
Abstract
Axially chiral biaryl amino-alcohols play a pivotal role in organic synthesis and drug discovery. However, only a very few enantioselective methods have been reported to synthesize chiral biaryl amino-alcohols. Therefore, the rapid enantioselective construction of optically active biaryl amino-alcohols still remains a formidable challenge. Here we report an N-heterocyclic carbene (NHC)-catalyzed atropoenantioselective acylation of biphenols triggered by a cooperative strategy consisting of desymmetrization followed by kinetic resolution. This protocol features broad substrate scope and good functional group tolerance, and allows for a rapid construction of axially chiral biaryl amino-alcohols in good to high yields and with excellent enantioselectivities. Furthermore, the structurally diverse axially chiral biaryl amino-alcohol derivatives provide multiple possibilities for chemists to develop catalysts or ligands for different chemical transformations.
Similar content being viewed by others

Introduction
Axially chiral biaryls1,2,3 have widely applied in many areas, including material science4,5 and drug discovery6. In addition, chiral biaryls have often played as ligands7 or catalysts8 in the development of enantioselective catalytic transformations. During the past two decades, BINOL and BINAM have proven to be representative examples in this category9,10,11,12,13,14. Afterward, NOBIN (Fig. 1) gradually grows up to the next privileged scaffold15 with numerous important applications, due to its perfect enantio-control and prominent bioactivity (Fig. 1, (R)-Streptonigrin, an antitumor agent).

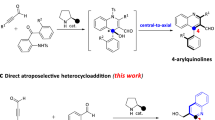
Representative molecules and synthetic protocols. a Two representative axially chiral molecules. b Asymmetric kinetic resolution of achiral biaryl amino alcohols. c Our synthetic proposal via a NHC-catalyzed atroposelective synthesis of axially chiral biaryl amino-alcohols via a cascade strategy of desymmetrization followed by kinetic resolution
In sharp contrast to the asymmetric preparation of BINOL or BINAM16,17,18,19, to date, only a very few enantioselective methods have been reported to synthesize NOBIN-type biaryl amino alcohols20,21,22,23. In 1992, Kocovsky24 and coworkers reported the asymmetric synthesis of NOBIN via oxidative of 2-naphthol with 2-naphylamine. However, the use of large excess amount of chiral auxiliary and required multistep crystallization sometimes restrict its further application. To face this issue, Tan’s group25 uncovered a phosphoric acid-catalyzed cross-coupling of 2-naphthylamines with iminoquinones, affording chiral biaryl amino alcohols in a concise and catalytic pattern. Shortly after, optical resolution rapidly developed into the next attractive method to isolate NOBIN enantiomers by leveraging the power of diastereoisomeric chiral salt formation, but was confined to unstable reproducibility in practical26,27. Getting the NOBIN derivatives through a direct transformation from chiral raw materials (e.g., BINOL28 or BINAM29) has also become an interesting way. Regrettably, this protocol is mostly applied to construct chiral binaphthyl-type amino-alcohols. Recently, kinetic resolution is recognized as an impresive technology to produce such biaryl structures. For example, the groups of Maruoka30 and Zhao31 reported a phase-transfer- or N-heterocyclic carbene-catalyzed asymmetric kinetic resolution to prepare enantioenriched chiral biaryl amino alcohols, independently (Fig. 1). However, no more than 50% theoretical yields inevitably affect the application of this approach. Overall, rapid synthesis of axially chiral biaryl amino-alcohols in a highly atropoenantioselective fashion is still in its infancy
Our group is interested in exploring carbene catalysis for the rapid assembling of axially chiral molecules. To date, we have successfully reported the N-heterocyclic carbene-catalyzed atropoenantioselective [3 + 3] annulation and kinetic resolution of anilides, affording valuable chiral α-pyrone-aryls and isoindolinones, respectively32,33. Despite aforementioned achievements, the unsolved challenges and the continuously growing demands of atropoenantiomers still drive us to develop more efficient and revolutionary protocols. We herein report a carbene-catalyzed34,35,36,37,38,39,40,41 atroposelective synthesis of axially chiral biaryl amino-alcohols via a cascade strategy of desymmetrization followed by kinetic resolution42 (Fig. 1). First, this approach can deliver nonclassical NOBIN derivatives (e.g., biphenyl- or phenyl-naphthyl-type amino-alcohols) in a high chemical yield. From the aspect of structural diversity, non-classical NOBIN-type derivatives will offer more possibilities for the exploration of new chiral catalysts or ligands. Second, in contrast to an independent desymmetrization or kinetic resolution method, the cooperation of desymmetrization43,44,45,46,47,48 with kinetic resolution49,50,51,52,53 has certain superiority in the control of enantioselectivity.
Results
Reaction optimization
We commenced our study by using biphenols (1a–c) as the model prochiral substrates, aldehyde (2a) as acylation reagent54,55,56,57,58,59,60,61,62 and DQ as oxidant63,64. Key results of reaction optimization are briefly summarized in Table 1. Building upon the indanol-derived triazolium scaffold, precatalysts with N-2,4,6-(Cl)3C6H265, or N-C6F566, substituents (Table 1, C2 and C3), derived from α-amino acids, were tested but exhibited low conversions and enantioselectivities (Table 1, entries 4 and 5). Interestingly, precatalyst C1 with N-2,4,6-(Me)3C6H2 (N-Mes)67 substituent provided 3a in 63% with 85% ee (Table 1, entry 1). If substrate 1b or 1c replaced 1a (R = NO2 or NH2; More information about changing R groups, see Supplementary Table 2), low ee and yield were observed (Table 1, entries 2 and 3). To our delight, further improved enantioselectivities were achieved with the N-2,4,6-(iPr)3C6H2 substituted catalyst C4 or C568 (Table 1, entries 6 and 7). Notably, the triazolium catalyst C5, which bears a strong electron-withdrawing group (NO2) at the remote aryl position, afforded 95% ee and 80% yield (Table 1, entry 7). We then chose catalyst C5, substrate 1a and 2a for further optimization. After extensive screening of solvents, bases and catalyst loading, an ideal result was obtained by using 10 mol% of C5 as catalyst, DCM as the solvent, and K2CO3 as the base (Table 1, entry 15). Meanwhile, nuclear magnetic resonance (NMR) spectrum confirmed that the main byproduct of this reaction was a bisadduct in which two hydroxyl groups were both acylated (see Supplementary Note 4).
Substrate scope
Having the optimal condition in hand, we turned our attention to the generality of aldehydes. As indicated in Fig. 2, a diverse set of aliphatic aldehydes underwent acylating reactions, affording their corresponding products in high yields with high to excellent ee values (Figs. 2 and 3b–i). Unambiguously, the steric effect of aldehydes has identified to be a critical factor for achieving high enantioselectivity. Aliphatic aldehydes bearing a steric bulky chain afforded a higher ee value (Figs. 2 and 3a, b, f, g). When aromatic aldehydes were used as substrates, reactions are generally messy, probably caused by the competitive benzoin reaction.

Scope of aldehydes. Reaction conditions: a mixture of 1a (0.10 mmol), 2 (0.15 mmol), K2CO3 (0.12 mmol), and DQ (0.12 mmol) in CH2Cl2 (1.0 mL) was stirred at room temperature under N2 for 12–24 h

Scope of biaryl biphenols. Reaction conditions: a mixture of 4a–4o (0.10 mmol), 2a (0.15 mmol), K2CO3 (0.12 mmol), and DQ (0.12 mmol) in CH2Cl2 (1.0 mL) was stirred at room temperature under N2 for 12–24 h. bent-cat. C5 was used
Encouraged by success with aliphatic aldehydes, we then planed to investigate the reactivity of biaryl-type biphenol substrates. N-Cbz protected compounds 4a–j having electron-donating groups (Me, MeO) and/or electron-withdrawing groups (Cl, CN) on the lower ring (Fig. 3, phenyl ring B) generated the coresponding products in high yields (85–94%) with excellent enantioselectivities (96– >99% ee). Cyclohexane ring fused biphenyl substrate also performed well as expected (Fig. 3, 4l, 91% yield and 96% ee). Pleasingly, the indole-based biaryl substrate also proceeded smoothly to afford product 4m with a promising ee value. Gratifyingly, the phenyl-naphthalenyl-type biaryl substrate was also tolerated to deliver the anticipated structure 4n with excellent enantioselectivity (Fig. 3, 99% ee). Incidentally, enantiomeric products can be achieved entirely through the enantiomer catalyst and similar reaction conditions (Fig. 3, 4o).
Mechanistic studies
To verify the mechanism, two control experiments were conducted. As indicated in Fig. 1a, 4), ent-3a was obtained in 49% yield with 93% ee in the presence of DQ (0.6 equiv). On the basis of these data, we can conclude that desymmetrization is a key contributor for enantio-control (Vfast/Vslow = 28:1)69. Meanwhile, we also wonder whether the second acylation is a kinetic resolution process, eventually resulting in an improved enantioselectivity. To approve this hypothesis, the control experiment of (±)-3a with 2a was designed and carried out, generating ent-3a in 47% yield with 76% ee (Fig. 1a, 4). This experiment result suggests that the conversion of 1a to major enantiomer 3a is much faster than the process between 1a and minor enantiomer (Vfast/Vslow = 5:1)69.

Postulated mechanistic pathways. a The control experiments (Eqs. (1) and (2)) show that the desymmetrization process is the main contributor to the observed ee of the product and the second acylation is a effective KR process that could improve the ee of (−)-3a. b The postulated mechanistic pathway to generate product (−)-3a
Synthetic transformations and applications
We anticipated that the biaryl amino-alcohol 7 (Fig. 5), prepared from 4n via a N-Cbz protected intermediate 6, could be directly utilized as a chiral catalyst in asymmetric alkylation reaction. As highlighted in Fig. 6, compound 7 successfully catalyzed the asymmetric alkylation of Ni-complex 8 with alkyl bromide 9–11 to generate complex 12–14 with promising er values. After a subsequent deprotection, 12–14 could efficiently transfer to valuable chiral α-amino acids70. To further expand synthetic utility, we conducted a Ru-catalyzed asymmetric reduction of ketone 18 by using chiral biaryl amino-alcohol derivatives 15–17 as ligands (for preparation of 15–17, see Supplementary Note 5). As indicated in Fig. 6, ligands 15–17 led to the corresponding product 19 in suggestable er values. In addition, a gram-scale synthesis (3.4 mmol) carried out under standard conditions afforded optically pure 3a in a pleasant result (Fig. 7, 89% yield, 99% ee). The absolute configuration of derivative 6 was determined by X-ray single crystal analysis (See Supplementary Fig. 1), and other structures were assigned by analogy.

Synthetic transformations. Reaction conditions: (1) 4n, TMSCHN2, CHCl3:MeOH/5:1, r.t., 24 h. (2) NaOMe, MeOH, r.t., 1.0 h. (3) Pd/C, H2, MeOH, r.t., 3.0 h

Synthetic applications. a Use of 7 as a chiral catalyst. b Utility of 15–17 as chiral ligands

Gram-scale synthesis. Reaction conditions: a mixture of 1a (3.4 mmol, 1.19 g), 2a (5.1 mmol, 0.55 mL), in CH2Cl2 (34.0 mL) was stirred at room temperature for 15 h
Discussion
In conclusion, we have developed an atropoenantioselective NHC-catalyzed acylation for the preparation of axially chiral biaryl amino-alcohols. The cascade strategy of desymmetrization followed by kinetic resolution could efficiently deliver axially chiral biaryl amino-alcohols with high to excellent ee values (up to >99% ee). Further studies on the exploration of other substrates and applications are ongoing projects in our laboratory.
Methods
Synthesis of racemic ¾
In a glovebox, a flame-dried Schlenk reaction tube equipped with a magnetic stir bar, were added racemic NHC precatalyst C13 (0.01 mmol,), K2CO3 (16.6 mg, 0.12 mmol), oxidant DQ (49.0 mg, 0.12 mmol), 1 (0.10 mmol), 2 (0.15 mmol), and freshly distilled CH2Cl2 (1.0 mL). The reaction mixture was stirred at room temperature for 12 h. The mixture was then filtered through a pad of Celite washed with CH2Cl2. After solvent was evaporated, the residue was purified by flash column chromatography to afford the racemic product 3/4.
Synthesis of ¾
In a glovebox, a flame-dried Schlenk reaction tube equipped with a magnetic stir bar, were added NHC precatalyst C5 (0.01 mmol,), K2CO3 (16.6 mg, 0.12 mmol), oxidant DQ (49.0 mg, 0.12 mmol), 1 (0.10 mmol), 2 (0.15 mmol), and freshly distilled CH2Cl2 (1.0 mL). The reaction mixture was stirred at room temperature until the starting material 1 was completely consumed (12–24 h). The mixture was then filtered through a pad of Celite washed with CH2Cl2. After solvent was evaporated, the residue was purified by flash column chromatography to afford the desired product 3/4.
Data availability
For 1H, 13C NMR and high-performance liquid chromatography spectra of compounds in this paper, see Supplementary Figs. 1–187. For details of the synthetic procedures, see Supplementary Notes. The supplementary crystallographic data for this paper could be obtained free of charge from The Cambridge Crystallographic Data Centre (6: CCDC 1880233) via www.ccdc.cam.ac.uk/data_request/cif.
References
Kozlowski, M. C., Morgan, B. J. & Linton, E. C. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev. 38, 3193–3207 (2009).
Bringmann, G., Gulder, T., Gulder, T. A. M. & Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 111, 563–639 (2011).
Tanaka, K. Transition-metal-catalyzed enantioselective [2 + 2 + 2] cycloadditions for the synthesis of axially chiral biaryls. Chem. Asian J. 4, 508–518 (2009).
Pu, Lin 1,1′-Binaphthyl dimers, oligomers, and polymers: molecular recognition, asymmetric catalysis, and new materials. Chem. Rev. 98, 2405–2494 (1998).
Hembury, G. A., Borovkov, V. V. & Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 108, 1–73 (2008).
LaPlante, S. R., Edwards, P. J., Fader, L. D., Kakalian, A. & Hucke, O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 6, 505–513 (2011).
Chen, Y., Yekta, S. & Yudin, A. K. Modified BINOL ligands in asymmetric catalysis. Chem. Rev. 103, 3155–3212 (2003).
Kočovský, P., Vyskočil, & & Smrcina, M. Non-symmetrically substituted 1, 1′-binaphthyls in enantioselective catalysis. Chem. Rev. 103, 3213-–33246 (2003).
Akiyama, T. Stronger brønsted acids. Chem. Rev. 107, 5744–5758 (2007).
Ding, K., Li, X., Ji, B., Guo, H. & Kitamura, M. Ten years of research on NOBIN chemistry. Curr. Org. Synth. 2, 499–545 (2005).
Ding, K., Guo, H., Li, X., Yuan, Y. & Wang, Y. Synthesis of NOBIN derivatives for asymmetric catalysis. Top. Catal. 35, 105–116 (2005).
Jacobsen, E. N., Pfaltz, A. & Yamamoto, H. (eds). Comprehensive Asymmetric Catalysis. 1, (Springer Science, New York, 2003).
De, C. K., Pesciaioli, F. & List, B. Catalytic asymmetric benzidine rearrangement. Angew. Chem. 125, 9463–9465 (2013).
Li, G. Q. et al. Organocatalytic aryl–aryl bond formation: an atroposelective [3, 3]-rearrangement approach to BINAM derivatives. J. Am. Chem. Soc. 135, 7414–7417 (2013).
Zhou, Q. L. (ed.). Privileged Chiral Ligands and Catalysts. (John Wiley, Hoboken, NJ, 2011).
Chen, Y. H. et al. Atroposelective synthesis of axially chiral biaryldiols via organocatalytic arylation of 2-naphthols. J. Am. Chem. Soc. 137, 15062–15065 (2015).
Wang, J. Z. et al. Symmetry in cascade chirality-transfer processes: a catalytic atroposelective direct arylation approach to BINOL derivatives. J. Am. Chem. Soc. 138, 5202–5205 (2016).
Moliterno, M. et al. Quinine-catalyzed asymmetric synthesis of 2, 2′-binaphthol-type biaryls under mild reaction conditions. Angew. Chem. Int. Ed. 55, 6525–6529 (2016).
Narute, S., Parnes, R., Toste, F. D. & Pappo, D. Enantioselective oxidative homocoupling and cross-coupling of 2-naphthols catalyzed by chiral iron phosphate complexes. J. Am. Chem. Soc. 138, 16553–16560 (2016).
Smrčina, M., Lorenc, M., Hanuš, V. & Kočovský, P. A facile synthesis of 2-amino-2′-hydroxy-1,1′-binaphthyl and 2,2′-diamino-1,1′-binaphthyl by oxidative coupling using copper (II) chloride. Synlett 4, 231–232 (1991).
Smrcina, M., Polakova, J., Vyskocil, S. & Kocovsky, P. Synthesis of enantiomerically pure binaphthyl derivatives. Mechanism of the enantioselective, oxidative coupling of naphthols and designing a catalytic cycle. J. Org. Chem. 58, 4534–4538 (1993).
Smrcina, M. et al. Selective cross-coupling of 2-naphthol and 2-naphthylamine derivatives. A facile synthesis of 2,2′,3-trisubstituted and 2, 2′,3,3′-tetrasubstituted 1,1′-binaphthyls. J. Org. Chem. 59, 2156–2163 (1994).
Ding, K. et al. Novel two-phase oxidative cross-coupling of the two-componentmolecular crystal of 2-naphthol and 2-naphthylamine. Chem. Commun. 7, 693–694 (1997).
Smrcina, M., Lorenc, M., Hanus, V., Sedmera, P. & Kocovsky, P. Synthesis of enantiomerically pure 2,2′-dihydroxy-1,1′-binaphthyl, 2,2′-diamino-1,1′-binaphthyl, and 2-amino-2′-hydroxy-1,1′-binaphthyl. Comparison of processes operating as diastereoselective crystallization and as second order asymmetric transformation. J. Org. Chem. 57, 1917–1920 (1992).
Chen, Y. H., Qi, L. W., Fang, F. & Tan, B. Organocatalytic atroposelective arylation of 2-naphthylamines as a practical approach to axially chiral biaryl amino alcohols. Angew. Chem. Int. Ed. 56, 16308–16312 (2017).
Mahmoud, H., Han, Y., Segal, B. M. & Cai, L. Chiral enrichment of 2-amino-2′-hydroxy-1,1′-binaphthyl. Tetrahedron 9, 2035–2042 (1998).
Smrčina, M. et al. Synthesis and resolution of racemic 2-amino-2′-hydroxy-1,1′-binaphthyl. Collect. Czech. Chem. Commun. 61, 1520–1524 (1996).
Singer, R. A. & Buchwald, S. L. Preparation of 2-amino-2′-hydroxy-1,1′-binaphthyl and N-arylated 2-amino-1,1′-binaphthyl derivatives via palladium-catalyzed amination. Tetrahedron Lett. 40, 1095–1098 (1999).
Patel, D. C. et al. Gram scale conversion of R-BINAM to R-NOBIN. J. Org. Chem. 81, 1295–1299 (2016).
Shirakawa, S., Wu, X. & Maruoka, K. Kinetic resolution of axially chiral 2-amino-1,1′-biaryls by phase-transfer-catalyzed N-allylation. Angew. Chem. Int. Ed. 52, 14200–14203 (2013).
Lu, S., Poh, S. B. & Zhao, Y. Kinetic resolution of 1,1′‐biaryl‐2, 2′‐diols and amino alcohols through NHC-Catalyzed atroposelective acylation. Angew. Chem. Int. Ed. 53, 11041–11045 (2014).
Zhao, C. et al. Enantioselective [3 + 3] atroposelective annulation catalyzed by N-heterocyclic carbenes. Nat. Commun. 9, 611 (2018).
Bie, J., Lang, M. & Wang, J. Enantioselective N-heterocyclic carbene-catalyzed kinetic resolution of anilides. Org. Lett. 20, 5866–5871 (2018).
Enders, D., Niemeier, O. & Henseler, A. Organocatalysis by N-heterocyclic carbenes. Chem. Rev. 107, 5606–5655 (2007).
Biju, A. T., Kuhl, N. & Glorius, F. Extending NHC-catalysis: coupling aldehydes with unconventional reaction partners. Acc. Chem. Res. 44, 1182–1195 (2011).
Ryan, S. J., Candish, L. & Lupton, D. W. Acyl anion free N-heterocyclic carbene organocatalysis. Chem. Soc. Rev. 42, 4906–4917 (2013).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485 (2014).
Mahatthananchai, J. & Bode, J. W. On the mechanism of N-heterocyclic carbene-catalyzed reactions involving acyl azoliums. Acc. Chem. Res. 47, 696–707 (2014).
Menon, R. S., Biju, A. T. & Nair, V. Recent advances in employing homoenolates generated by N-heterocyclic carbene (NHC) catalysis in carbon–carbon bond-forming reactions. Chem. Soc. Rev. 44, 5040–5052 (2015).
Flanigan, D. M., Romanov-Michailidis, F., White, N. A. & Rovis, T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 115, 9307–9387 (2015).
Wang, M. H. & Scheidt, K. A. Cooperative catalysis and activation with N-heterocyclic carbenes. Angew. Chem. Int. Ed. 55, 14912–14922 (2016).
Mori, K. et al. Enantioselective synthesis of multisubstituted biaryl skeleton by chiral phosphoric acid catalyzed desymmetrization/kinetic resolution sequence. J. Am. Chem. Soc. 135, 3964–3970 (2013).
Liu, Q. & Rovis, T. Asymmetric synthesis of hydrobenzofuranones via desymmetrization of cyclohexadienones using the intramolecular Stetter reaction. J. Am. Chem. Soc. 128, 2552–2553 (2006).
Wadamoto, M., Phillips, E. M., Reynolds, T. E. & Scheidt, K. A. Enantioselective synthesis of α,α-disubstituted cyclopentenes by an N-heterocyclic carbene-catalyzed desymmetrization of 1,3-diketones. J. Am. Chem. Soc. 129, 10098–10099 (2007).
Li, B. S., Wang, Y., Proctor, R. S., Jin, Z. C. & Chi, Y. R. Carbene-catalyzed desymmetrization of 1,3-diols: access to optically enriched tertiary alkyl chlorides. Chem. Commun. 52, 8313–8316 (2016).
Lu, S. et al. Access to enantiopure triarylmethanes and 1,1‐diarylalkanes by NHC‐catalyzed acylative desymmetrization. Chem. Eur. J. 23, 2275–2281 (2017).
Huang, Z. et al. Access to P-stereogenic phosphinates via N-heterocyclic carbene-catalyzed desymmetrization of bisphenols. J. Am. Chem. Soc. 138, 7524–7527 (2016).
Wu, Z. & Wang, J. Enantioselective medium-ring lactone synthesis through an NHC-catalyzed intramolecular desymmetrization of prochiral 1, 3-diols. ACS Catal. 7, 7647–7652 (2017).
Suzuki, Y., Yamauchi, K., Muramatsu, K. & Sato, M. First example of chiral N-heterocyclic carbenes as catalysts for kinetic resolution. Chem. Commun. 23 2770–2771 (2004).
Kano, T., Sasaki, K. & Maruoka, K. Enantioselective acylation of secondary alcohols catalyzed by chiral N-heterocyclic carbenes. Org. Lett. 7, 1347–1349 (2005).
De Sarkar, S., Biswas, A., Song, C. H. & Studer, A. Kinetic resolution of secondary alcohols by NHC-catalyzed oxidative esterification. Synthesis 2011, 1974–1983 (2011).
Lu, S., Poh, S. B., Siau, W. Y. & Zhao, Y. Kinetic resolution of tertiary alcohols: highly enantioselective access to 3-hydroxy-3-substituted oxindoles. Angew. Chem. Int. Ed. 52, 1731–1734 (2013).
Binanzer, M., Hsieh, S. Y. & Bode, J. W. Catalytic kinetic resolution of cyclic secondary amines. J. Am. Chem. Soc. 133, 19698–19701 (2011).
Chow, K. Y. K. & Bode, J. W. Catalytic generation of activated carboxylates: direct, stereoselective synthesis of β-hydroxyesters from epoxyaldehydes. J. Am. Chem. Soc. 126, 8126–8127 (2004).
Reynolds, N. T., Read de Alaniz, J. & Rovis, T. Conversion of α-haloaldehydes into acylating agents by an internal redox reaction catalyzed by nucleophilic carbenes. J. Am. Chem. Soc. 126, 9518–9519 (2004).
Burstein, C. & Glorius, F. Organocatalyzed conjugate umpolung of α, β-unsaturated aldehydes for the synthesis of γ-butyrolactones. Angew. Chem. Int. Ed. 43, 6205–6208 (2004).
Sohn, S. S., Rosen, E. L. & Bode, J. W. N-heterocyclic carbene-catalyzed generation of homoenolates: γ-butyrolactones by direct annulations of enals and aldehydes. J. Am. Chem. Soc. 126, 14370–14371 (2004).
Chan, A. & Scheidt, K. A. Conversion of α,β-unsaturated aldehydes into saturated esters: an umpolung reaction catalyzed by nucleophilic carbenes. Org. Lett. 7, 905–908 (2005).
Mahatthananchai, J., Zheng, P. & Bode, J. W. α,β-Unsaturated acyl azoliums from N-heterocyclic carbene catalyzed reactions: observation and mechanistic investigation. Angew. Chem. Int. Ed. 50, 1673–1677 (2011).
Delany, E. G. et al. Aerobic oxidation of NHC-catalysed aldehyde esterifications with alcohols: benzoin, not the Breslow intermediate, undergoes oxidation. Chem. Commun. 49, 6513–6515 (2013).
Wang, X., Wu, Z. & Wang, J. α-Fluoroallenoate synthesis via N-heterocyclic carbene-catalyzed fluorination reaction of alkynals. Org. Lett. 18, 576–579 (2016).
Wu, Z. & Wang, J. N-Heterocyclic carbene-catalyzed chemoselective S–O bond cleavage of benzenesulfonic carbamate. Org. Lett. 20, 7607–7610 (2018).
Guin, J., De Sarkar, S., Grimme, S. & Studer, A. Biomimetic carbene-catalyzed oxidations of aldehydes using TEMPO. Angew. Chem. Int. Ed. 47, 8727–8730 (2008).
De Sarkar, S., Biswas, A., Samanta, R. C. & Studer, A. Catalysis with N-heterocyclic carbenes under oxidative conditions. Chem. Eur. J. 19, 4664–4678 (2013).
Wheeler, P., Vora, H. U. & Rovis, T. Asymmetric NHC-catalyzed synthesis of α-fluoroamides from readily accessible α-fluoroenals. Chem. Sci. 4, 1674–1679 (2013).
Kerr, M. S. & Rovis, T. Enantioselective synthesis of quaternary stereocenters via a catalytic asymmetric Stetter reaction. J. Am. Chem. Soc. 126, 8876–8877 (2004).
He, M., Struble, J. R. & Bode, J. W. Highly enantioselective azadiene Diels–Alder reactions catalyzed by chiral N-heterocyclic carbenes. J. Am. Chem. Soc. 128, 8418–8420 (2006).
Zhao, C., Li, F. & Wang, J. N-Heterocyclic carbene catalyzed dynamic kinetic resolution of pyranones. Angew. Chem. Int. Ed. 55, 1820–1824 (2016).
Hayashi, T. et al. Catalytic asymmetric synthesis of axially chiral biaryls by palladium-catalyzed enantioposition-selective cross-coupling. J. Am. Chem. Soc. 117, 9101–9102 (1995).
Belokon, Y. N. et al. Synthesis of α-amino acids via asymmetric phase transfer-catalyzed alkylation of achiral nickel (II) complexes of glycine-derived Schiff bases. J. Am. Chem. Soc. 125, 12860–12871 (2003).
Acknowledgements
Generous financial support for this work is provided by: the National Natural Science Foundation of China (Nos. 21672121 and 21871160), the “Thousand Plan” Youth Program of China, the Tsinghua University, the Bayer Investigator fellow, the Fellowshio of Tsinghua-Peking Centre for Life Sciences (CLS).
Author information
Authors and Affiliations
Contributions
G.M.Y. conducted the main experiments; D.H.G. and D.M. prepared the several starting materials, including substrates. J.W. conceptualized and directed the project, and drafted the paper with the assistance from co-authors. All authors contributed to the discussions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks Yong Huang and other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, G., Guo, D., Meng, D. et al. NHC-catalyzed atropoenantioselective synthesis of axially chiral biaryl amino alcohols via a cooperative strategy. Nat Commun 10, 3062 (2019). https://doi.org/10.1038/s41467-019-10878-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-10878-7
This article is cited by
-
N-Heterocyclic carbene-catalyzed enantioselective (dynamic) kinetic resolutions and desymmetrizations
Science China Chemistry (2024)
-
Atroposelective desymmetrization of 2-arylresorcinols via Tsuji-Trost allylation
Communications Chemistry (2023)
-
Conformational enantiodiscrimination for asymmetric construction of atropisomers
Nature Communications (2022)
-
Carbene-catalyzed atroposelective synthesis of axially chiral styrenes
Nature Communications (2022)
-
Asymmetric synthesis of binaphthyls through photocatalytic cross-coupling and organocatalytic kinetic resolution
Science China Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
