PH2/PCO ratios. As seen in this Fig. (and also in Supplementary Fig. 5), only pure metallic cobalt phases (mainly represented by the (111) plane of Co fcc at 2θ ~44.3°, JCPDS 15–0806) are observed for virgin Co4Mn1K0.1 after treatment in pure H2. Almost identical diffraction patterns occur after exposing the catalyst to H2-rich syngas (PH2/PCO = 30/1) for 10 h. While metal diffraction remains the main phase when increasing the CO partial pressure to adjust PH2/PCO = 9/1 and 5/1, trace amounts of Co2C diffraction are observed at 2θ ~42.5° (JCPDS 05–0704). Interestingly, metallic cobalt diffraction fades away when further increasing the CO partial pressure to reach PH2/PCO = 3/1; after 8 h time-on-stream at this pressure ratio hardly any metallic Co diffraction persists (for a detailed evolution of cobalt and Co2C diffraction lines as a function of time-on-stream see Supplementary Fig. 5). The diffraction lines (blue) centered at 2θ of 37.1, 41.4, 42.5, and 45.8° correspond to the (011), (020), (111), and (210) planes of Co2C (JCPDS 05–0704). For a partial pressure ratio PH2/PCO = 1/1, Co2C is definitely the only diffraction phase left. Strikingly, when decreasing the CO partial pressure from high back to low again (PH2/PCO = 9/1 and 30/1) no change in the diffraction pattern is encountered. We therefore conclude that the back transformation of the Co2C phase to metallic Co is slow under H2-rich syngas conditions. However, after exposing the catalyst to pure H2 at 220 °C for 2 h, both fcc and hcp cobalt phases are retrieved. This behavior is reminiscent of reports on the Co2C metastability in the presence of H218 and the decomposition of Co2C to hcp cobalt and graphite32. On the other hand, cobalt carbide formation under H2-rich (H2/CO = 9) F–T synthesis conditions was reported by Ducreux et al.33. Different from these earlier reports, our measurements clearly establish a hysteresis effect in the Co–Co2C reversible transition. Closed hysteresis loops involving the complete back transformation of Co2C into metallic Co are obtained after treatment in pure H2 (or at very high PH2/PCO ratios >30).

In situ XRD of a Co4Mn1K0.1 catalyst. The results refer to a reaction temperature of 220 °C and atmospheric pressure. The same batch of catalyst was sequentially (from low to up) exposed to pure H2 or syngas (at various pCO/pH2) for different times-on-stream (TOS), see also Fig. S5. Red peaks indicate fcc cobalt (111), blue and green peaks are due to Co2C and hcp cobalt, respectively. Orange peaks at 2θ~41.3° are either Co2C (020) or hcp Co
The correlation of a reversible chemical reconstruction of Co particles and the catalytic performance in terms of activity and selectivity ultimately lead to a remarkable hysteresis behavior, in which Co metallic phases correlate with high paraffin and Co2C with high oxygenate/olefin selectivity. However, questions as to the influence of the MnOx phase as a dispersant and/or promoter persist and will be tackled by high-resolution transmission electron microscopy (HRTEM), along with energy-dispersive X-ray spectroscopy (EDS). To allow an as close as possible correlation of the catalytic test procedures preceding HRTEM/EDS measurements with those leading to kinetic hysteresis effects as shown in Fig. 1, we have investigated the same batch of Co4Mn1K0.1 catalyst after reaction with PH2/PCO of 30 (Fig. 4a–c) and 1.5 (panels (d–f) of the same Fig.) for 12 h using a fresh charge of catalyst in both cases. To mimic possible “memory” effects in the chemical composition of the catalyst while changing the PH2/PCO ratio from low to high, we have subjected another sample charge to low such ratios (PH2/PCO = 1.5) before adjusting the end-of-the-loop value PH2/PCO = 30. As can be seen in Fig. 4, both metallic and oxidic phases undergo significant structural and chemical alterations while varying the PH2/PCO ratio. The upper panel demonstrates Co metal particles to be mainly present in fcc and (to somewhat lesser extent) hcp phases under hydrogen-rich reaction conditions. It is interesting to note that bulk XRD has allowed identifying fcc Co rather than hcp, see Fig. 2. Running the catalytic reaction under CO-rich conditions, PH2/PCO = 1.5 (Fig. 4d–f), causes metallic Co to entirely transform into Co2C and MnOx to develop lath-like Mn5O8 (Mn(II)2Mn(IV)3O8). The occurrence of the latter is in agreement with our previous studies under similar reaction conditions22. Most surprisingly, when moving from low PH2/PCO ratios back to higher ones (Fig. 4g–i), major amounts of Co2C persist these strongly reducing conditions while Mn5O8 decomposes to develop smaller MnO aggregates with cubic rock-salt structure. Metallic Co phases reappear very slowly in mainly hcp and lesser fcc crystal structure at PH2/PCO = 30.

HRTEM images along with EDS chemical mapping. The TEM image of the Co4Mn1K0.1 catalyst was obtained after CO hydrogenation at 220 °C, Ptotal = 40 bar. The partial pressure ratio is cycled between a–c: H2/CO = 30, d–f: H2/CO = 1.5, and g–i: back to H2/CO = 30. Sample history and time-on-stream (TOS): a–c fresh catalyst after 12 h TOS, d–f: fresh catalyst after 24 h TOS, g–i: catalyst subjected to H2/CO = 1.5 for 12 h first (as in d–f), then to successively increasing H2/CO ratios up to 30 (for another 12 h TOS). Scale bar for HAADF 50 nm, scale bar for HRTEM 1 nm
Discussion
We have shown in this communication that prominent structural changes of a potassium-promoted Co/MnOx catalyst occur during the Fischer–Tropsch reaction. The systematic variation of the H2/CO partial pressure ratios has allowed us to follow the reversible Co–Co2C bulk phase transformation as a function of time using in situ XRD, complemented by representative ex situ HRTEM studies. Once being formed at low such ratios, Co2C has proven to be rather perseverant with regard to its reduction to metallic Co. This observation, which is not in accordance with earlier reports claiming the Co2C phase to be “metastable”17,18, seems to be a key factor responsible for the activity–selectivity hysteresis of the CO hydrogenation. Ignoring in a first approximation the influence of the MnOx phase and that of the potassium promoter, it seems that the occurrence of Co2C provides a positive feedback on chain-lengthened oxygenates and olefins formation. It is remarkable that at stoichiometric (PH2/PCO = 2/1) as well as under-stoichiometric reaction conditions the combined selectivities of functionalized hydrocarbons range between ~70 and ~83%, at rather low CO conversion, while under H2-rich conditions, after entering the high-reactivity hysteresis branch, the product spectrum simplifies to ~35% 1-alcohols and 65% straight paraffins at a CO conversion of nearly 75% (PH2/PCO = 9/1).
In a previous study with Mn(II)-promoted Co/SiO2, Johnson et al.30,31 considered the high selectivity of C5+ formation with their catalyst as being due to an increase of COad concentrations, i.e., a decrease of the Had/COad ratios, following the strong Lewis acid–base interaction between Mn(II) and COad. While the present paper provides clear evidence for the partial pressure ratio PH2/PCO to have a profound influence on the selectivity of the reaction, the amounts of Mn(II) (in MnO) or Mn(II)–Mn(IV) (in Mn5O8) employed in our study are far beyond the low Mn(II) promoter concentrations in Co/SiO2. To the best of our knowledge, neither Co/SiO2 nor Co/Al2O3 catalysts have ever been reported to produce significant amounts of chain-lengthened oxygenates during CO hydrogenation. Such catalysts have also not been observed to undergo reaction-induced structural changes involving a Co–Co2C bulk phase transition. We therefore conclude, in agreement with others23,24,25,26,27, that this phase transition is MnOx mediated. Different from Si(IV) or Al(III), the valence fluctionality between Mn(II) and Mn(IV) seems to play an important role in the bulk transformation of Co into Co2C and is therefore key to understanding the rich variety of functionalized products in the CO hydrogenation over CoMn-based catalysts.
The significant activity–selectivity hysteresis reported in this communication may raise the question for the possible occurrence of oscillatory behavior. So far, we have not yet observed any. If such periodic change in activity and selectivity occurs for the present system, under isothermal conditions and in the absence of mass transport limitations, we would anticipate a rather long-period behavior due to the slowness of the Co–Co2C phase transition (speculating that this process would provide a suitable feedback for autonomous oscillations). Long-period oscillations are not unusual34 and were, for example, detected for the NO/hydrogen reaction over Rh single-crystal surfaces35. In this case, the reversible oxidation of the Rh surface region was identified to provide the necessary feedback. A recent paper by Suchorski et al.36 reported intriguing results on the occurrence of multifrequential oscillations in the O2–H2 reaction over Rh and demonstrated the importance of reversible subsurface oxygen diffusion.
We finally note that nonisothermal self-sustained rate oscillations in the F–T reaction were previously reported by Tsotsis et al.37. Both the reactor design as well as the catalyst system (Fe/ZSM-5) were very different from ours, though. Due to the exothermicity of the reaction, the authors observed peak-to-peak amplitudes of about 150 °C in the reactor temperature. Such large jumps in temperature are clearly absent under our experimental conditions. The discovery of rate-selectivity hysteresis, as reported in this paper, opens new avenues to tune the catalytic performance of the Co-based Fischer–Tropsch reaction. We anticipate bistability not to be limited to potassium-promoted Co/MnOx catalysts but to apply to other systems with different selectivity signatures as well.
Methods
Catalyst preparation
Co4Mn1K0.1 catalyst was prepared via oxalate co-precipitation followed by thermal activation. To prepare the Co–Mn–-K mixed oxalate precursor, first a solution of both Co(NO3)2.6H2O and Mn(NO3)2.4H2O in acetone (100 ml), an aqueous solution of KNO3 (5 ml), and an acetone solution of H2C2O4. 2H2O (150 ml) were prepared in three separated beakers. Then the mixed acetone solution of Co(NO3)2.6H2O and Mn(NO3)2.4H2O together with the aqueous solution of KNO3 were added fast and simultaneously, under vigorous stirring, to the solution of H2C2O4. 2H2O. Stirring was kept for at least 5 min until the color of the precipitates appeared homogeneous. Then the slurries were kept overnight for aging. After removal of the supernatant acetone, the precipitate was centrifuged and dried overnight at 110 °C. The obtained oxalate precursor was then treated thermally in the presence of hydrogen through stripping of CO2 under H2 (30 ml min−1) at 370 °C for 1 h. After activation, the BET surface area of the catalyst was ~50 m2/g.
Catalyst characterization
X-ray diffraction (XRD) patterns of the catalysts were collected with a Cu Kα source using a Rigaku Miniflex-600 X-ray diffractometer operating at 40 mA and 35 kV in the continuous-scan mode with steps of 1 degree/min in a wide 2θ angle range from 20 to 80°.High resolution Transmission electron microscopy (TEM) studies were performed with aberration corrected FEI Titan 80-300 operated at 300 kV. The instrument is equipped with a CEOS GmbH double-hexapole aberration corrector for the probe forming lens, which allows angstrom level resolution in scanning imaging modes. The present observations were performed in scanning mode using an HAADF detector. The probe convergence angle was 18 mrad, with an inner collection angle of 52 mrad. The EDS maps were collected with an aberration corrected Scanning Transmission Electron Microscope (STEM) (JEOL-ARM200F) operated at 200 kV. The instrument is equipped with a 100mm2 Silicon Drift Detector (~0.7srad, JEOL Centurio), allowing for high efficiency Energy Dispersive X-ray Spectroscopy. Acquisition and evaluation of the spectra was performed by NSS Thermo Scientific software package. In general, the STEM sample preparation involved mounting powder samples on copper grids covered with lacey carbon support films, and then immediately loading them into the TEM airlock.
Catalytic testing
High-pressure catalytic tests were performed in a fixed-bed plug-flow reactor (Φinner = 7 mm). Typically, 1.3 g of oxalate were diluted with up to 2 g of SiC to achieve isothermal plug-flow conditions followed by in situ TPDec in H2 at 0.1 MPa (30 ml min−1) and 370 °C for 1 h (after oxalate decomposition, the amount of activated catalyst is around 0.5 g). The reactor was subsequently cooled to temperatures below 100 °C in flowing hydrogen before adding CO to produce a syngas feed with a specific H2/CO pressure ratio. Metal carbonyls (mainly Ni(CO)4) were removed by passing the CO feed through a zeolite 4A trap at high temperature before introduction into the reactor. The total flow rates (H2 + CO) were fixed at 40 ml min−1 providing GHSV = 3600 h−1 (Gas Hourly Space Velocity). After pressurizing the system to 40 bar, the temperature for the catalytic tests was raised using low heating rates of 1 °C min−1 to 220 °C. Catalytic activities and product selectivities were determined after stabilization for at least 12 h, and measured by an online GC-MS (Agilent 7890A GC/5975 MS).
Data availability
Any data that support the plots and calculations within this paper as well as other findings of this study can be available from the corresponding author upon reasonable request.
References
Imbihl, R. & Ertl, G. Oscillatory kinetics in heterogeneous catalysis. Chem. Rev. 95, 697–733 (1995).
Fischer, F. & Tropsch, H. Über die Herstellung synthetischer Ölgemische (Synthol) durch Aufbau aus Kohlenoxyd und Wasserstoff. Brennst. Chem. 4, 276–285 (1923).
Fischer, F. & Tropsch, H. Die Erdölsynthese bei gewöhnlichem Druck aus den Vergasungsprodukten der Kohle. Brennst. Chem. 7, 97–104 (1926).
Bahr, H. A. & Jessen, V. Die Kohlenoxyd-Spaltung am Kobalt. Ber. Dtsch Chem. Ges. 63, 2226–2237 (1930).
Bukur, D. B. et al. Activation studies with a promoted precipitated iron Fischer-Tropsch catalyst. Ind. Eng. Chem. Res. 28, 1130–1140 (1989).
Amelse, J. A., Butt, J. B. & Schwartz, L. H. Carburization of supported iron synthesis catalysts. J. Phys. Chem. 82, 558–563 (1978).
Niemantsverdriet, J. W., Van der Kraan, A. M., Van Dijk, W. L. & Van der Baan, H. S. Behavior of metallic iron catalysts during Fischer-Tropsch synthesis studied with Moessbauer spectroscopy, x-ray diffraction, carbon content determination, and reaction kinetic measurements. J. Phys. Chem. 84, 3363–3370 (1980).
Jung, H. J., Vannice, M. A., Mulay, L. N., Stanfield, R. M. & Delgass, W. N. The characterization of carbon-supported iron catalysts: chemisorption, magnetization, and Mössbauer spectroscopy. J. Catal. 76, 208–224 (1982).
Weller, S., Hofer, L. J. E. & Anderson, R. B. The role of bulk cobalt carbide in the Fischer—Tropsch Synthesis. J. Am. Chem. Soc. 70, 799–801 (1948).
Anderson, R. B., Hall, W. K., Krieg, A. & Seligman, B. Studies of the Fischer–Tropsch Synthesis. V. Activities and surface areas of reduced and carburized cobalt catalysts. J. Am. Chem. Soc. 71, 183–188 (1949).
Gruver, V. et al. Deactivation of a Fischer-Tropsch catalyst through the formation of cobalt carbide under laboratory slurry reactor conditions. Prepr. Pap. -ACS, Div. Pet. Chem. 49, 192–194 (2004).
Xiong, J. M. et al. The formation of Co2C species in activated carbon supported cobalt-based catalysts and its impact on Fischer-Tropsch reaction. Catal. Lett. 102, 265–269 (2005).
Tavasoli, A., Malek Abbaslou, R. M. & Dalai, A. K. Deactivation behavior of ruthenium promoted Co/γ-Al2O3 catalysts in Fischer–Tropsch synthesis. Appl. Catal. A 346, 58–64 (2008).
Karaca, H. et al. Structure and catalytic performance of Pt-promoted alumina-supported cobalt catalysts under realistic conditions of Fischer–Tropsch synthesis. J. Catal. 277, 14–26 (2011).
Mohandas, J. C. et al. Fischer–Tropsch synthesis: characterization and reaction testing of cobalt carbide. ACS Catal. 1, 1581–1588 (2011).
Pan, Z. & Bukur, D. B. Fischer–Tropsch synthesis on Co/ZnO catalyst—Effect of pretreatment procedure. Appl. Catal. A 404, 74–80 (2011).
Hofer, L. J. E. & Peebles, W. C. X-Ray diffraction studies of the action of carbon monoxide on cobalt-thoria-kieselguhr catalysts. J. Am. Chem. Soc. 69, 2497–2500 (1947).
Weller, S. Kinetics of carbiding and hydrocarbon synthesis with cobalt Fischer-Tropsch catalysts. J. Am. Chem. Soc. 69, 2432–2436 (1947).
Claeys, M. et al. In situ magnetometer study on the formation and stability of cobalt carbide in Fischer–Tropsch synthesis. J. Catal. 318, 193–202 (2014).
Jalama, K. Fischer–Tropsch synthesis over Co/TiO2 catalyst: effect of catalyst activation by CO compared to H2. Catal. Commun. 74, 71–74 (2016).
Paterson, J., Peacock, M., Ferguson, E., Purves, R. & Ojeda, M. In situ diffraction of Fischer–Tropsch catalysts: cobalt reduction and carbide formation. ChemCatChem 9, 3463–3469 (2017).
Xiang, Y. & Kruse, N. Tuning the catalytic CO hydrogenation to straight- and long-chain aldehydes/alcohols and olefins/paraffins. Nat. Commun. 7, 13058 (2016).
Zhong, L. et al. Cobalt carbide nanoprisms for direct production of lower olefins from syngas. Nature 538, 84–87 (2016).
Li, Z. et al. Effects of sodium on the catalytic performance of CoMn catalysts for Fischer–Tropsch to olefin reactions. ACS Catal. 7, 3622–3631 (2017).
Zhao, Z. et al. Insight into the formation of Co@Co2C catalysts for direct synthesis of higher alcohols and olefins from syngas. ACS Catal. 8, 228–241 (2017).
An, Y. et al. Morphology control of Co2C nanostructures via the reduction process for direct production of lower olefins from syngas. J. Catal. 366, 289–299 (2018).
An, Y. et al. Effect of reaction pressures on structure–performance of Co2C-based catalyst for syngas conversion. Ind. Eng. Chem. Res. 57, 15647–15653 (2018).
Xiang, Y. et al. Long-chain terminal alcohols through catalytic CO hydrogenation. J. Am. Chem. Soc. 135, 7114–7117 (2013).
Paterson, J. et al. Manipulation of Fischer-Tropsch synthesis for production of higher alcohols using manganese promoters. ChemCatChem 10, 5154–5163 (2018).
Johnson, G. R., Werner, S. & Bell, A. T. An investigation into the effects of Mn promotion on the activity and selectivity of Co/SiO2 for Fischer-Tropsch synthesis: evidence for enhanced CO adsorption and dissociation. ACS Catal. 5, 5888–5903 (2015).
Johnson, G. R. et al. Investigations of element spatial correlation in Mn-promoted Co-based Fischer–Tropsch synthesis catalysts. J. Catal. 328, 111–122 (2015).
Denzil, J. M., Jan van de, L., Hans, J. W. N. & Abdool, M. S. in Advances in Fischer-Tropsch Synthesis, Catalysts, and Catalysis Chemical Industries (CRC Press, 2009).
Ducreux, O., Lynch, J., Rebours, B., Roy, M. & Chaumette, P. in Studies in Surface Science and Catalysis. Vol. 119 (eds Parmaliana, A., Sanfilippo, D., Frusteri, F., Vaccari, A., & Arena, F.) 125–130 (Elsevier, 1998).
in Studies in Surface Science and Catalysis Vol. 86 (eds Slin’ko, M. M., & Jaeger N. I.) 47–120 (Elsevier, 1994). https://www.sciencedirect.com/science/article/abs/pii/S0167299108615396?via%3Dihub.
Heinze, S., Schmatloch, V. & Kruse, N. Hysteresis effects and long-period oscillations of the D2NO reaction on Rh(110). Surf. Sci. 341, 124–132 (1995).
Suchorski, Y. et al. Visualizing catalyst heterogeneity by a multifrequential oscillating reaction. Nat. Commun. 9, 600 (2018).
Tsotsis, T. T., Rao, V. U. S. & Polinski, L. M. Reaction rate oscillations during Fischer-Tropsch synthesis on Fe-precipitated Nu-1 zeolite-type catalysts. AIChE J. 28, 847–851 (1982).
Acknowledgements
This work was supported by National Science Foundation under contract No. CBET-1438227. A portion of the research was performed at EMSL, a national scientific user facility sponsored by the Department of Energy's Biological and Environmental Research and located at PNNL. PNNL is a multiprogram national laboratory operated for the U.S. DOE by Battelle.
Author information
Authors and Affiliations
Contributions
Y.X. performed the catalytic measurements, XRD characterization, and contributed to drafting the paper. L.K. performed HAADF-STEM and HRTEM characterization. N.K. initiated the research project and drafted the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiang, Y., Kovarik, L. & Kruse, N. Rate and selectivity hysteresis during the carbon monoxide hydrogenation over promoted Co/MnOx catalysts. Nat Commun 10, 3953 (2019). https://doi.org/10.1038/s41467-019-11836-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11836-z
This article is cited by
-
Metastable nickel–oxygen species modulate rate oscillations during dry reforming of methane
Nature Catalysis (2024)
-
Alcohol Synthesis via Fischer–Tropsch Synthesis over Activated Carbon Supported Alkaline Earth Modified Cobalt Catalyst
Catalysis Letters (2021)
-
Chemical Transient Kinetics in Studies of the Fischer–Tropsch Reaction and Beyond
Catalysis Letters (2021)
-
Nanoscale Perspectives of Metal Degradation via In Situ Atom Probe Tomography
Topics in Catalysis (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
