
Abstract
Measurement of electron transfer at single-molecule level is normally restricted by the detection limit of faraday current, currently in a picoampere to nanoampere range. Here we demonstrate a unique graphene-based electrochemical microscopy technique to make an advance in the detection limit. The optical signal of electron transfer arises from the Fermi level-tuned Rayleigh scattering of graphene, which is further enhanced by immobilized gold nanostars. Owing to the specific response to surface charged carriers, graphene-based electrochemical microscopy enables an attoampere-scale detection limit of faraday current at multiple individual gold nanoelectrodes simultaneously. Using the graphene-based electrochemical microscopy, we show the capability to quantitatively measure the attocoulomb-scale electron transfer in cytochrome c adsorbed at a single nanoelectrode. We anticipate the graphene-based electrochemical microscopy to be a potential electrochemical tool for in situ study of biological electron transfer process in organelles, for example the mitochondrial electron transfer, in consideration of the anti-interference ability to chemicals and organisms.
Similar content being viewed by others
Introduction
Electron transfer is of great interest in many research of basic chemical and biological phenomena1,2,3, molecular electronics4,5 and energy materials6,7,8. In particular, measurement of electron transfer at single-molecule level is critical to the in situ study of biological processes9,10,11. Electrochemical detection methods provide a powerful analytical tool for understanding and tracing local electron transfer in molecules adsorbed on the electrode, and substantial advances have been made for this aim. Common electrochemical detection strategies for local electron transfer reactions fall into two main categories: scanning electrochemical microscopy (SECM)12,13,14 and plasmonic-based electrochemical current imaging (P-ECi)15,16,17,18. SECM measures the local current by scanning a microelectrode across the surface, and found abundant applications. P-ECi offers a faster image rate and an extremely sensitivity to the refractive index change of chemical species between oxidized and reduced states. The above electrochemical technologies show the potential to be used to measure local electron transfer reactions at single-molecule level. However, one of the challenges involved in this aim is the detection limit. The key issue is the signal-to-noise ratio determined by the background current in circuit or interference from chemical species. Many efforts have been made to resolve the current at lower ranges, such as ultramicroelectrodes19,20 and surface-enhanced Raman spectroscopy21,22; however, the detection limit is normally restricted in the picoampere to nanoampere range13,15. It is highly desirable to develop a new electrochemical detection strategy avoiding above interferences.
Graphene is an ideal two-dimensional material for developing abundant photonics and optoelectronics devices, such as displays, optical modulators, and plasmonic devices23,24. Furthermore, graphene has been widely used to fabricate working electrodes in a variety of electrochemical methods because of its appealing flexible, transparent properties, and low capacitance17,25,26,27. However, the intrinsic Fermi level-controlled optoelectronic properties of graphene have barely been studied for the measurement of electrochemical reactions. One of possible reasons is the weak scattering (<0.1%) and absorption (~2.3%) of single-layer graphene23,28, making it invisible with most conventional microscopies. The optical conductivity of graphene in the visible region can be efficiently modulated by the Fermi level and charged carrier density, involving variation of interband transitions28,29,30. Interband transitions correlate with the absorption and scattering cross section, offering a potential way to directly measure electron transfer reactions with imaging technologies.
In this work, our observation highlights the Fermi level-responsive Rayleigh scattering of graphene and attached plasmonic nanoparticles. We construct a theoretical model that converts the scattering intensity to the local current density based on experimental results. Accordingly, we develop a unique graphene-based electrochemical microscopy (GEM) technique that makes a straightforward advance in the detection limit. Contrary to the conventional optical electrochemical methods using the change in refractive index as probes, for example, P-ECi, GEM directly measure the in situ-transferred electron charges, avoiding interferences from background current noise and chemical species. In order to enhance the Rayleigh scattering and electron transfer rate, plasmonic gold nanostars (GNS) are immobilized on graphene surface and act as nanoelectrodes. Results reveal that GEM illustrates an ultralow faraday current detection limit (4.5 × 10−18 A) at single nanoelectrodes. Using GEM, we successfully show the potential to measure electron transfer in single cytochrome c molecules, which is an essential redox protein involved in the mitochondrial electron transfer.
Results
Construction of graphene-based electrochemical microscopy
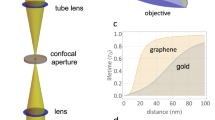
The construction of a three-electrode electrochemical cell for GEM is schematically illustrated in Fig. 1a. A 47-nm-thick gold film is deposited on a cover slide with a 4-mm-diameter hole in center. A graphene layer is transferred onto the hole and acts as the working electrode, which is immersed in electrolyte (0.1 M KNO3) with a Ag/AgCl reference and a platinum counter electrode. All potentials mentioned in this work are relative to the reference. Single-gold nanostars, which demonstrate a uniform scattering over a broad range of wavelength, from visible to near infrared region (Supplementary Fig. 1), are immobilized on the graphene and act as nanoelectrodes. Scattering images are captured by our homemade total internal reflection dark-field microscope31. As shown in Fig. 1b, the fabricated electrochemical cell is placed above the objective, where a collimated white light from a laser-driven light source is directed onto the cover slide via the objective, and then scattered by the graphene and GNS. A barrier is placed at the back focus plane of the objective to stop the reflected light and only the scattering light is directed to a camera to form dark-field scattering images (Fig. 1c). The uniform white background reveals the Rayleigh scattering of graphene. Such weak scattering is not suitable for following experiments. In contrast, bright red scattering spots are assigned to individual GNS, contributed by the far-field incident light and the near-field scattering from underneath graphene together. According to this, GNS may have the ability to enhance the scattering of underneath graphene, and the effective enhanced area is the near-field scattering cross section of GNS32,33.

Schematic illustration of construction of the optical setup. a Schematic illustration of the construction of electrochemical cell, where WE, RE, and CE are working electrode, reference, and counter electrodes, respectively. A graphene layer is transferred onto a gold-coated cover slide with a 4-mm-diameter hole in center, on which attaching a 3.5-mm-diameter PDMS electrochemical cell to avoid reactions on the gold film. b Schematic illustration of the total internal reflection dark-field microscope. c Scattering image of single gold nanostars on the graphene layer. Scale bar is 10 μm. Inset is the transmission electron microscopy image of a gold nanostar. Scale bar is 50 nm
Correlation of surface charge and scattering intensity
The hypothesis of plasmonic enhancement of GNS is verified by investigating the scattering intensity of the graphene and GNS modulated by applied potentials. Figure 2a displays the relative scattering change (ΔI/I) of a single GNS and neighboring graphene (Fig. 1c, open squares) at different potentials (E). Note that the background scattering has been deducted before for clarity. Simultaneously, we measure the optical conductivity (σ) of the graphene sample (details in “Methods” section). Notably, the strong E-dependent ΔI/I of GNS and the graphene are both in good agreement with the change in σ, which can be qualitatively understood from different electronic band structures34,35,36,37. At low potentials (E < −0.2 V), the Fermi level of graphene (EF) is close to the Dirac point, leaving the graphene with low carrier density. The optical conductivity remains constant at a high level, and interband transitions occur when electrons are excited by incident photons (ℏω), resulting in a strong absorption and scattering36,37. When E is higher than −0.2 V, a strong E-dependent scattering is observed. In this range, EF gets close to the transition threshold (EF = 1/2ℏω) due to the positive holes accumulation, and interband transitions start to be forbidden. Higher than 0.8 V, EF is far away from the transition threshold (EF > 1/2ℏω), and the strongly hole-doping leaves the lowest optical conductivity, as well as the scattering intensity. We plot ΔI/I to σ in Fig. 2b, and find a good linear relationship, ΔI/I = σ/σ0 − 1, where the quantum conductivity σ0 is defined as e2/4ℏ. The EF-dependent ΔI/I consequently yields the correlation of graphene’s carrier density (nc) and ΔI/I, since we have EF = ℏνF(πnc)1/2. We calculate nc-dependent ΔI/I (details in “Methods” section) and ΔI/I scales down with nc in the range from 5 × 1017 to 1 × 1018 m−2 (Fig. 2b, black line),
where A is calculated to be −6.9 × 1017 m−2 from the slope of fitting curve.

Correlation of the scattering and optical conductivity of graphene. a Potential dependence of relative scattering change of a gold nanostar (red), neighboring graphene (blue), and the optical conductivity of the graphene (black). Insets show the band structures of graphene at corresponding potentials. b Correlation of the relative scattering change and the optical conductivity (red and blue), and the theoretical correlation of graphene’s carrier density and relative scattering change (black line). c Introduction of excess charges doping in the graphene layer with hydroxyl and hydroxonium ions. Insets show the corresponding energy diagrams. d Potential dependence of relative scattering change of gold nanostars with different surface condition: from negative (top) to positive (bottom) charge
The correlation of ΔI/I and nc offers a direct way to measure a redox reaction taking place on graphene that involves electron transfer processes. For example, in an inner-sphere electrode reaction, such as redox reaction of Fe(CN)63−/4−, there is a strong interaction of the reactant and the electrode38,39. It introduces excess charged carriers and hence a shift of Fermi level to the graphene27,40. In order to further understand the contribution of excess charges to the scattering intensity, we change the electrolyte pH from 2.5 to 10 with phosphate buffered saline (PBS) (see “Methods” section), leaving the graphene with excess negative and positive charged carriers, respectively41. Negative charges drive EF of graphene shift above the Dirac point, and with positive charges EF is expected to shift below the Dirac point (Fig. 2c). For comparison, we investigate E-dependent ΔI/I of GNS with different charges (Fig. 2d). Indeed, we found an apparent shift of the threshold potential from 0.7 V (negative charge) to 0.3 V (positive charge). That is to say, when an inner-sphere redox reaction takes place on the graphene, both the electric double layer (EDL) charging and electron transfer processes contribute to nc. However, the contribution from EDL charging is exceedingly smaller than electron transfer because of the low capacitance of graphene (Supplementary Fig. 2).
Imaging the current density at single nanoelectrodes
We have known the linear relationship of nc and ΔI/I according to Eq. (1). The carrier charge density in graphene is the opposite of charge density in solution42, thus the charging current density ic is given by
where e is the charge of a single carrier. Equation (2) reveals that the charging current density can be obtained by time derivative of ΔI/I (Supplementary Fig. 2). When an inner-sphere electron transfer reaction38,39, such as Fe(CN)63−/4−, takes place on the graphene surface, nc is contributed by two types of charges: nc = nEDL + nET, where nEDL and nET is the carrier density induced by charging and electron transfer, respectively. Thus, the faraday current i can be easily calculated from ΔI/I via Fick’s law (details in “Methods” section), which can be expressed by
where B is \(\left( {z_{\mathrm{O}}D_{\mathrm{O}}^{ - 1/2} - z_{\mathrm{R}}D_{\mathrm{R}}^{ - 1/2}} \right)^{ - 1}\), where zO and zR are the charges of the oxidized and reduced molecules, DO and DR are the diffusion coefficients of the redox species. n is the number of electrons involved in one redox reaction, e is the elementary charge, Ka defines the adsorption of redox molecules, L−1 is the inverse Laplace transform, and \(\overline {\Delta I/I} \left( s \right)\) is the Laplace transform of ΔI/I.
GEM allows for imaging the local faraday current density by performing Eq. (3) to scattering image sequence. To demonstrate it, we studied the cyclic voltammograms (CV) of 1 mM Fe(CN)63−/4− in 0.1 M KNO3 with conventional electrochemical method and GEM, simultaneously. Figure 3a–d show snapshots of current density movie (Supplementary Movie 1) at different potentials. At −0.6 V, far away from the redox potential, the current density is near zero everywhere (Fig. 3a). When potential increases, oxidation of Fe(CN)64− takes place and a growing current density is observed where GNS are located, which reaches maximum at 0.12 V (Fig. 3b). As the potential cycles back, the current is inverted, attributed to the reduction of Fe(CN)63−, and the maximum negative current is located at 0.02 V (Fig. 3c). The current eventually disappears when the potential cycles back to −0.6 V. The big contrast makes it possible to exclusively study electron transfer reactions at a single GNS without interference of graphene around, similar to ultramicroelectrodes.

Electrochemical reaction of potassium ferricyanide at single gold nanostars. a–d Electrochemical current images of multiple gold nanostars on the graphene at different potentials during continuous cycling of the potential between −0.6 V and 0.6 V at a rate of 0.1 V s−1 (see Supplementary Movie 1). Scale bar is 5 μm. The electrolyte is 0.1 M KNO3 containing 1.0 mM K3[Fe(CN)6]. e Cyclic voltammograms of 5-labeled gold nanostars extracted from the electrochemical current images. f Averaged cyclic voltammogram of the 5 gold nanostars (red), cyclic voltammogram of graphene area (black), and the conventional cyclic voltammogram measured with a potentiostat (blue)
Figure 3e displays CVs of multiple GNS (labeled with 1–5) extracted from the current density movie, and similar shapes are presented among these GNS in spite of some small deviations in the amplitudes of oxidation and reduction peaks due to their heterogeneity. We calculate the averaged CV of these GNS and compare it with the graphene area and the conventional CV recorded with a potentiostat (Fig. 3f). The CV of graphene area is indeed in good agreement with the conventional CV. Surprisingly, the CV of GNS shows ~10 times larger peak current density and a higher signal-to-noise ratio than that of graphene. We attribute it to the higher surface area of GNS, which offers more adsorption sites for reactive molecules. Moreover, the peak separation (ΔEp) of GNS (100 mV) is much closer to the ideal CV than that of the conventional CV (250 mV). It is well known that noble metals will give better electron transfer kinetics than graphene surfaces for inner-sphere redox couples, driven by the local density of states of the electrode near its Fermi level and the reorganization energy of the molecules43,44. Such better electron transfer kinetics induces a faster accumulation of charges on the surface of GNS. As a result, a faster change in the scattering intensity is observed.
We now examine the anti-interference performance and the detection limit of GEM. The main interference chiefly arises from the change in refractive index, which is commonly the detection signal of other optical electrochemical strategies, such as P-ECi15. As shown in Fig. 4a, when we add 1% (v/v) ethanol to the electrolyte, the scattering intensity of a single GNS keeps steady, though a small fluctuation is induced by injection. For comparison, the total internal reflection intensity of the same area is recorded, which shows a stepwise decrement following the change in refractive index (Fig. 4b). Thus, the contribution of refractive index to GEM can be neglected. The current detection limit of GEM is determined by the background charging current noise level. As shown in Supplementary Fig. 2, the noise level is evaluated to be 7.2 × 10−4 A m−2. Thus, we estimate a current detection limit of 4.5 × 10−18 A at a single GNS, since the near-field scattering cross section of GNS is 2.10 × 10−15 m2 (see “Methods” section). Such attoampere scale detection limit offers a considerable advance for electrochemical detection methods, which is currently in the picoampere to nanoampere range13,15.

Refractive index dependence of scattering and reflected intensity. a Time course of the scattering intensity of a single gold nanostar. b Time course of the total internal reflection intensity of the same area
Measuring electron transfer of cytochrome c molecules
Measuring electron transfer at single-molecule level demands for both high spatial resolution and sensitivity, and the attoampere scale detection limit allows GEM to meet these demands. We take a redox protein cytochrome c for example to demonstrate this capability. Cytochrome c is adsorbed on 3-mercaptopropionic acid-modified GNS via the electrostatic attraction18. Scanning transmission electron microscopy (STEM) imaging of adsorbed proteins is shown in Fig. 5a. The dash line describes the outline of a single GNS with sharp tips, which is coated by a uniform 2-nm-thick uranyl acetate negative staining layer (see “Methods” section). Multiple bulges are located at the two tips in the focal plane, representing adsorbed cytochrome c molecules. Small bulges (~3 nm) reveal individual molecules and big bulges (~6 nm) are induced by aggregates of several molecules (more examples in Supplementary Fig. 3). We count the number of cytochrome c molecules at each tip of at least 20 GNS, and found a concentrated distribution of histograms in range from 0 to 3 molecules (Supplementary Fig. 4). In consideration that only half area of a tip is visible, the amount of cytochrome c molecules at each tip is estimated to be 0–6. Thus a single GNS with around 8 tips should have 0–48 cytochrome c molecules. Measuring the electron transfer of such few cytochrome c molecules is barely to be achieved by current electrochemical technologies. For GEM, when electron transfer reactions take place on the graphene electrode without any diffusion, the current density could be simply measured by (details in “Methods” section)

Electron transfer measurement in cytochrome c at single gold nanostars. a Scanning transmission electron microscopy imaging of a gold nanostar with adsorbed cytochrome c. Scale bar is 50 nm. b Current density imaging of multiple gold nanostars with and without (open circles) cytochrome c modification at 0.16 V during continuous cycling of the potential between −0.4 V and 0.5 V at a rate of 0.1 V s−1. Scale bar is 5 μm. c Cyclic voltammograms of single gold nanostars with (blue) and without (black) cytochrome c modification, where the dash line is an ideal CV. The electrolyte is 70 mM PBS (pH 7.0) and the scan rate is 0.1 V s−1. d Scattering imaging of gold nanostars in b, where open circles indicate the location of gold nanostars without cytochrome c modification. Scale bar is 5 μm
Figure 5b shows the current image of multiple GNS at 0.16 V where the current reaches the maximum during a continuous cycling. Some of them have been immobilized with cytochrome c, demonstrating the clear contrast. However, the rest GNS with only 3-MPA modification, whose positions are marked by open circles (Fig. 5d), show no contrast. The CV of cytochrome c adsorbed on a single GNS (labeled with 1) is demonstrated in Fig. 5c (blue line), where a pair of well-defined reduction and oxidation peaks are found at around 0.16 V. The shape and peak current are similar to the theoretic CV (dash line) of a fully reversible one electron transfer reaction for 60 redox molecules immobilized on the graphene electrode38. In remarkable contrast, the CV of GNS without cytochrome c (labeled with 6) only shows background noise (Fig. 5c, black line) because the charging current is decreased by 3-MPA modification18.
Further insight to the fast electron transfer events can be obtained by increasing the frame rate to 500 Hz, while the potential scan rate is set to be 10 mV s−1 in order to reduce the charging background. We measure and compare the first (Fig. 6a) and second (Fig. 6b) CV cycles of a GNS with cytochrome c. Intriguingly, the broad reduction and oxidation peaks of cytochrome c become discrete spikes (magnifications in Fig. 6c, d), which are assigned to individual reduction and oxidation events. Deviations in the amplitudes reveal the different amount of electrons transferred in each event. Furthermore, spikes in different cycles occur at different potentials near the standard redox potential of cytochrome c during successive cycles, even at the same GNS. We attribute such stochastic spikes to dynamic states of cytochrome c molecules, arising from the lateral molecular interaction, variation in redox-site/electrode electronic coupling, or microenviromental variance45. To investigate whether the stochastic spikes from single electron transfer events can reproduce the ideal CV, we measure CVs of abundant GNS (Supplementary Fig. 5). As shown in Fig. 6e, histograms of reduction (blue) and oxidation (red) events both show distributions near the standard redox potential. The good correlation reveal that the apparent CV is the statistical result of stochastic electron transfer events.

Stochastic electron transfer events measurement in cytochrome c. The first (a) and second (b) cycle of cyclic voltammograms of a single gold nanostar with cytochrome c modification. The electrolyte is 70 mM PBS (pH 7.0) and the scan rate is 0.01 V s−1. c, d Magnifications of the region marked with dash squares in a and b, respectively. Insets showing current density images (1 by 1 μm) of the gold nanostar at corresponding potentials. e Histograms showing the distribution of single oxidation (red) and reduction (blue) events with the applied potential. The ideal CV of cytochrome c (dash line) is also shown. f Histograms showing the distribution of the electron transfer number of oxidation (red) and reduction (blue) events
We calculate the electron transfer number of these reduction and oxidation events to estimate the number of cytochrome c at a GNS. As shown in Fig. 6f, histograms of the electron transfer number during reduction (blue) and oxidation (red) events both show concentrated distributions in range from 10 to 30 electrons, despite a rare distribution up to 115 is also observed. Note that ten electrons is the detection limit of our method due to the background noise level. Thus, the possibility of reduction and oxidation events with <10 transferred electrons should not be excluded. That is, one reduction or oxidation event involves varying number of cytochrome c, predominantly ranging from several to dozens of molecules, matching the number measured with STEM images.
Discussion
In summary, we have proposed a universal electrochemical microscopy GEM based on the Fermi level-responsive optical conductivity of graphene to attain an ultrasensitive electron transfer measurement. Instead of measuring the current in circuit or the change in refractive index, GEM directly determines the change in local charge density. Ultrasensitive detection of local faraday current makes it possible to trace the electron transfer process in cytochrome c at single-molecule level. Although the detection limit of GEM is excellent relative to other electrochemical detection methods, the speed could be further improved by using high speed camera to trace individual dynamic electron transfer processes, which is in the range of nanoseconds to microseconds. Besides, the gold nanostars used here could be replaced by other scattering nanoparticles, such as Ag, TiO2, and Au-Pt alloy nanoparticles. Since electron transfer reactions always involve variation in charge density, GEM provides a universal tool for study in many fields, such as basic chemical and biological phenomena, molecular electronics and energy materials. For example, Shewanella species use a direct electron transfer mechanisms to produce electricity through outer-membrane cytochrome c46, and it is of great importance to exclusively study such process apart from electron shuttle-based indirect electron transfer mechanisms.
Methods
Measuring the optical conductivity of graphene
In this work, a white light is used for imaging the scattering of graphene. Optical conductivity σ(ω) of graphene at frequency in visible region, where the interband transitions dominate, can be calculated simply by including the Fermi–Dirac distribution30,34:
where EF is graphene’s Fermi level, which is tuned by carrier density nc by EF = ℏνF(πnc)1/2, σ0 is the quantum conductivity defined as e2/4ℏ. When potentials in range from −0.8 to 1.0 V are applied, we measure σ(ω) of the graphene layer with corresponding transmittance T by:
where N is the number of layers, \({\it{\epsilon }}_0\), \({\it{\epsilon }}_1\), and \({\it{\epsilon }}_2\) are the vacuum permittivity, relative permittivities of medias below and above the graphene layer, respectively. In our work, the graphene layer is placed between glass \(\left( {{\it{\epsilon }}_1 = 2.25} \right)\) and the electrolyte \(\left( {{\it{\epsilon }}_2 = 1.77} \right)\).
The transmittance of graphene is measured with the microscope setup shown in Fig. 1b, except the normal incident light source. We move the area of interest to the border of the graphene layer, and measure the reflected intensity of the graphene (Igraphene) and the glass area (Iglass). The transmittance is calculated by:
Combining Eqs. (6) and (7), we have the local optical conductivity of the graphene at different potentials (Fig. 2a, black line).
Measuring electrochemical current from scattering intensity
We have shown that the charging current density can be measured by eq. (2). Now we focus on the contribution of nET to describe electron transfer reactions. Electron transfer-induced charge density qET can be expressed in terms of the oxidized and reduced product concentrations, CO and CR, and given by
where F is Faraday constant, Ka = Csurf(t)/C(0,t) defines the adsorption of redox molecules, zO and zR are the charges of oxidized and reduced molecules, \(C_{\mathrm{O}}^0\) and \(C_{\mathrm{R}}^0\) are the concentrations of oxidized and reduced molecules in bulk solution, respectively.
Conventional electrochemical methods measure current density versus potential or time, which is related to CO(0,t) and CR(0,t)15,38:
where n is number of electrons transferred per reaction, and DO and DR are the diffusion coefficients of oxidized and reduced molecules, respectively.
During a redox reaction, the diffusion equation of oxidized species obeys Fick’s laws:
where only the diffusion in vertical direction is considered because of the thin diffuse layer.
By performing Laplace transform on Eq. (10), we have
where A'(s) is a function to be determined from boundary conditions at the electrode surface. To relate the concentrations to current density, we perform Laplace transform on Eq. (9) and then combine it with Eq. (11), leading to
and a similar relation could be obtained for the reduced species
Combining Eqs. (8), (12), and (13), we have
Substituting Eq. (1) into Eq. (14), we have
Performing Laplace transform on Eq. (15), and we have
Thus, the faraday current density can be given by:
where L−1 is the inverse Laplace transform. For Fe(CN)63−/4−, n is 1, zO, and zR are −3 and −4, and DO and DR are 7.2 × 10−10 and 6.67 × 10−10 m2 s−1, respectively47. Ka is determined to be 1 × 10−7 m (see below).
When electrochemical reactions occur only on the graphene electrode, such as redox reactions of adsorbed proteins, the current density can be simply given by17:
where CO(t) and CR(t) are surface concentrations of the oxidized and reduced products. Combining Eqs. (6) and (18), and we have
Note that Eq. (19) shows the same express as Eq. (2). That is to say, the faraday current density could be measured together with the charging current in this model.
Calibration of K a
To calibrate the adsorption constant Ka, defined as Ka = Csurf(t)/C(0,t), we measure the change in ΔI/I of gold nanostars induced by the addition of 1 mM Fe(CN)63−/4−. To simplify the model, a +0.6 V potential is applied, which is more positive than the standard oxidation potential (Fig. 3e). The positive potential leaves almost only Fe(CN)63− in the diffusion layer (hundreds of micrometers thick) near the graphene surface. The electrolyte is 0.1 M KNO3, thus the addition of 1 mM K3Fe(CN)6 will not affect the ion strength and the electrical double layer. Thus ΔI/I is only contributed by the adsorption of Fe(CN)63− ions, and Ka is expressed by:
where Na is Avogadro’s constant, and Ka is found to be 1 × 10−7 m.
Calculation of near-field scattering cross section
The near-field scattering cross section Csca of a gold nanostar, which determines the scattering intensity and therefore the current at single gold nanostars, is calculated by the effective polarizability by32,33:
where k = 2π/λ is the wave number, and \(\alpha _ \bot ^{{\mathrm{eff}}}\) is the effective polarizability, governed by:
where εg and εAu are the dielectric constant of graphene and gold, respectively and r = 40 nm is the radius of gold nanostars, which is considered as gold nanospheres for simplification because of the similar scattering cross-section of two types of nanoparticles (Supplementary Fig. 6). According to Eqs. (21) and (22), Csca is calculated to be 2.10 × 10−15 m2.
Chemicals and general techniques
Poly(methyl methacrylate) and gold etchant were purchased from Sigma-Aldrich (Shanghai, China). Gold nanostars (80-nm core diameter) and gold nanospheres (80-nm diameter) were purchased from NanoSeedz Ltd CVD Graphene (3–5 layers) on copper foil is purchased from Nanjing XFNANO Materials Tech Co., Ltd Absolute ethanol, acetone, cysteamine, 3-mercaptopropionic acid, and purified bovine heart cytochrome c were purchased from Aladdin Reagent Inc. PBS was purchased from Nanjing KeyGen Biotech. Co. Ltd. All other reagents are of analytical grade. Ultrapure water with a resistivity of 18.2 MΩ cm was produced using a Milli-Q apparatus (Millipore) and used in the preparation of all solutions. Cover slides were purchased from Thorlabs Co., Ltd PDMS was prepared using Sylgard 184, Dow Corning. Copper etchant was prepared by dissolving 10 g CuSO4 in 50 mL deionized water and 50 mL 37% hydrochloric acid.
UV-vis spectra were recorded on a UV-1750 spectrophotometer (Shimadzu, Kyoto, Japan). Scanning transmission electron micrographs were captured on a JEOL 2800 transmission electron microscope. Dark-field images and spectra measurements were carried out on Nikon Ti-E microscope. A broadband light source (EQ-99XFC LDLS, Energetiq Technology) was used for incident illumination. True-color dark-field images are captured by a color-cooled digital camera (DS-RI1, Nikon), and the scattering spectra of single nanoparticles was measured by a monochromator (Acton SP2300i, PI) equipped with a spectrograph CCD (PIXIS 400BR_excelon, PI) and a grating (grating density: 300 L mm−1; blazed wavelength: 500 nm). The conventional electrochemical experiments were carried out on a potentiostat (ACFBP1, Pine Research Instrumentation).
Preparation of gold nanostars
Gold nanostars were prepared via a typical seed-mediated growth process. The seed solution is prepared by dissolving 0.25 mL citrate (0.01 M) and 0.125 mL HAuCl4 (0.01 M) to 9.625 mL DI water, followed by the addition of 150 μL fresh cold NaBH4 (0.01 M). The solution is then shaken for 3 h. To prepare the growth solution, 42.75 mL Tetradecyltrimethylammonium bromide (0.1 M), 1.8 mL HAuCl4 (0.01 M), 270 μL AgNO3 (0.01 M), and 300 μL ascorbic acid (0.1 M) were mixed. Finally, 60 μL seed solution was added to the growth solution, followed by incubation at room temperature overnight.
Fabrication of the electrochemical cell
A 47-nm-thick gold film was coated on cover slide, followed by treatment of gold etchant for 1 min in the center. The remaining gold film was used for connection between the graphene and the potentiostat. A CVD graphene sample was transferred onto the etched hole of the gold substrate with a PMMA-mediated approach. Simply, a layer of PMMA was spin-coated onto the graphene, and the metal below it was etched away completely. The PMMA/graphene stack was then transferred onto the Au surface. After the graphene was transferred onto the gold substrate, the PMMA layer was dissolved and removed by acetone. An electrochemical cell (with 3.5 mm inner diameter) made of PDMS on was placed on top of the graphene sample, and KNO3 or PBS solution was used as electrolyte. The potential of graphene was controlled with respect to Ag/AgCl reference electrode with the potentiostat using a platinum wire as counter electrode. Gold nanostars were then deposited on the graphene for following experiments. To immobilize individual gold nanostars on the graphene, 100 μL of ultrapure water was added into the cell, followed by the addition of 10 μL 20 pM gold nanostars. After sedimentation for 10 min, excess gold nanostars were removed by pipet and the electrochemical cell was thoroughly rinsed with the ultrapure water.
Optical measurement and imaging processing
For dark-field scattering imaging, the electrochemical cell was placed on the 100× oil immersion objective (NA = 1.49) equipped by a Nikon Ti-E inverted microscope. A barrier was placed at the back focus plane of the objective to stop the reflected light and only the scattering light was directed to a CMOS camera (AVT Pike F-032B). For total internal reflection imaging, the barrier was removed. In order to calculate the relative scattering change (ΔI/I), the background scattering was initially removed by subtracting the scattering intensity far away from the Dirac point. The pure potential dependent scattering was then normalized by dividing the scattering intensity at the Dirac point. The relative scattering change at each pixel was processed to produce a current density image of the surface. The frame rate is 10 Hz for all optical measurements except that shown in Fig. 6, Supplementary Fig. 5 (500 Hz).
Charge doping and cytochrome c modification
The doping charges of graphene were modulated by adding 0.1 M PBS (pH from 2.5 to 10) to the electrolyte (0.1 M KNO3). For cytochrome c modification, gold nanostars were immersed in 5 mM 3-mercaptopropionic acid for 2 h and subsequently 50 μM cytochrome c for 1 h. In order to observe the negative contrast of single cytochrome c molecules with STEM, gold nanostars with cytochrome c modification were incubated in 2% uranyl acetate for 2 s.
Data availability
The data and computer codes supporting the findings of this study are available from the authors upon reasonable request.
References
Sikes, H. D. et al. Rapid electron tunneling through oligophenylenevinylene bridges. Science 291, 1519–1523 (2001).
Pascher, T., Chesick, J. P., Winkler, J. R. & Gray, H. B. Protein folding triggered by electron transfer. Science 271, 1558–1560 (1996).
Light, S. H. et al. A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria. Nature 562, 140–144 (2018).
Li, Y. et al. Mechanical stretching-induced electron-transfer reactions and conductance switching in single molecules. J. Am. Chem. Soc. 139, 14699–14706 (2017).
Li, Y. et al. Transition from stochastic events to deterministic ensemble average in electron transfer reactions revealed by single-molecule conductance measurement. Proc. Natl Acad. Sci. 116, 3407–3412 (2019). 201814825.
Jailaubekov, A. E. et al. Hot charge-transfer excitons set the time limit for charge separation at donor/acceptor interfaces in organic photovoltaics. Nat. Mater. 12, 66 (2012).
Xiang, B., Li, Y., Pham, C. H., Paesani, F. & Xiong, W. Ultrafast direct electron transfer at organic semiconductor and metal interfaces. Sci. Adv. 3, e1701508 (2017).
Tisdale, W. A. et al. Hot-electron transfer from semiconductor nanocrystals. Science 328, 1543–1547 (2010).
Zhang, Z. et al. Electron transfer by domain movement in cytochrome bc1. Nature 392, 677 (1998).
Birsoy, K. et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015).
Blumberger, J. Recent advances in the theory and molecular simulation of biological electron transfer reactions. Chem. Rev. 115, 11191–11238 (2015).
Bard, A. J. et al. Chemical imaging of surfaces with the scanning electrochemical microscope. Science 254, 68–74 (1991).
Polcari, D., Dauphin-Ducharme, P. & Mauzeroll, J. Scanning electrochemical microscopy: a comprehensive review of experimental parameters from 1989 to 2015. Chem. Rev. 116, 13234–13278 (2016).
Kuss, S., Polcari, D., Geissler, M., Brassard, D. & Mauzeroll, J. Assessment of multidrug resistance on cell coculture patterns using scanning electrochemical microscopy. Proc. Natl Acad. Sci. 110, 9249–9254 (2013).
Shan, X., Patel, U., Wang, S., Iglesias, R. & Tao, N. Imaging local electrochemical current via surface plasmon resonance. Science 327, 1363–1366 (2010).
Shan, X. et al. Imaging the electrocatalytic activity of single nanoparticles. Nat. Nanotechnol. 7, 668–672 (2012).
Wang, Y., Shan, X., Wang, H., Wang, S. & Tao, N. Plasmonic imaging of surface electrochemical reactions of single gold nanowires. J. Am. Chem. Soc. 139, 1376–1379 (2017).
Wang, Y. et al. Fast electrochemical and plasmonic detection reveals multitime scale conformational gating of electron transfer in cytochrome c. J. Am. Chem. Soc. 139, 7244–7249 (2017).
Brasiliense, V. et al. Correlated electrochemical and optical detection reveals the chemical reactivity of individual silver nanoparticles. J. Am. Chem. Soc. 138, 3478–3483 (2016).
Hafez, M. E., Ma, H., Ma, W. & Long, Y.-T. Unveiling the Intrinsic Catalytic Activities of Single-gold-nanoparticle-based Enzyme Mimetics. Angew. Chem. 131, 6393–6398 (2019).
Cortés, E. et al. Monitoring the electrochemistry of single molecules by surface-enhanced raman spectroscopy. J. Am. Chem. Soc. 132, 18034–18037 (2010).
Zrimsek, A. B. et al. SinglE-molecule Chemistry with Surface- and Tip-enhanced Raman Spectroscopy. Chem. Rev. 117, 7583–7613 (2017).
Koppens, F. H. L. et al. Photodetectors based on graphene, other two-dimensional materials and hybrid systems. Nat. Nanotechnol. 9, 780 (2014).
Chen, Z. et al. Imaging local heating and thermal diffusion of nanomaterials with plasmonic thermal microscopy. ACS Nano 9, 11574–11581 (2015).
Bae, S. et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotechnol. 5, 574 (2010).
Kim, K. S. et al. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 457, 706 (2009).
Shan, X. et al. Mapping local quantum capacitance and charged impurities in graphene via plasmonic impedance imaging. Adv. Mater. 27, 6213–6219 (2015).
Nair, R. R. et al. Fine structure constant defines visual transparency of graphene. Science 320, 1308–1308 (2008).
Bonaccorso, F., Sun, Z., Hasan, T. & Ferrari, A. C. Graphene photonics and optoelectronics. Nat. Photon 4, 611 (2010).
Mak, K. F. et al. Measurement of the optical conductivity of graphene. Phys. Rev. Lett. 101, 196405 (2008).
Chen, X. et al. Imaging the transient heat generation of individual nanostructures with a mechanoresponsive polymer. Nat. Commun. 8, 1498 (2017).
Knoll, B. & Keilmann, F. Near-field probing of vibrational absorption for chemical microscopy. Nature 399, 134 (1999).
Bohren, C. F. & Huffman, D. R. Absorption and Scattering of Light by Small Particles, (Wiley Interscience, 1983).
Stauber, T., Peres, N. M. R. & Geim, A. K. Optical conductivity of graphene in the visible region of the spectrum. Phys. Rev. B 78, 085432 (2008).
Kim, J. et al. Electrical control of optical plasmon resonance with graphene. Nano Lett. 12, 5598–5602 (2012).
Liu, M. et al. A graphene-based broadband optical modulator. Nature 474, 64 (2011).
Sfeir, M. Y. et al. Probing electronic transitions in individual carbon nanotubes by rayleigh scattering. Science 306, 1540–1543 (2004).
Bard, A. J. & Faulkner, L. R. Electrochemical Methods: Fundamentals and Applications, (Wiley, 1980).
McCreery, R. L. Advanced carbon electrode materials for molecular electrochemistry. Chem. Rev. 108, 2646–2687 (2008).
Nakatsuka, N. et al. Aptamer–field-effect transistors overcome Debye length limitations for small-molecule sensing. Science 362, 319–324 (2018).
Ang, P. K., Chen, W., Wee, A. T. S. & Loh, K. P. Solution-gated epitaxial graphene as pH sensor. J. Am. Chem. Soc. 130, 14392–14393 (2008).
Chen, F., Qing, Q., Xia, J., Li, J. & Tao, N. Electrochemical gate-controlled charge transport in graphene in ionic liquid and aqueous solution. J. Am. Chem. Soc. 131, 9908–9909 (2009).
Friis, S. D., Taaning, R. H., Lindhardt, A. T. & Skrydstrup, T. Silacarboxylic acids as efficient carbon monoxide releasing molecules: synthesis and application in palladium-catalyzed carbonylation reactions. J. Am. Chem. Soc. 133, 18114–18117 (2011).
Young, S. L., Kellon, J. E. & Hutchison, J. E. Small gold nanoparticles interfaced to electrodes through molecular linkers: a platform to enhance electron transfer and increase electrochemically active surface area. J. Am. Chem. Soc. 138, 13975–13984 (2016).
Salverda, J. M. et al. Fluorescent cyclic voltammetry of immobilized azurin: direct observation of thermodynamic and kinetic heterogeneity. Angew. Chem. Int. Ed. 49, 5776–5779 (2010).
Lovley, D. R. Bug juice: harvesting electricity with microorganisms. Nat. Rev. Microbiol. 4, 497 (2006).
Konopka, S. J. & McDuffie, B. Diffusion coefficients of ferri- and ferrocyanide ions in aqueous media, using twin-electrode thin-layer electrochemistry. Anal. Chem. 42, 1741–1746 (1970).
Acknowledgements
This research is supported by the National Natural Science Foundation of China (Grants nos. 21834004, 21327902, 21427807, and 21605081), the Natural Science Foundation of Jiangsu Province (Grants no. BK20160638), the International Cooperation Foundation from the Ministry of Science and Technology (Grants no. 2016YFE0130100), the Fundamental Research Funds for the Central Universities (Grants no. 020514380173), and the Excellent Research Program of Nanjing University (Grants no. ZYJH004).
Author information
Authors and Affiliations
Contributions
Z.C. and J.J.Z. conceived the study. Q.X., P.X. and M.W. performed the experiments. Z.C. and X.C. built the microscope setup. Z.C., Q.X. and J.J.Z. designed the experiments. J.R.Z. and H.Y.C. advised the experiments. Z.C. analyzed the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communication thanks Kian Ping Loh and other, anonymous, reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xia, Q., Chen, Z., Xiao, P. et al. Fermi level-tuned optics of graphene for attocoulomb-scale quantification of electron transfer at single gold nanoparticles. Nat Commun 10, 3849 (2019). https://doi.org/10.1038/s41467-019-11816-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11816-3
This article is cited by
-
Amplification of oxidative stress with lycorine and gold-based nanocomposites for synergistic cascade cancer therapy
Journal of Nanobiotechnology (2021)
-
Single-entity electrochemistry at confined sensing interfaces
Science China Chemistry (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
