
Abstract
Efficient electroreduction of carbon dioxide to multicarbon products in aqueous solution is of great importance and challenging. Unfortunately, the low efficiency of the production of C2 products limits implementation at scale. Here, we report reduction of carbon dioxide to C2 products (acetic acid and ethanol) over a 3D dendritic copper-cuprous oxide composite fabricated by in situ reduction of an electrodeposited copper complex. In potassium chloride aqueous electrolyte, the applied potential was as low as −0.4 V vs reversible hydrogen electrode, the overpotential is only 0.53 V (for acetic acid) and 0.48 V (for ethanol) with high C2 Faradaic efficiency of 80% and a current density of 11.5 mA cm−2. The outstanding performance of the electrode for producing the C2 products results mainly from near zero contacting resistance between the electrocatalysts and copper substrate, abundant exposed active sites in the 3D dendritic structure and suitable copper(I)/copper(0) ratio of the electrocatalysts.
Similar content being viewed by others

Introduction
Carbon dioxide (CO2) is an abundant C1 resource1. Water, as a cheap resource, is a suitable electrochemical reaction medium and hydrogen source. In the quest for developing techniques for CO2 utilization, electrochemical reduction offers a potential route to economical conversion of CO2 and water into value-added chemicals and fuels2. Until now, extensive efforts have been devoted to the development of catalysts for electrocatalytic reduction of CO2 in water3,4,5. Despite the notable progress, it is still highly desirable to develop efficient electrocatalysts to steer the reaction pathways toward higher value products and reduce reaction overpotential.
Products with two or more carbons (C2+ product), such as acetic acid and ethanol, are very useful chemicals or fuels. Efficient electrochemical reduction of CO2 to C2+ products is of great importance6,7,8,9. Literature survey shows that Cu-based catalysts are the most efficient material for electrocatalytic reduction of CO2, and Cu is a key component for forming multicarbon products10,11,12,13,14,15,16,17,18,19,20. On Cu-derived oxide electrocatalysts, C2 or C2+ products could be formed with Faradaic efficiency (FE) of ~70%11,12. Cu nanoparticles, Cu nanocubes and Cu nanoclusters show selectivity up to 60% for transformation of CO2 to C2+ products9,13,14,15. Plasma-activated Cu or Cu nanocube catalysts exhibit high FE to ethylene (60%) or ethylene/ethanol (45%/22%)16,17. Recent study indicates that the FE of ethylene on Boron-modified Cu-based electrocatalysts could reach 79%6. Cu gas diffusion electrode was found to be efficient for CO2 reduction to ethylene with a FE of 70% at high current density8. Besides, Cu2S–Cu core–shell–vacancy catalyst could boost CO2 reduction to C2 product with a FE of 32% at −0.95 V vs RHE18. Metal-free catalysts, such as nitrogen-doped mesoporous carbon, are also used as efficient catalysts for CO2 reduction to ethanol (44%)19. It was also demonstrated that bimetallic catalysts have improved activity and selectivity for CO2 reduction to hydrocarbons20. Comparing with production of C1 chemicals or fuels, the reports on electrochemical reduction of CO2 to C2+ products is very limited. Especially, exploration of electrochemical systems to mediate multiple proton transfers with low overpotential is still a challenge for CO2 reduction to C2+ products because the C–C coupling is difficult. It is both scientifically and practically appealing to explore efficient electrolysis systems for CO2 reduction to C2+ product under heterogeneous catalysis conditions in aqueous media.
Designing robust electrocatalysts is very important for efficient CO2 reduction21,22. Previous reports indicate that heterogeneous metal-based catalysts derived from molecular complexes can be favorable electrocatalysts23,24. The catalytic properties of the catalysts can be tailored by adjusting and regulating the composition of the complexes, and they exhibit excellent catalytic activity toward water splitting and oxygen reduction25,26,27,28,29. So far, it is also an approach toward the fabrication of electrocatalysts, such as additive controlled electrodeposited Cu catalysts for CO2 reduction30,31,32. However, as a precursor, the complex was generally prepared ex situ or used as a homogeneous additive in decomposition of the new structure in CO2 electroreduction.
Electrodeposition is a very useful technique that requires simple equipment with precise control of the growth processes, purity, structures, and morphologies of the deposits. The deposits are well suited for many applications, such as fuel cells, batteries, and sensors33,34,35. This method has also been used to prepare electrocatalysts for CO2 reduction36,37,38,39. For example, electrodeposited In or Bi electrode can promote CO2 reduction to CO with high selectivity36,37. Cu2O prepared by electrodeposition method can control the catalytic selectivity of CO2 to hydrocarbons11. In situ deposited Cu nanodendrites as gas diffusion layer can reach high FE for C2 products38. In general, it is easy to produce two-dimensional (2D) materials using the direct electrodeposition method. While an electrodeposition process can also create a 3D material, it is difficult to control the microstructure with desired features33. Therefore, it is very interesting to explore more practical methods for obtaining stable 3D structures with desirable structures for CO2 electrocatalytic reduction.
The immobilization of molecular catalysts onto surfaces provides some obvious advantages over heterogeneous catalysts, such as controllable 3D structure, fast electron transfer rate, etc. Recently, an electrosynthesis method has been used to prepare metal organic complex films supported on conducting substrates40,41. This route constructs the metal organic complex films by assembling the reduced organic linkers and oxidized metal ions on a conductive substrate. The synthetic method is currently emerging as a promising approach for producing films of specific configurations, particularly in sensors and electric devices40.
Herein, we report a more direct and facile method to electrodeposit a high-surface area Cu-complex film onto conductive substrates. A 3D dendritic Cu–Cu2O composite catalyst (denoted as Cu–Cu2O/Cu) can be obtained via in situ growth and decomposition of the corresponding Cu-complex film. By using the Cu-complex electrodes as precursors and templates, the Cu–Cu2O/Cu electrode has unique characteristics, such as dendritic 3D structure as well as near zero contact resistance between the catalyst and the substrate. The in situ synthesized electrode has outstanding catalytic performance for CO2 reduction to C2 products in KCl aqueous electrolyte. At −0.4 V vs RHE, the FE of C2 products could reach 80% with a current density of 11.5 mA cm−2. The respective overpotentials for acetic acid and ethanol are 0.53 and 0.48 V, respectively.
Results
Electrosynthesis and characterization of Cu-complex/Cu
The electrodes were fabricated via two steps, including in situ electrodeposition of Cu-complex film on Cu substrate (denoted as Cu-complex/Cu) and in situ electroreduction of the CuII in Cu-complex film to obtain Cu–Cu2O/Cu electrode, which are discussed in the following.
In this work, six Cu-complexes with different ligands, which are denoted as Complex-1 to Complex-6 (Supplementary Scheme 1), were in situ deposited on Cu substrate by electrosynthesis method40,41, where the numbers correspond to that of the ligands shown in Supplementary Scheme 1. The procedures to prepare the Cu-complex/Cu is shown schematically in Fig. 1a taking Complex-1 (pyromellic acid, 1,2,4,5-H4BTC) as the example. Briefly, the electrosynthesis of Complex-1/Cu was carried out in a two-electrode system, which consisted mainly of Cu foil anode, Cu foil cathode, ligand, and electrolyte solution. The CuII ions produced from electro-oxidation of the Cu anode (Cu substrate) coordinated with the electro-deprotonated organic ligands generated at the cathode, and then the complex film grew in situ on Cu substrate surface.

In situ anodic electrodeposition of copper complex on copper foil. a Schematic illustration of the process to prepare Cu-Complex-1 film on Cu substrate. H4BTC = pyromellic acid, BTC4− = tetraanion of pyromellitic acid; b SEM images of Complex-1 formed on Cu substrate at electrodeposition times of 0, 0.5, 5, 15, 30, 60 min. The side-view of the film obtained at 60 min is also given in the figure as an insert
The growth of Complex-1 film on Cu substrate at different electrodeposition times were investigated using scanning electron microscopy (SEM, Fig. 1b). It is demonstrated that Complex-1 film with 3D hierarchical structure was formed with increasing electrodeposition time. The side-view SEM image of the Complex-1/Cu with electrodeposition time of 60 min is also given (Fig. 1b, Supplementary Figs. 1–6), indicating that the complex film of about 60 µm compacted tightly with the Cu substrate without any noticeable crack. This is understandable because the Cu ions were generated from the Cu substrate and the complexes grew on the substrate surface.
The complex with electrodeposition time of 60 min was further characterized by Fourier transform infrared (FT-IR) and X-ray diffraction (XRD) methods (Supplementary Figs. 7 and 8). The results show that the complexes were synthesized successfully and there was no evident impurity in the complexes. The X-ray photoelectron spectroscopy (XPS) spectra indicate that CuII was the major metallic species in the complex (Supplementary Fig. 9). N2 adsorption/desorption method was used to determine the surface areas and porous properties of the complex, and the results (Supplementary Fig. 10 and Supplementary Table 1) demonstrate that the complex has porous structure, and the molar ratio of Cu and ligand in the complex was 2.69, which was obtained from the Cu and C contents in the complexes determined by induced coupled plasma (ICP) methods (Supplementary Table 2) and the complex loading was 0.67 mg cm−2 after 60 min deposition (Supplementary Table 3). Small angle X-ray scattering (SAXS) technique was employed to analyze the fractal structure. The surface fractal (Ds) values were obtained from the lnI(h) vs ln(h) plots (Supplementary Fig. 11). It can be seen that the surface of the complexes was coarse. The surface roughness factors of the complex film and Cu substrate were also determined by measuring the double layer capacitance using the reported method4,42. The results (Supplementary Fig. 12 and Supplementary Table 4) indicate that the surface of the complex film was much rougher than that of Cu substrate. All the results above confirm that the Cu-complex was successfully synthesized on the Cu substrate by the electrodeposition method. This is understandable because multibenzenecarboxylates are commonly used ligands in preparation of metal complexes43,44,45,46,47,48. The formation of the complexes was further proved by successful synthesis of the single crystal with 3D supramolecular structure (Supplementary Figs. 13−18 and Supplementary Tables 15–21) using interface growth method.
Decomposition and characterization of Cu–Cu2O/Cu
We found that the CuII in Cu-complex/Cu electrode was reduced quickly in N2 or CO2-saturated 0.1 M KCl aqueous electrolyte at applied potential of −0.4 V vs RHE. The current density at different electroreduction times are given in Fig. 2a. The current density was reduced to near zero after 5 min in N2-saturated electrolyte. The current density in CO2-saturated electrolyte became constant after 5 min, and the current after 5 min was originated from the CO2 electroreduction, which will be discussed in detail in the following sections. The change of electrode properties with electroreduction times was studied by SEM, XRD, XPS, and X-ray absorption fine structure spectroscopy (XAFS) methods. SEM images (Fig. 2b and Supplementary Fig. 19) show clearly that the 3D flower-like morphology of the complex was transformed into the dendritic 3D structure completely in initial 5 min of electroreduction. The morphology did not change notably after 5 min, and the thickness of the dendritic layer was about 40 µm. The XPS spectra of the catalyst film at different electroreduction times given in Fig. 2c, d indicate that the valence states of Cu changed from CuII to CuI and Cu0 during the initial stage of electroreduction. The film after electroreduction was composed of Cu and Cu2O as can be known from the XRD patterns (Supplementary Fig. 20). The ratio of CuI/Cu0 became constant after 5 min of electroreduction (Supplementary Fig. 21 and Supplementary Table 5). The XAFS modeling and analysis results (Supplementary Figs. 22, 23 and Supplementary Table 6) further revealed the existence of Cu–O and Cu–Cu in the catalyst after electroreduction, and the CuI/Cu0 ratio was unchanged after 5 min, which is consistent with the results of XPS study (Supplementary Fig. 21 and Supplementary Table 5). The transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM) images of the catalyst with an electroreduction time of 5 min are presented in Fig. 2e–g, demonstrating that high density of grain boundaries (GBs) exist between Cu and Cu2O. The oxygen vacancies on the interface of Cu and Cu2O create new defect states located in the band gap, and the electrons on the defect states are easily excited, leading to the enhanced conductivity49,50,51. All the results above show that the Complex-1 changed into Cu and Cu2O composite denoted as Cu–Cu2O-1 (the number corresponds to the number of the complex) on Cu substrate after electroreduction, and the composition and morphology were stable after 5 min of reduction. As expected, the Cu–Cu2O-1 film and Cu substrate in the Cu–Cu2O-1/Cu also compacted tightly without any noticeable crack (Fig. 2b). In this work, the Cu-complexes on the Cu substrate acted as the precursor and template for the formation of the Cu–Cu2O-1 catalyst with 3D structure on the Cu substrate.

Decomposition of copper–cuprous oxide electrodes. a Reduction current density in 0.1 M KCl solution saturated with CO2 and N2 at applied potential of −0.4 V vs RHE, and the inserts show the current density at initial stage; b SEM images of the electrode with different electroreduction times (0 min, 2 min, 5 min, 10 min, 5 h); c, d Quasi in situ XPS spectra of the catalyst with different electroreduction times (0 min, 2 min, 5 min, 10 min, 5 h). Cu 2p photoelectron peak distribution: (i) CuII of Complex-1 structure; (ii) CuI/Cu0 of Cu–Cu2O-1 structure; O 1s photoelectron peak distribution: (i) Oxygen of CuxO on Complex-1 structure; (ii) Oxygen of CuxO on Cu–Cu2O-1 structure; and (iii) oxygen defect species; e TEM and f, g HR-TEM images of the Cu–Cu2O-1 with electrorduction time of 5 h
The electrocatalytic performance of Cu–Cu2O/Cu
The linear sweep voltammograms (LSVs) over Cu–Cu2O-1/Cu electrode in CO2 or N2 saturated 0.1 M KCl aqueous electrolyte were determined (Supplementary Fig. 24), which shows reduction of CO2 clearly. The dependence of current density on concentration of KCl solution saturated by CO2 over Cu–Cu2O-1/Cu electrode is provided in Supplementary Fig. 25, demonstrating that 0.1 M KCl solution was the optimal electrolyte.
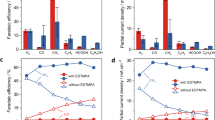
The electrocatalytic performance of the Cu–Cu2O-1/Cu electrode was further investigated by electrolysis of CO2 at different applied potentials in 0.1 M KCl aqueous solution using a typical H− type cell, in which water acted as a proton source36,52, and the results are given in Fig. 3. The experiment at each condition was repeated three times and the average values are used. In this work, the applied potential is referencing to the reversible hydrogen electrode (RHE) and the current density is calculated by geometric surface area. Under the reaction conditions, acetic acid and ethanol were the only liquid products detected by NMR spectroscopy, and H2, CO and CH4 were the gaseous product determined using gas chromatography (GC) (Supplementary Fig. 26 and Supplementary Table 7). The electrocatalytic performances of neat Cu foil, Cu foam obtained by direct electrodeposition without using ligand, Cu2O and CuO electrodes prepared using reported method11,40,41,53 were also studied for comparison (Supplementary Fig. 27), and the results are also given in Fig. 3. Clearly, Cu–Cu2O−1/Cu electrode had much better performance for C2 product (acetic acid and ethanol) generation than other Cu-based electrodes. The applied potential and the overpotential over the Cu–Cu2O-1/Cu electrode were much lower (Fig. 3a), and the FE was much higher (Fig. 3b). At applied potential of −0.4 V vs RHE, the overpotential on Cu–Cu2O-1/Cu electrode was as low as 0.53 V (for acetic acid) and 0.48 V (for ethanol) (Fig. 3c, d). The current density and FE for the C2 product were 11.5 mA cm−2 and 80.7% (48% for acetic acid and 32% for ethanol), respectively. Supplementary Table 8 presents the recent advances in reduction of CO2 to C2+ products. Previous report indicates that dendritic Cu catalysts are capable of reducing CO2 to C2 products. These desired structures can be synthesized through different methods7,38,54. However, the characteristics of the catalyst such as structure and oxidation state are difficult to be controlled, which lead to high overpotential of the C2 products. Using molecular complex as an additive is another method to promote reduction of CO2 to C2 product30,31,32. However, as a precursor, the complex was generally prepared ex situ and used as a homogeneous additive in decomposition of the new structure. Compared with these methods, the Cu–Cu2O catalyst synthesized in this work through in situ deposition/decomposition method exhibited controllable 3D structure, high charge transfer rate and lower overpotential toward liquid C2 products. By comparing with the data reported (Supplementary Table 8), it can be known that this in situ electrosynthesized Cu–Cu2O-1/Cu electrode had significantly lower applied potential and overpotential, higher current density, and higher FE for liquid C2 products, especially in aqueous electrolyte. In addition, the Cu–Cu2O-1/Cu electrode exhibited long-term stability in the electrolysis, which was known from the fact that the current density and FE did not change considerably with electrolysis time in 24 h, as shown in Supplementary Fig. 28. The result in Supplementary Table 9 indicates that the amounts of the C2 products changes with applied potential and the amounts of the generated C2 products increased almost linearly with increasing of electrolysis time (Supplementary Fig. 29). The turnover frequency (TOF) for CO2 reduction reaction over the catalysts at different applied potentials are provided in Supplementary Table 10. The Cu–Cu2O-1/Cu displayed best performance and the TOFs of acetic acid and ethanol were 17.0 and 15.6 h−1, respectively.

Carbon dioxide reduction over various copper-based electrocatalysts. a The total current density at different applied potentials; b Faradaic efficiencies of the C2 products at different applied potentials; c, d Dependence of partial current density of acetic acid and ethanol on overpotential. Data were collected at room temperature and ambient pressure; electrolyte, 0.1 M KCl; CO2 stream, 5 sccm
Electrolyte also plays an important role in the CO2 reduction. Recently, researchers have reported the reduction of CO2 to C2+ products in KCl aqueous electrolyte6,55,56,57, and it was found to decrease the overpotential and increase the C2 selectivity. The rise in local pH facilitated a higher amount of adsorbed *CO, which promotes their C–C coupling to C2 products56,58,59,60,61,62. To investigate whether the local pH affected the product selectivity, we have also carried out experiments using KHCO3 electrolyte (Supplementary Figs. 30–32 and Supplementary Table 11). The linear sweep voltammograms using CO2 saturated KHCO3 aqueous electrolytes of different concentrations (Supplementary Fig. 31a) illustrate that maximum current density was obtained in 0.5 M KHCO3 solution. The electrolysis experiments showed that the current densities and selectivity to C2 products in KCl electrolyte were higher than those in KHCO3 electrolyte at all applied potentials (Supplementary Fig. 31c), indicating that the electrolytes influence the electrochemical reaction significantly. One of the main reasons may be that in concentrated KHCO3 solution, sufficient HCO3− is present to neutralize OH−. Therefore, the formation of H2 is favored and C2 product is less preferred58. The result suggests that local pH influenced the selectivity of the reaction, which is consistent with the results in the recent studies6,54,55,56,57.
To confirm that CO2 was carbon source of the product, we also conducted the blank experiments using N2 to replace CO2 in the electrolysis. The experiments showed that no product was formed in the electrolysis when using N2. To further verify that the product was derived from CO2 reduction, isotope−labeled 13CO2 was used to study the reaction over Cu–Cu2O-1/Cu. From 1H NMR spectra in Supplementary Figs. 33 and 34, we can only see the H signal of 13CH3 group on the acetic acid and ethanol, which splits into two peaks by the coupling with 13C atom. Moreover, the 13C NMR spectra showed strong 13C signals as 13CO2 was used as the feedstock (Supplementary Fig. 35). The results further confirm that all the carbon atoms in the product were from CO2, which is consistent with the conclusion obtained from the blank experiment using N2.
Investigation into the catalytic ability of Cu–Cu2O/Cu
There are several reasons for the outstanding performance of the Cu–Cu2O-1/Cu electrode in the electrocatalytic reduction of CO2 to the C2 products. First, the Cu–Cu2O-1 catalyst and Cu substrate contacted very well in the electrode. This structure can reduce or eliminated the contacting resistance between the catalyst and the Cu substrate, which is favorable to reducing the overpotential and applied potential. In order to get some evidence to support this point, some control experiments were designed. We prepared Complex-1 by solvothermal method using the same ligand and fabricated the complex electrode by loading the complex suspension onto the carbon paper (CP) using conventional drop-casting method (Supplementary Figs. 36 and 37), and the complex was also reduced to Cu–Cu2O composite by electroreduction to obtain Cu–Cu2O-1/CP electrode as known from the XPS characterization (Supplementary Fig. 38). Supplementary Fig. 39 compared the CV curves obtained using the Cu–Cu2O-1/Cu and Cu–Cu2O-1/CP electrodes prepared by the two methods. Supplementary Fig. 40 and Supplementary Table 12 show the electrolysis results over the two electrodes at the optimized potential. The applied potential and overpotential over the Cu–Cu2O-1/Cu electrode was much lower than that over Cu–Cu2O-1/CP electrode. Electrochemical impedance spectroscopy (EIS) study showed that the film resistance (Rf) between the catalyst and substrate in the Cu–Cu2O-1/Cu electrode was much smaller than that in the Cu–Cu2O-1/CP. Moreover, the charge transfer resistance (Rct) between the electrolyte and electrode surface of Cu–Cu2O-1/Cu electrode was also much smaller than that of Cu–Cu2O-1/CP electrode system (Supplementary Fig. 41 and Supplementary Table 13). The smaller Rf and Rct result in the lower applied potential and overpotential. Second, the Cu–Cu2O-1 catalyst in Cu–Cu2O-1/Cu had 3D dendritic structure. The 3D structure results in abundant exposed active sites (i.e., grain boundaries, oxygen vacancies), leading to high activity of the Cu–Cu2O-1 electrocatalyst. To verify the importance of 3D structure, we performed the CO2 reduction using the Cu–Cu2O-1/Cu electrodes obtained from in situ reduction of the Complex-1/Cu prepared at different electrodeposition times (Fig. 1b and Supplementary Fig. 42). Supplementary Fig. 42 shows that the current density increased continuously with electrodeposition time. This can be explained by the fact that the amount of Cu–Cu2O-1 catalyst in the Cu–Cu2O-1/Cu electrodes increased with electrodeposition time. The FE increased with electrodeposition time at beginning, and then became nearly independent of electrodeposition time. Figures 1b and 2b show that the 3D structure gradually formed with increasing deposition time. A reasonable interpretation of the result is that 3D structure provides more opportunity for the C–C coupling because of the longer residue time of the reaction species. When the film of 3D structure was thick enough, the residue time was long enough for the intermediates to form the C2 product, and thus the FE was not changed obviously with the thickness of the Cu–Cu2O-1 film. Third, the ligand in the complex is crucial for the formation of precursor, which is important for the construction of Cu–Cu2O structure (Supplementary Fig. 19). This can be known from the fact that the FE over other Cu-based electrodes, such as metallic Cu, electrodeposited Cu foam, Cu2O, and CuO was much lower (Fig. 3, Supplementary Fig. 27). In addition, the density functional theory (DFT) calculation and experimental study also showed that Cu–Cu2O is preferred electrocatalyst for production of C2 products57,63. The synergy between the active surfaces of CuI/Cu0 improves significantly both CO2 activation and CO dimerization to generate C2 products. The results also showed that use of the Cu-complex as the precursor is crucial for the outstanding performance because the efficiency of Cu catalyst obtained by direct electrodeposition was much poorer.
The electrocatalytic performance of other Cu–Cu2O/Cu electrodes
In order to verify the versatility of the method, we also prepared Cu–Cu2O/Cu electrodes using other ligands, including 1,2,4-H3BTC (2), 1,3,5-H3BTC (3), 1,2,3-H3BTC (4), 2,6-H2PyDC (5), and 1,4-H2BDC (6). The complexes on Cu substrate are denoted as Complex-2, Complex-3, Complex-4, Complex-5, Complex-6, respectively. The complexes were also characterized using different methods, and the results are given in Supplementary Figs. 1–11, Supplementary Figs. 13–18, and Supplementary Tables 1–4. The corresponding electrodes are represented by Cu–Cu2O-2/Cu, Cu–Cu2O-3/Cu, Cu–Cu2O-4/Cu, Cu–Cu2O-5/Cu, Cu–Cu2O-6/Cu, and the characterization results are provided in Supplementary Figs. 19–21 and Supplementary Table 5. All the results showed that 3D dendritic Cu2O–Cu films on Cu electrodes could also be prepared using these ligands, and the detailed discussion on this are provided in the supporting information. Subsequently, the as-prepared Cu–Cu2O/Cu electrodes were used for CO2 reduction in 0.1 M KCl aqueous electrolyte, and the results are given in Fig. 4, Supplementary Figs. 43–45. Generally, these Cu–Cu2O/Cu electrodes could also promote CO2 electroreduction to C2 products at very low applied potential and overpotential with high selectivity in the aqueous electrolyte. At −0.4 V vs RHE, the FEs of C2 products over Cu–Cu2O-2/Cu, Cu–Cu2O-3/Cu, Cu–Cu2O-4/Cu, Cu–Cu2O-5/Cu, and Cu–Cu2O-6/Cu were 67.6%, 56.7%, 54.4%, 50.2%, and 41.8%, respectively. The results suggest that the strategy to prepare 3D dendritic Cu–Cu2O/Cu electrodes for highly efficient electroreduction of CO2 to C2 product is versatile. Among all the electrodes, Cu–Cu2O-1/Cu has the best activity, which may results from the lowest charge transfer resistance between electrolyte and electrode (Supplementary Table 13B). In addition, the largest surface roughness factor of the Complex-1 precursor (Supplementary Fig. 11 and Supplementary Table 4) favored the generation of Cu–Cu2O with abundant exposed active sites. All these factors can enhance the catalytic activity of the electrode.

Carbon dioxide reduction over copper–cuprous oxide electrodes prepared using ligands two to six. a The total current density at different applied potentials; b Faradaic efficiencies of the C2 products at applied potential of −0.4 V vs RHE. c, d Dependence of partial current density of acetic acid and ethanol on overpotential, respectively. Data were collected at room temperature and ambient pressure; electrolyte, 0.1 M KCl; CO2 stream, 5 sccm
Mechanism
With the aim to understand the mechanistic pathway toward the formation of acetic acid and ethanol by CO2 reduction, some control experiments were conducted in the presence of the possible reaction intermediates, such as CO, formic acid, formaldehyde, acetaldehyde, and acetic acid. Supplementary Table 14 presents the production rates of acetic acid and ethanol obtained in these experiments. The results indicate that CO, formaldehyde and acetaldehyde are three important intermediates in the C2 product pathway. Supplementary Fig. 46A shows the IR spectra of the electrolyte after different electrolysis times. The intensity of acetic acid and ethanol increased with increasing electrolysis time, indicating that the amount of C2 products generated increased with electrolysis time, suggesting the formation of acetic acid and ethanol from electroreduction of CO2. We also conducted the IR study in the presence of possible reaction intermediates (Supplementary Fig. 46B). The result indicates that CO, formaldehyde, and acetaldehyde are the possible intermediate in the mechanistic pathway. This conclusion is consistent with the result in Supplementary Table 14. Supplementary Scheme 2 gives the possible mechanistic pathway for the electrocatalytic production of acetic acid and ethanol. The three key steps are (i) CO2 activation; (ii) C1 product formation, which was found to compete with C2 products; (iii) CO dimerization, which can be important *COCHO or *COCO intermediate for C2 products formation. Subsequently, the reaction is divided into two paths. For formation of ethanol, both *COCHO and *COCO can be considered as the precursors. The C–C bond is subsequently formed and ethanol is generated via reduction of CH3CHO intermediate. For acetic acid, the intermediate *COCHO is further reduced at the electrode surface to form –CH3COO− species. This tentative mechanistic pathway is able to explain the experimental observations discussed above.
Discussion
In summary, 3D dendritic Cu–Cu2O/Cu electrodes for CO2 reduction can be prepared by electroreduction of in situ deposited Cu-complex film on Cu substrate successfully. They had outstanding performance for electrochemical reduction of CO2 to C2 product in KCl aqueous solution. The results show that reducing contacting resistance between the catalysts and the substrate in the electrodes, increasing exposed active sites in the 3D structure and chemical composition of Cu–Cu2O composition are favorable to enhance the efficiency for the reduction of CO2 to C2 products. This work opens a simple way to efficient transformation of CO2 into valuable chemicals or liquid fuels, and the findings are helpful for designing other electrodes for efficient electroreduction of CO2. We believe that this work will trigger many interesting work in the future.
Methods
Materials
CO2 (Beijing Beiwen Gas Chemical Industry Co., Ltd., research grade) had a purity of 99.999% and used as received. 13CO2 (99 atom% 13C, <1 atom%) was purchased from Beijing Gaisi Chemical Gases Center. Pyromellic acid (1,2,4,5-H4BTC, 96%), 1,2,4-benzenetricarboxylic acid (1,2,4-H3BTC, 98%), 1,3,5-benzenetricarboxylic acid (1,3,5-H3BTC, purity > 99%), 1,2,3-benzenetricarboxylic acid (1,2,3-H3BTC, 95%), 2,6-pyridinedicarboxylic acid (2,6-H2PyDC, 99%), tetramethylammonium bromide (TMAB, 99%), and tetrabutyl ammonium perchlorate (TBAP, 99.9%) were obtained from J&K Scientific Ltd. 1,4-Bezenedicarboxylic acid (1,4-H2BDC, 99%) was purchased from TCI. Cu(NO3)2·3H2O (A. R. grade), acetonitrile (AcN, A. R. grade), methanol (A. R. grade), dimethylformamide (DMF, A. R. grade), acetone (A. R. grade), and Cu foil (0.2 mm in thickness, purity > 99.99%) were provided by Sinopharm Chemical Reagent Co., Ltd, China. Potassium chloride (KCl, 99.997%) was purchased from Alfa Aesar China Co., Ltd, which was purified by recrystallization three times before use. Toray carbon paper (CP, TGP-H-60, 19 × 19 cm) and Nafion N-117 membrane (0.180 mm thick, ≥ 0.90 meg/g exchange capacity) were purchased from Alfa Aesar China Co., Ltd. Polytetrafluoroethylene (PTFE, 60 wt% aqueous solution) was purchased from Sigma-Aldrich Co. LLC. Tetrabutylammonium hexfluorophosphate (TBAPF6, >98%) was provided by the Centre of Green Chemistry and Catalysis, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences.
In situ electrosynthesis of the Complex-1 to 5 on Cu substrate
The structures of the ligands used are shown in Supplementary Scheme 1. The procedures were similar to that used for the electrosynthesis of metal organic frameworks40,41. Typically, two Cu foils with dimension of 0.2 mm × 10 mm × 10 mm were used for the working and counter electrode with a gap of 1 cm and the electrochemical experiments could be controlled by a DC mode on a galvanostat/potentiostat (CS310, Wuhan Corrtest Instrument Co., China). The electrolyte consisted of 25 mL of ethanol/water (75:25 vol%) solution, 5–15 mg MTAB (99.9%, supporting electrolyte), and 5 mg Cu-complex-1 (or 15 mg Complex-2, or 10 mg Complex-3, or 5 mg Complex-4, or 15 mg Complex-5, or 15 mg Complex-6). The electrosynthesis was performed by applying a potential difference of 9.0 V between the Cu foil electrodes in 70 °C electrolyte. Cu-Complex was in situ formed at the anode.
In situ electrosynthesis of Complex-6 on Cu substrate
The procedures for the synthesis of Complex-6 film with organic linker 1,4-BDC was similar to that discussed above. The electrolyte was DMF solvent with 20 g/L of linkers and 15 mg of MTAB was used as the supporting electrolyte. Before electrodeposition, the electrolyte was stirred at 70 °C for several minutes before a clear solution was obtained. The anodic deposition was performed by applying a potential difference of 9.0 V between the two Cu foils.
Synthesis of Complex-1 by solvothermal method
The procedures to synthesize Complex-1 by thermal method were similar to that for preparing HKUST-145. A ethanol/water (75:25 vol%) solution was used as the solvent. In a typical experiment, 1,2,4,5-H4BTC (0.12 g, 0.5 mM) was dissolved in 20 mL of ethanol, and Cu(NO3)2·3H2O (0.242 g, 1.0 mM) was dissolved in 5 mL of H2O. The Cu(NO3)2·3H2O solution was slowly added to the 1,2,4,5-H4BTC solution with stirring at room temperature. Then 2 mL of DMF was added to the mixture solution with stirring. The mixture was transferred into a Teflon-lined autoclave and the reaction occurred at 90 °C under hydrothermal condition for 48 h. After cooling to room temperature, the solid was collected and washed with H2O and ethanol. The obtained Cu-Complex-1 was dried in a vacuum oven at 80 °C for 24 h.
Synthesis of Complex-3 by solvothermal method
The procedures to synthesize the Complex-3 with organic linker 1,3,5-H3BTC were similar to that reported previously45. In a typical experiment, Cu(NO3)2·3H2O (2.6 g, 10.7 mM) was dissolved in 30 mL of H2O in a flask. 1,3,5-H3BTC (0.68 g, 3.2 mM) was dissolved in 30 mL of ethanol. The Cu(NO3)2·3H2O solution was slowly added to the 1,3,5-H3BTC solution with stirring at room temperature. The solution became turbid with formation of precipitate. DMF (2 mL) was added to the mixture, and then the combination was transferred to a Teflon-lined autoclave and allowed to be reacted at 80 °C for 24 h. After cooling to room temperature, the solid was collected and washed with H2O and ethanol. The obtained Cu-Complex was dried in a vacuum oven at 80 °C for 24 h.
Fabrication of the electrodes using Cu-Complexes synthesized by solvothermal method
The complex electrodes were prepared by loading Cu-Complex suspension onto the carbon paper (CP) using drop-casting method. Briefly, the as-prepared Cu-Complex was dispersed in acetone, then suitable amount of 6.0 wt% PTFE aqueous solution was added to the as prepared Cu-Complex dispersion. The final mixture contained 10 mg/mL complex and 0.6 wt% PTFE, which was ultrasonicated for 30 min to form a uniform suspension. Then 0.1 mL of the suspension was loaded on the 1.0 cm × 1.0 cm CP. The electrodes were dried in an oven at 60 °C for 1 h before the electrochemical experiment.
Electrodeposition of Cu2O on Cu foil
Electrochemical growth of Cu2O film was accomplished by using a conventional three-electrode cell using the method reported11. The copper foil substrate was used as working electrode, a platinum sheet as counter electrode, and Ag/AgCl as reference electrode. The electrodeposition was carried out cathodically using the solution containing 0.02 M CuSO4 and 0.34 M DL-Lactic acid (85%) at pH = 9 adjusted using 2 M NaOH. Deposition were carried out at 70 °C and −0.95 V vs Ag/AgCl for 30 min.
Electrodeposition of CuO on Cu foil
CuO thin films were grown by electrodeposition technique using potentiostatic method reported53. A standard three-electrode cell was used for the electrodeposition. The copper foil substrate was used as working electrode, a platinum sheet as counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. The electrodeposition of CuO was carried out cathodically from an aqueous bath composed of CuSO4 (0.03 M) and L(+) tartaric acid (0.03 M). The deposition of CuO thin film was carried out at a deposition potential of −650 mV vs SCE. The solution pH was adjusted to 10–12 by the addition of 2 M NaOH solution. The deposition was carried out at 75 °C for 30 min.
Direct electrodeposition of Cu foam on Cu foil
The method was similar to that for synthesis of Cu-Complex40,41, and the main difference was that the ligand was not used. A Cu foam on Cu foil was electrodeposited in a solution containing 0.25 M CuSO4, 25 mL of ethanol/water (75:25 vol%) solution, 5–15 mg MTAB (99.9%, supporting electrolyte) at ambient temperature (25 °C). Prior to experiment, the electrolyte was deoxygenated by high-purity nitrogen for at least 30 min. Cu foam was deposited onto Cu foil by applying a constant potential of 9.0 V between two Cu foil electrodes in the electrolyte mentioned above.
Material characterization
Powder X-ray diffraction (XRD) patterns were acquired with a X-ray diffractometer (Model D/MAX2500, Rigaka) with Cu-Kα radiation, and the scan speed was 5°/min. X-ray photoelectron spectroscopy (XPS) analysis was conducted on the Thermo Scientific ESCALab 250Xi (USA) using 200 W monochromatic Al Kα radiation. The 500 µm X-ray spot was used for XPS analysis. The base pressure in the analysis chamber was about 3 × 10−10 mbar. The contents of Cu and C in the complex were determined by induced coupled plasma optical emission spectrometer (ICP-OES, VARIN VISTA-MPX). The morphologies of materials were characterized by a HITACHI S-4800 scanning electron microscope (SEM) and a JEOL JEM-2100F high-resolution transmission electron microscopy (HR-TEM).The porosity properties and surface areas of the materials were obtained from nitrogen adsorption–desorption isotherms determined using a Micromeritics ASAP 2020 M system. For XRD, ICP-OES, gas adsorption–desorption measurements, in order to get enough sample, at least 10 electrodes were prepared at the same conditions and the catalyst films were scraped and collected for characterization.
The FT-IR spectra were collected at a resolution of 4 cm−1 on a Bruker Vector 27 spectrophotomfeter in the 400–4000 cm−1 region. The IR spectra of samples were measured by the conventional KBr pellet method. To get enough sample, at least 10 electrodes were prepared at the same conditions and the complex films were scraped and collected.
The Quasi in situ X-ray photoelectron spectra (XPS) were measured on an AXIS ULTRA DLD spectrometer with AlKα resource (hv = 1486.6 eV). The protective sample transfer procedure was similar to that reported in the literature64. For investigating the evolution of Cu species in the reaction process, catalysts were electrolyzed with different times in the CO2-saturated electrolytes. After that, the samples were immediately immersed in Ar-prepurged acetone and then transferred into an Ar-filled glove box to protect the catalyst from directly contacting of air. Then, the samples was cleaned, dried and cut into 3 × 3 mm and glued on a stage with a double sided adhesive. The stage was evacuated into vacuum to prevent the sample to be oxidized in the air. The subsequent testing processes were the same as that of the common X-ray photoelectron spectroscopy. The contaminated carbon C 1s signal at 284.8 eV was used to calibrate binding energies. The spectra were deconvolved with XPS PEAK 4.1 software by subtracting the Shirley background and applying the Lorentzian–Gaussian function. The modified Auger parameter (αCu) was calculated according to the following equation:
where EB is the binding energy of the Cu 2p3/2 core level and EK is the kinetic energy of the Cu LMM Auger electron.
X-ray absorption fine structure spectroscopy (XAFS) measurements were taken at 1W2B beamline of Beijing Synchrotron Radiation Facility (BSRF), China. The Cu K-edge (8979 eV) XAFS spectra were collected under electrochemical environment using the fluorescence detection method. The EXAFS data were processed according to the standard procedures using the ATHENA module implemented in the IFEFFIT software packages. The Cu K-edge X-ray absorption spectra of the catalysts were collected after reaction at different times. The Cu K-edge X-ray absorption spectra of Cu-complexes, CuO, Cu2O, and Cu foil were collected for reference.
Small angle X-ray scattering (SAXS) experiments were carried out at Beamline 1W2A at the Beijing Synchrotron Radiation Facility. The apparatus and the procedures were similar to that used in previous work52,65. The data were collected using a CCD detector (MAR) with maximum resolution of 3450 × 3450 pixels. The wavelength of the X-ray was 1.54 Å, and the distance of the sample to detector was 1.31 m. In a typical experiment, the sample was added into the sample cell, and the X-ray scattering data were recorded. The 2-D SAXS images were obtained from the detector and then transformed into the profiles of intensity (I) vs wavevector (q) by the software FiT2D52,65. To get enough sample, at least 10 electrodes were prepared at the same conditions and the complex films were scraped and collected.
For XRD, ICP-OES, N2 adsorption–desorption, FT-IR, XAFS, and SAXS measurements, in order to get enough sample, at least 10 electrodes were prepared at the same conditions and the catalyst films were scraped and collected for characterization.
Crystal data and structure refinement for Complex-1, Complex-4, and Complex-5
In total, 0.05 mmol of 1,2,4,5-H4BTC, or 1,2,3-H3BTC or 2,6-H2PyDC was dissolved in 20 mL ethanol, and then a solution of 0.05 mmol Cu(NO3)2 in 5 mL H2O was added to the mixture. As the solvent evaporating, blue crystals were obtained after solvent evaporation. Then, it was washed with acetone and air dried. The product was recrystallized from ethanol–H2O (4:1).
Crystallographic measurements for Complex-1, Complex-4, and Complex-5 were made using a Bruker APEX area-detector diffractometer. The intensity data were collected using graphite monochromataed Mo kα radiation. The structures were solved by direct methods and refined by full-matrix least-squares techniques on F2. Structure solution and refinement was accomplished using SIR97, SHELXL97, and WINGX. The all H atoms were positioned geometrically and allowed to ride on their parent atoms. The molecular structure plots were prepared by using ORTEPIII.
Crystal data and structure refinement are given in Supplementary Figs. 13–18. Anisotropy thermal parameters, structure factors, full lists of bond distances, bond angles, and torsion angles are given in Supplementary Table 15. Selected bond lengths and angles are given in Supplementary Tables 16–21. The CCDC number of Complex-1, Complex-4, and Complex-5 were 1936102, 1936103, and 193104, respectively.
pH value measurements
The pH values of gas-free, N2-satuarated, and CO2-saturated electrolyte solutions (0.1 M KCl) were determined by an electronic pH meter (METTLER TOLEDO FE-20K). To determine the pH value of the CO2-saturated electrolyte solution at ambient temperature, CO2 was bubbled into 20 mL solution in a beaker (25 mL/min) under stirring. The pH value of the solution was monitored at different times, and the value was recorded when the value became unchanged with time.
Linear sweep voltammetry and pH value measurements
An electrochemical workstation (CHI 6081E, Shanghai CH Instruments Co., China) was used for all CO2 reduction experiments. The apparatus and procedures were similar to that used previously52. The linear sweep voltammetry (LSV) measurements were carried out in a single compartment cell with three-electrode configuration, which consisted of a working electrode, a platinum gauze auxiliary electrode and Ag/AgCl reference electrode. Aqueous solution of KCl or KHCO3 was used as cathode electrolyte. Before each set experiment, the electrolyte was bubbled with N2 or CO2 for at least 30 min to form N2 or CO2 saturated solution. The LSV measurement in gas-saturated electrolyte was conducted at a sweep rate of 20 mV/s in the potential range of −0.2 to −1.1 V vs Ag/AgCl. For a catalyst prepared using the Cu-complex as the precursor, the complex was first electroreduced for 5 min at 0.4 V vs RHE to form Cu–Cu2O catalyst.
In this work, all potentials were referencing to RHE by considering the pH of the solution by Eq. (2)66.
Thermodynamic potentials
The overpotential is the difference between the applied potential and the thermodynamic potential for the reaction of interest. Thermodynamic potentials for the reactions of CO2 to acetic acid and CO2 to ethanol were calculated, and the values are 0.125 and 0.084 V, respectively. The data are calculated from the standard molar Gibbs energy of formation at 298.15 K67,68 and the method is discussed below. The calculation assumes that gases are at 1 atm and liquids are in their pure form.
Therefore, compared with hydrogen evolution reaction (HER) in aqueous electrolyte solutions (Eq. (5)), the primary reactions that occurred on the electrode in aqueous solution at pH 7.0 vs standard hydrogen electrode (SHE) (Eq. (6)) is:
Applying the Nernst equation (Eq. (7)), the equilibrium potential is estimated to be:
Therefore, using the same method, E0 for the half reaction of CO2 to acetic acid and CO2 to ethanol are:
Electrochemical impedance spectroscopy measurements
The experimental apparatus was the same as that for LSV measurements. The experiment was carried out in CO2 saturated 0.1 M KCl solution. The impedance spectra was recorded at open circuit potential (OCP) with an amplitude of 5 mV of 10−2–10−5 Hz. The data obtained from the electrochemical impedance spectroscopy (EIS) measurements were fitted using the Zview software (Version 3.1, Scribner Associates, USA).
Electrochemical capacitance measurements
Cyclic voltammetry (CV) was used for the electrochemical capacitance measurements. The experiments were performed by collecting cyclic voltammograms in 0.1 M TBAPF6 in MeCN solution at scan rates from 50 to 400 mV/s. The experiment was performed in three-electrode configurations, in which a silver wire was used as quasi-reference electrode and Pt gauze was used as conunter electrode. Data obtained over a potential range of 40 mV around the open circuit potential of Cu–Cu2O-1/Cu electrode is shown in Fig. S13. Plot of the current density from the quasi-square CV curves, calculated at the open circuit potential, as a function of scan rate. The surface roughness factor was calculated from the capacitance data4,42.
CO2 reduction electrolysis and product analysis
Controlled potential electrolysis (CPE) was carried out. The electrolysis experiments were conducted at 25 °C in a typical H-type cell, which was similar to that used in our previous works for CO2 electrochemical reduction52. The as synthesized Cu-Complex electrodes were used as the working electrode. The Ag/AgCl (saturated KCl) was used as the reference electrode and the Pt gauze was used as counter electrode. The cathode and anode compartments were separated through a Nafion 117 proton exchange membrane. A KCl or KHCO3 aqueous solution was used as cathode electrolyte. H2SO4 aqueous solution (0.5 M) was used as anodic electrolyte. Electrochemical reduction of CO2 was carried out at ambient temperature. Under the continuous stirring, CO2 was bubbled into the catholyte (25 mL/min) for 60 min before electrolysis. After that, potentiostatic electrochemical reduction of CO2 was carried out with CO2 bubbling (5 mL/min). The gaseous product was collected and analyzed by gas chromatography (GC, HP 4890D), which was equipped with FID and TCD detectors using helium as the internal standard. 1H NMR and 13C NMR measurements of products were performed on a Bruker Avance III 400 HD spectrometer. The samples were measured using DMSO-d6 as a lock solution and TMS as an internal standard. To identify the reduction products, 1H NMR spectroscopy was carried out to quantify the liquid products after electrolysis of desired time at each given potential. The current density and Faradaic efficiencies (FEs) of the products were calculated using the amounts of the products obtained from GC and 1H NMR analysis.
Similarly, all potentials were referencing to RHE using the pH of the CO2-saturated KCl solution.
After the quantification, the Faradaic efficiency (FE) toward each product were calculated as follows58:
where n is number of moles of electrons to participate in the Faradaic reaction, F is the Faraday constant (96485 C mol−1), and C is the amount of charge passed through the working electrode.
Turnover frequency measurements
TOF for CO2 reduction reaction over the catalysts was calculated on the basis of amounts of products, electrolysis time, the moles of Cu species on the electrode determined gravimetrically. The moles of Cu species were assumed to the same as that in the complexes precursors. To get the reliable amount of the complex, at least 10 Cu-complex/Cu electrodes were prepared, and the mass of the complex was calculated from the mass difference before and after electrodeposition. The moles of Cu was known from the molecular weight of the complex (Supplementary Table 15) and mass of the complex in the complex/Cu electrode (Supplementary Table 3).
Tafel analysis
The Tafel plots were constructed from the partial current density and the overpotentail. The overpotential was the difference between the applied potential and the thermodynamic potential for the reaction of interest4. As mentioned above thermodynamic potentials for the reactions of CO2 to acetic acid and CO2 to ethanol are 0.125 and 0.084 V, respectively.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available from the corresponding author on request.
References
He, M. Y., Sun, Y. H. & Han, B. X. Green carbon science: scientific basis for integrating carbon resource processing, utilization, and recycling. Angew. Chem. Int. Ed. 52, 9620–9633 (2013).
Qiao, J. L., Liu, Y. Y., Hong, F. & Zhang, J. J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014).
Gao, S. et al. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 529, 68–71 (2016).
Li, C. W. & Kanan, M. W. CO2 Reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 134, 7231–7234 (2012).
Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208–1213 (2015).
Zhou, Y. S. et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 10, 974–980 (2018).
Luna, P. D. et al. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 1, 103–110 (2018).
Dinh, C. T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Loiudice, A. et al. Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 55, 5789–5792 (2016).
Hori, Y., Takahashi, I., Koga, O. & Hoshi, N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A: Chem. 199, 39–47 (2003).
Ren, D. et al. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(I) oxide catalysts. ACS Catal. 5, 2814–2821 (2015).
Lum, Y., Yue, B., Lobaccaro, P., Bell, A. T. & Ager, J. W. Optimizing C-C coupling on oxide-derived copper catalysts for electrochemical CO2 reduction. J. Phys. Chem. C. 121, 14191–14203 (2017).
Roberts, F. S., Kuhl, K. P. & Nilsson, A. High selectivity for ethylene from carbon dioxide reduction over copper nanocube electrocatalysts. Angew. Chem. Int. Ed. 54, 5179–5182 (2015).
Song, Y. et al. High-selectivity electrochemical conversion of CO2 to ethanol using a copper nanoparticle/N-doped graphene electrode. Chem. Sel. 1, 6055–6061 (2016).
Jiang, K. et al. Metal ion cycling of Cu foil for selective C-C coupling in electrochemical CO2 reduction. Nat. Catal. 1, 111–119 (2018).
Mistry, H. et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123 (2016).
Gao, D. F. et al. Plasma-activated copper nanocube catalysts for efficient carbon dioxide electroreduction to hydrocarbons and alcohols. ACS Nano 11, 4825–4831 (2017).
Zhuang, T. T. et al. Steering post-C-C coupling selectivity enable high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 1, 421–428 (2018).
Song, Y. F. et al. Metal-free nitrogen-doped mesoporous carbon for electroreduction of CO2 to ethanol. Angew. Chem. Int. Ed. 56, 10840–10844 (2017).
Clark, E. L., Hahn, C., Jaramillo, T. F. & Bell, A. T. Electrochemical CO2 reduction over compressively strained CuAg surface alloys with enhanced multi-carbon oxygenate selectivity. J. Am. Chem. Soc. 139, 15848–15857 (2017).
Costentin, C., Robert, M. & Savéant, J. M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 42, 2423–2436 (2013).
Zhu, D. D., Liu, J. L. & Qiao, S. Z. Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 28, 3423–3452 (2016).
Andreiad is, E. S. et al. Molecular engineering of a cobalt-based electrocatalytic nanomaterial for H2 evolution under fully aqueous conditions. Nat. Chem. 5, 48–53 (2013).
Blasco-Ahicart, Soriano-López, Z., Carbó, J. J., Poblet, J. M. & Galan-Mascaros, G. Polyoxometalate electrocatalysts based on earth-abundant metals for efficient water oxidation in acidic media. Nat. Chem. 10, 24–30 (2018).
Schley, N. D. et al. Distinguishing homogeneous from heterogeneous catalysis in electrode-driven water oxidation with molecular iridium complexes. J. Am. Chem. Soc. 133, 10473 (2011).
Stracke, J. J. & Finke, R. G. Electrocatalytic water oxidation beginning with the cobalt polyoxometalate [Co4(H2O)2(PW9O34)2]10−: Identification of heterogeneous CoOx as the dominant catalyst. J. Am. Chem. Soc. 133, 14872–14875 (2011).
Cobo, S. et al. A janus cobalt-based catalytic material for electro-splitting of water. Nat. Mater. 11, 802–807 (2012).
Kaeffer, N. et al. The dark side of molecular catalysis: Diimine-dioxime cobalt complexes are not the actual hydrogen evolution electrocatalyst in acidic aqueous solutions. ACS Catal. 6, 3727–3737 (2016).
Kanan, M. W. & Nocera, D. G. In situ formation of an oxygen-evoluing catalyst in neutral water containing phosphate and Co2+. Science 321, 1072–1075 (2008).
Huan, T. N. et al. From molecular copper complexes to composite electrocatalytic materials for selective reduction of CO2 to formic acid. J. Mater. Chem. A 3, 3901–3907 (2015).
Han, Z., Kortlever, R., Chen, H. Y., Peters, J. C. & Agapie, T. CO2 reduction selective for C≥2 products on polycrystalline copper with N-substituted pyridinium additives. ACS Cent. Sci. 3, 853–859 (2017).
Hoang, T. T. H., Ma, S., Gold, J. I., Kenis, P. J. A. & Gewirth, A. A. Nanoporous copper films by additive-controlled electrodeposition: CO2 reduction catalysis. ACS Catal. 7, 3313–3321 (2017).
Shin, H. C., Dong, J. & Liu, M. Nanoporous structures prepared by an electrochemical deposition process. Adv. Mater. 15, 1610–1614 (2003).
Lu, X. Y. & Zhao, C. Electrodeposition of hierarchically structured three-dimensional nickel-iron electrodes for efficient oxygen evolution at high current densities. Nat. Commun. 6, 6616 (2015).
Sun, Y. J. et al. Electrodeposited cobalt-sulfide catalyst for electrochemical and photoelectrochemical hydrogen generation from water. J. Am. Chem. Soc. 135, 17699–17702 (2013).
Medina-Ramos, J., DiMeglio, J. L. & Rosenthal, J. Efficient reduction of CO2 to CO with high current density using in situ or ex situ prepared Bi-based materials. J. Am. Chem. Soc. 136, 8361–8367 (2014).
Ding, C. M., Li, A. L., Lu, S. M., Zhang, H. F. & Li, C. In situ electrodeposition indium nanocrystals for efficient CO2 reduction to CO with low overpotential. ACS Catal. 6, 6438–6443 (2016).
Reller, C. et al. Selective electroreduction of CO2 toward ethylene on nano dendritic copper catalysts at high current density. Adv. Energy Mater. 7, 1602114 (2017).
Chen, Y. H. & Kanan, M. W. Tin oxide dependence of the CO2 reduction efficiency on tin electrodes and enhanced activity for tin/tin oxide thin-film catalysts. J. Am. Chem. Soc. 134, 1986–1989 (2012).
Li, W. J., Tu, M., Cao, R. & Fischer, R. A. Metal-organic framework thin films: electrochemical fabrication techniques and corresponding applications & perspectives. J. Mater. Chem. A 4, 12356–12369 (2016).
Yadnum, S. et al. Site-selective synthesis of Janus-type metal-organic framework composites. Angew. Chem. Int. Ed. 53, 4001–4005 (2014).
Yang, J. et al. A multifunctional biphasic water splitting catalyst tailored for integration with high-performance semiconductor photoanodes. Nat. Mater. 16, 335–341 (2017).
Köferstein, R. & Robl, C. Synthesis and crystal structure of two CuII-benzene-1,2,4,5-tetracarboxylates with three-dimensional open frameworks. Z. Anorg. Allg. Chem. 640, 310–316 (2014).
Qin, C., Wang, X. L., Wang, E. B., Hu, C. W. & Xu, L. [Cu2(HBTC)2(H2O)2(μ2-H2O)]·2H2O: a new arm-shaped two-dimensional copper coordination polymer having both rhombic cavities and helical-like channels. Inorg. Chem. Commun. 7, 788–791 (2004).
Jeong, N. C., Samanta, B., Lee, C. Y., Farha, O. K. & Hupp, J. T. Coordination-chemistry control of proton conductivity in the iconic metal-organic framework material HKUST-1. J. Am. Chem. Soc. 134, 51–54 (2011).
Ma, L. F., Li, C. P., Wang, L. Y. & Du, M. CoII and ZnII coordination frameworks with benzene-1,2,3-tricarboxylate tecton and flexible dipyridyl co-ligand: a new type of entangled architecture and a unique 4-connected topological network. Cryst. Growth Des. 11, 3309–3312 (2011).
van Albada, G. A., Ghazzali, M., Al-Farhan, K., Bouwman, E. & Reedijk, J. Three new pyridine-2,6-dicarboxylate copper(II) compounds with coordinated pyrimidine-based ligands: synthesis, characterization and crystal structures. Polyhedron 52, 1059–1064 (2013).
Carson, C. G. et al. Synthesis and structure characterization of copper terephthalate metal-organic frameworks. Eur. J. Inorg. Chem. 2009, 2338–2343 (2009).
Puigdollers, A. R., Schlexer, P., Tosoni, S. & Pacchioni, G. Increasing oxide reducibility: the role of metal/oxide interface in the formation of oxygen vacancies. ACS Catal. 7, 6493–6531 (2017).
Cavalca, F. et al. Nature and distribution of stable subsurface oxygen in copper electrodes during electrochemical CO2 reduction. J. Phys. Chem. C. 121, 25003–25009 (2017).
Kumar, B. et al. Reduced SnO2 porous nanowires with a high density of grain boundaries as catalysts for efficient electrochemical CO2-into-HCOOH conversion. Angew. Chem. Int. Ed. 56, 3645–3649 (2017).
Zhu, Q. G. et al. Efficient reduction of CO2 into formic acid on a lead or tin electrode using an ionic liquid catholyte mixture. Angew. Chem. Int. Ed. 55, 9012–9016 (2016).
Dhanasekaran, V., Mahalingam, T., Chandramohan, R., Rhee, J. K. & Chu, J. P. Electrochemical deposition and characterization of cupric oxide thin films. Thin Solid Films 520, 6608–6613 (2012).
Rahaman, M., Dutta, D., Zanetti, A. & Broekmann, P. Electrochemical reduction of CO2 into multicarbon alcohols on activated Cu mesh catalysts: an indentical location (IL) study. ACS Catal. 7, 7946–7956 (2018).
Nakata., K., Ozaki, T., Terashima, C., Fujishima, A. & Einaga, Y. High-yield electrochemical production of formaldehyde from CO2 and seawater. Angew. Chem. Int. Ed. 53, 871–874 (2014).
Huang, Y., Ong, C. W. & Yeo, B. S. Effects of electrolyte anions on the reduction of carbon dioxide to ethylene and ethanol on copper (100) and (111) surface. ChemSusChem 11, 3299–3306 (2018).
Lee, S., Kim, D. & Lee, J. Electrocatalytic production of C3-C4 compounds by conversion of CO2 on a chloride-induced Bi-phasic Cu2O-Cu catalyst. Angew., Chem. Int. Ed. 54, 14701–14705 (2015).
Hori, Y., Murata, A. & Takahashi, R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electroe in aqueous solution. J. Chem. Soc., Faraday Trans. 1: Phys. Chem. Condens. Phases 85, 2309–2326 (1989).
Varela, A. S., Ju, W., Reier, T. & Strasser, P. Tuning the catalytic activity and selectivity of Cu for CO2 electroreduction in the presence of halides. ACS Catal. 6, 2136–2144 (2016).
Gao, D. F., Arán-Ais, R., Jeon, H. S. & Cuenya, B. R. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2, 198–210 (2019).
Gao, D. F., Scholten, F. & Cuenya, B. R. Improved CO2 electroreduction performance on plasma-activated Cu catalysts via electrolyte design: halide effect. ACS Catal. 7, 5112–5120 (2017).
Raciti, D. R., Mao, M., Park, J. H. & Wang, C. Local pH effect in the CO2 reduction reaction on high-surface-area copper electrocatalysts. J. Electrochem. Soc. 165, F799–F804 (2018).
Xiao, H., Goddard, W. A. III, Cheng, T. & Liu, Y. Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. PNAS 114, 6685–6688 (2017). 201702405.
Wu, Z. S., Gan, Q., Li, X. L., Zhong, Y. R. & Wang, H. L. Elucidating surface restructuring-induced catalytic reactivity of cobalt phosphide nanoparticles under electrochemical conditions. J. Phys. Chem. C. 122, 2848–2853 (2018).
Kang, X. C. et al. Mesoporous inorganic salts with crystal defects: unusual catalysts and catalyst supports. Chem. Sci. 6, 1668–1675 (2015).
Asadi, M. et al. Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquid. Science 353, 467–470 (2016).
Li, C. W., Ciston, J. & Kanan, M. W. Electroreduction of carbon monoxide to liquid fuel on oxide –derived nanocrystalline copper. Nature 508, 504–507 (2014).
CRC. Handbook of Chemistry and Physics 9th edn, section 5 (CRC, 2013).
Acknowledgements
The authors thank the National Key Research and Development Program of China (2017YFA0403101), National Natural Science Foundation of China (21773267, 21673248, 21733011) and the Chinese Academy of Sciences (QYZDY-SSW-SLH013). SAXS experiments were carried out at Beijing Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
Q.G.Z., J.M. and B.X.H. proposed the project, designed the experiments and wrote the paper; Q.G.Z. performed the whole experiments. X.F.S. conducted the 13C measurements. D.X.Y. performed FT-IR, XAFS and the XPS tests. L.R.Z. and J.Z. assisted in analyzing the XAFS results. X.C.K. and Z.H.W. assisted in analyzing the SAXS data. X.F.S., D.X.Y. and X.C.K. also performed the analysis of experimental data; B.X.H. supervised the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications would like to thank Hailiang Wang and the other, anonymous, reviewers for their contributions to the peer review of this work. Peer review reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, Q., Sun, X., Yang, D. et al. Carbon dioxide electroreduction to C2 products over copper-cuprous oxide derived from electrosynthesized copper complex. Nat Commun 10, 3851 (2019). https://doi.org/10.1038/s41467-019-11599-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11599-7
This article is cited by
-
Dinuclear Cu(I) molecular electrocatalyst for CO2-to-C3 product conversion
Nature Catalysis (2024)
-
Ambient temperature CO2 fixation to pyruvate and subsequently to citramalate over iron and nickel nanoparticles
Nature Communications (2023)
-
Stabilizing copper sites in coordination polymers toward efficient electrochemical C-C coupling
Nature Communications (2023)
-
Interfacial engineering of SnO2/Bi2O2CO3 heterojunction on heteroatoms-doped carbon for high-performance CO2 electroreduction to formate
Nano Research (2023)
-
Cu2+1O/CuOx heterostructures promote the electrosynthesis of C2+ products from CO2
Nano Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
