
Abstract
Nickel-catalyzed asymmetric cross-coupling of secondary alkyl electrophiles with different nucleophiles represents a powerful strategy for the construction of chiral tertiary carbon centers. Yet, the use of aryl Grignard reagents or aryl zinc halides in many reactions typically resulted in low enantioselectivity, mainly due to their slow transmetalation step in the catalytical cycle and consequently the requirement of relatively high temperature. Here we report that the use of lithium aryl zincate [Ph2ZnBr]Li facilitates the transmetalation step of the nickel-catalyzed cross-coupling reaction. Based on this discovery, a highly enantioselective construction of fluoroalkyl-substituted stereogenic center by a nickel-catalyzed asymmetric Suzuki-Miyaura coupling of α-bromobenzyl trifluoro-/difluoro-/mono- fluoromethanes with a variety of lithium aryl zincates [Ph2ZnBr]Li that were in situ generated from the reaction of lithium organoboronate with 1.0 equivalent of ZnBr2 was described.
Similar content being viewed by others

Introduction
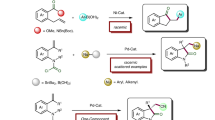
Over the past two decades, nickel-catalyzed asymmetric cross-coupling of secondary alkyl electrophiles with different nucleophiles has emerged as powerful methods for the construction of chiral tertiary carbon centers1,2,3,4. Since the seminar work by Fu and co-workers in 20055, a number of activated racemic alkyl halides such as α-bromoamides5, α-bromoketones6, benzylic bromides and chlorides7,8, allylic chlorides9 or 1-bromo-1-fluoroalkane10 and unactivated racemic alkyl halides such as β- or γ-ether, amide or sulfonyl-substituted alkyl bromides11,12, and α-haloboronates13 were effectively employed as the coupling partners, while the choice of nucleophiles was originally mainly focused on alkyl zinc halides. Only recently, the nickel-catalyzed asymmetric couplings of racemic alkyl halides were successfully extended to other nucleophiles such as alkyl-9-BBN, aryl Grignard reagents, aryl zinc halides, aryl/vinyl silicates vinyl/alkynyl indium/zirconium/aluminum reagents (Fig. 1a)6,14,15,16,17,18,19.

Ni-catalyzed asymmetric cross-coupling of racemic secondary alkyl halides. a State-of-the-art. b Coupling of fluoroalkylated secondary alkyl bromides with Aryl zinc chloride. c Coupling with lithium borates. d Coupling with in situ generated zincate (this work)
Organoboron reagents are one of the most widely studied and applied reagents that allows for the efficient construction of carbon-carbon and carbon-heteroatom bonds20,21,22. Non-asymmetric couplings of secondary alkyl bromides with aryl boronic acids under nickel catalysis have been reported in early 200423. Yet, mainly due to the slow transmetalation step of the aryl boronic acid to the active nickel intermediate, theses reactions typically required 60 °C to occur to full conversion. To facilitate the transmetalation step, Fu and co-workers15 turned to alkyl-9-BBN and found that the reaction could be conducted at 5 °C-room temperature to ensure high enantioselectivity. Nevertheless, alkyl boranes are generally air and moisture sensitive and should be prepared in situ by hydroboration of alkene before use, which hampered their widespread applications.
In 2017, we discovered that the transmetalation step in nickel-catalyzed asymmetric Suzuki-Miyaura coupling of CF3O-substituted secondary benzylic bromide was much faster when easily available, air-insensitive lithium organoboronate instead of aryl boronic acid was used as the nucleophile24. In this case, the reaction occurred smoothly at 0 °C to give the coupled product with a CF3O-stustituted stereogenic center with excellent enantioselectivity (Fig. 1c). Inspired by this discovery and considering the fact that fluoroalkyl groups, including trifluoromethyl (CF3-), difluoromethyl (HCF2-) and monofluoromethyl (CH2F-) group are important structural motifs in refining the lead compound’s selectivity and pharmacokinetics for new drug discovery25,26,27,28, we envisaged that the same strategy might work if a fluoroalkylated secondary benzylic bromide was allowed to react with lithium organoboronate. One main problem for the transition-metal-catalyzed coupling reactions of fluoroalkylated secondary benzylic bromides is the fluoride elimination from the fluoroalkylated secondary benzylic metal species if the subsequent transmetalation step is too slow29,30. The key for the success of such a coupling reaction, therefore, is to accelerate the transmetalation step. Herein, we report the use of lithium aryl zincate [Ph2ZnBr]Li facilitates the transmetalation step of the nickel-catalyzed cross-coupling reaction. Consequently, a nickel-catalyzed highly enantioselective coupling reaction for the construction of the optically active fluoroalkylated benzhydryl derivatives from easily available racemic α-bromobenzyl trifluoro-/difluoro-/monofluoromethanes and lithium organoboronates was developed (Fig. 1d).

Ni-catalyzed asymmetric cross-coupling reaction with different nucleophiles. a Coupling with lithium phenyl boronate. b Coupling with Grignard reagent. c Coupling with aryl zinc halide
Results
Screening of reaction conditions
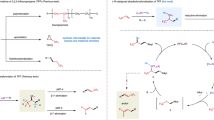
Initially, we tried the reaction of racemic trifluoromethylated benzylic bromide 1a and lithium organoboronate 2a as a model reaction to optimize the reaction conditions. Surprisingly, the reaction did not take place at all when it was conducted in tetrahedrofuran (THF) at 0 °C for 8.0 h using a combination of 20 mol% NiBr2•DME and 25 mol% L1 as the catalyst, which is the condition for the construction of trifluoromethoxylated stereogenic center (Fig. 2a). Notably, when 1.0 equivalent of ZnBr2 was used as additive, the reaction occurred after 8 h at 0 °C to afford the coupled product in 65% yield with 85:15 e.r. (Fig. 2a). As a comparison, we studied the reaction of other nucleophiles such as Grignard reagent PhMgBr or PhZnBr. As summarized in (Fig. 2b, c), reaction of substrate 1a with PhMgBr, under the identical conditions, mainly afforded the undesired defluorinated side product in 51% yield, while the reaction of substrate 1a with PhZnBr were slow and the formation of the coupled product was not observed.
A quick further survey of the reaction conditions disclosed that a combination of NiBr2•DME with ligand L2 was the most efficient catalyst and the desired product 3a was obtained in 62% yield with 95.5:4.5 e.r. along with the undesired defluorinated side product 3a’ in 5% yield when the reaction was conduct at −15 °C for 12 h (Table 1, entry 1). Switching the additive to ZnCl2 gave slightly inferior results, while using MgBr2 as additive was not effective at all (Table 1, entries 2–3). Further studied showed that reactions in DME or diglyme occurred in moderate yields and good enantioselectivity, while reactions in other solvents such as THF, DMA, or DMF were less effective and reaction in toluene was completely shut down (Table 1, entries 4–8). Notably, using a combination of DME/diglyme (v/v = 1/1) as the solvent gave slightly improved yield and enantioselectivity (Table 1, entry 9). The amount of ZnBr2 was also important for the reaction. While the reaction with 2.0 equivalents of ZnBr2 gave comparative yield and enantioselectivity, reaction conducted with 0.5 equivalent of ZnBr2 occurred in much lower yield although the enantioselectivity was high (Table 1, entries 11–12). We next studied the effect of different nickel precursors and ligands. It was found that reaction using different nickel precursors such as NiCl2•DME or Ni(OAc)2 had little effect on the efficiency of the reaction. Yet, the choice of the ligand plays a key role in delivering the good yields and high enantioselectivity. Pyridine-oxazoline ligand with either an electron-donating group (-OMe) or an electron-withdrawing group (-CF3) at 5-position, as well as a methyl group at 3-position of the pyridyl moiety were less effective (Table 1, entries 15–17). Likewise, two commonly used dinitrogen ligands for nickel-catalyzed asymmetric coupling reaction were also ineffective under these conditions (Table 1, entries 18–19). Control experiment showed that reaction in the absence of nickel catalyst did not occur at all (Table 1, entry 20). Furthermore, efforts to decreasing the catalyst loadings disclosed that the amount of side products increased to 9–15% when a combination of 10 mol% NiBr2•DME and 12.5 mol% L2 or 5.0 mol% NiBr2•DME and 6.25 mol% L2 was used (Table 1, entries 21–22).
Mechanistic investigation
During the optimization of the reaction conditions, it was found that addition of 1.0 equivalent of ZnBr2 dramatically accelerated the reaction rate. Presumptively, mixing lithium aryl boronate with ZnBr2 might generate several different arylated zinc species that could accelerate the transmetalation step and the overall catalytic reaction. To probe which arylated zinc species was involved in the reaction, we did several control experiments (Fig. 3). First, reaction of compound 1a with 3.0 equivalents of PhZnBr in the presence/absence of 3.0 equivalents of LiBr occurred under standard conditions in less than 5% yield of the coupled product. Likewise, reaction of compound 1a with 3.0 equivalents of Ph2Zn, again, gave the desired product in less than 5% yield. These results clearly excluded the possibility of the involvement of PhZnBr and Ph2Zn in the current reaction. Interestingly, addition of 3.0 equivalent of LiBr to the reaction of compound 1a with Ph2Zn led to full conversion of the starting material and gave the coupled compound 3a in 78% yield with 95.5:4.5 e.r. These experimental results suggest that an anionic zincate [Ph2ZnBr]- might be involved in the reaction, consistent with the observation from Ingleson and co-workers31 that mixing 2.0 equivalents of lithium aryl boronate with ZnBr2 at room temperature generated an anionic [PhxZnBry]- (x + y = 3).

Control experiments. The effects of different nucleophiles
To gain more support about the formation of lithium zincate from lithium aryl boronate with ZnBr2, we studied and compared the 13C nuclear magnetic resonance (NMR) spectra of the species generated from mixing lithium aryl boronate with ZnBr2 and Ph2Zn with LiBr. As shown in Fig. 4, mixing equimolar amount of Ph2Zn with LiBr at room temperature in THF-d8 for 0.5 h cleanly generated [Ph2ZnBr]Li, as evidence by a peak with a chemical shift at 161.0 ppm in 13C NMR spectrum, which corresponds to the ipso-carbon of the phenyl group in [Ph2ZnBr]Li. Likewise, the same species was formed after 0.5 h at room temperature for the reaction of 3.0 equivalvents of lithium phenyl boronate 2a with ZnBr2. These results suggest an anionic arylated zincate [Ph2ZnBr]Li could facilitate the transmetalation step, and consequently, accelerates the overall catalytic reaction. However, we could not exclude an alternative pathway in which ZnBr2 may abstract a bromide from nickel benzylic bromide complex to generate a cationic η3-benzyl nickel species32 that could more rapidly participate in a transmetalation reaction with lithium organoboronate 2a.

NMR detection of intermediates. 13C NMR spectra of Ph2Zn, Ph2Zn + LiBr, 2a and 2a + ZnBr2 in THF-d8

Substrate scope of asymmetric coupling of α-bromobenzyl trifluoromethane with lithium aryl boronates. All reaction were conducted with compound 1 (0.3 mmol), phenylboronic pinacol ester 2 (0.9 mmol), NiBr2•DME (20 mol%), ligand L2 (25 mol%) and ZnBr2 (0.3 mmol) in DME/diglyme (v/v = 1/1) at −15 °C for 12 h. Isolated yields and e.r. was determined by chiral HPLC analysis
Substrate scope investigation
Having identified the optimized conditions and the likelihood role of ZnBr2, we next investigated the generality of the nickel-catalyzed coupling reaction for the preparation of enantio-enriched benzhydryl trifluoromethane derivatives. As summarized in Fig. 5, in general, trifluoromethylated benzylic bromides (1a–e) with electron-withdrawing substituted groups such as ester, cyano, nitro, trifluoromethyl or trifluoromethoxy group reacted smoothly with lithium phenyl boronates 2a–f to afford the coupled products in moderate to good yields and enantioselectivities (Figs. 5 and 3 a–h, j–m). For example, reactions of both α-bromo-4-nitrobenzyl trifluoromethane and α-bromo-3-trifluoromethyl benzyl trifluoromethane with lithium phenyl boronate 2a gave the corresponding products 3k and 3 m in 53% and 75% yields with excellent enantioselectivities 96:4 and 97:3 e.r., respectively, (Figs. 5 and 3 k, m). Notably, trifluoromethylated benzylic bromides with a halogen group such as chloride, bromide, and fluorine were compatible and reacted with lithium phenyl boronates 2a to give the corresponding products 3n–p in 65%, 68%, and 52% yields, with 96:4, 95:5 and 96:4 e.r., respectively (Figs. 5, 3 n–p ). Furthermore, α-bromobenzyl trifluoromethyl with para-, meta-, and ortho-substituents are all compatible coupling partners, affording the desired products in moderate to good yields and enantioselectivities. For example, both α-bromo-3,5-dibromide benzyl trifluoromethane and α-bromo-2-fluorine-4-cyano benzyl trifluoromethane reacted to afford compounds 3v, 3y in 58% and 71% yield with 96:4 and 98:2 e.r., respectively (Figs. 5, 3 v, y). Nevertheless, trifluoromethylated benzylic bromides with electron-donating groups occurred in much less yields and moderate enantioselectivities (Figs. 5, 3 ab–ac ). In these cases, the formations of two side products including the homocoupling side products and the defluorinated side products were observed. Next, we investigated the scope of lithium aryl boronates. It was found that reactions of lithium aryl boronates with meta-substituted aryl groups occured in moderate to good yields and enantiolectivities (Figs. 5, 3 b–e ), while reactions of para-methyl or fluoride substituted lithium aryl boronates occurred to generate the corresponding products in moderate yields and enantioselectivities (Figs. 5, 3 g, h ). However, the formation of the desired coupling product was not observed when lithium aryl boronates with ortho-methyl group (2i) was subjected to the reaction conditions (Scheme 2, 3i). Previously reported method for the preparation of enantio-enriched benzhydryl trifluoromethane derivatives typically required to use optically secondary α-(trifluoromethyl)benzyl tosylates to react with various aryl boronic acids in the presence of a palladium catalyst19,29,33,34,35,36, while Fu and coworker37 reported a highly enantioselective nickel-catalyzed coupling of fluoroalkylated secondary alkyl bromide with aryl zinc chlorides (Fig. 1c). Thus, the current method provided an alternative, more efficient method to access this family of compounds.
Encouraged by the high enantioselectivity in nickel-catalyzed coupling of α-bromobenzyl trifluoromethane with lithium aryl boronates, we next tried to extend this reaction to other fluoroalkyl-substituted benzyl bromides. After a quick screen of the reaction conditions, it was found that when a more sterically hindered ligand L7 was used as the ligand and the reaction temperature was decreased to −40 °C, good enantioselectivities could be achieved (Fig. 6). For example, reactions of 4-(1-bromo-2,2-difluoroethyl)−3-fluorobenzonitrile with lithium phenyl boronate 2a and lithium 4-fluorophenyl boronate 2 h occurred smoothly after 12 h to afford the corresponding products in 94:6 e.r. (Figs. 6 and 4a, b). Since few methods for the construction of difluoromethyl-substituted stereogenic carbon center have been reported previously38,39, the current method represents an attractive approach for the preparation of optically active difluoromethylated benzhydryl derivatives.

Substrate scope for reactions with α-bromobenzyl di-/mono-fluoromethane. All reaction were conducted with compound 1 (0.3 mmol), phenylboronic pinacol ester 2 (0.9 mmol), NiBr2•DME (20 mol%), ligand L7 or L8 (25 mol%) and ZnBr2 (0.3 mmol) in DME/diglyme (v/v = 1/1) at −10 or −40 °C for 12 h. Isolated yields and e.r. was determined by chiral HPLC analysis
On the other hand, reaction of monofluoromethylated substrates were much more challenging. After carefully screening of the combination of nickel salts and ligands, it was found that using a combination of NiBr2•DME with ligand L8 could catalyze the reactions of 4-(1-bromo-2-fluoroethyl)arenes with lithium phenyl boronate 2a to give the corresponding coupled products 4j–l after 12 h at −10 °C in moderate enantioselectivities (79:21 ∼ 83:17 e.r.).
Synthetic application
To showcase the applicability of the nickel-catalyzed asymmetric coupling reaction of racemic trifluoromethylated benzylic bromide with lithium organoboronate, we applied this protocol for the synthesis of trifluoromethylated mimic of an inhibitor for the histone lysine methyltransferase enhancer of Zeste Homolog 2 (EZH2)40. As shown in Fig. 7, compound 5 was generated in 55% overall yield with 94:6 e.r. via a four-step transformation from easily available α-bromo-4-tert-butoxycarbylbenzyl trifluoromethane.

Synthetic applications. Application in the preparation of fluoroalkylated derivatives of drug candidates
Owing to the slightly acidic proton in the difluoromethyl group, which allows it to act as a lipophilic hydrogen-bond donor, the difluoromethyl group (CHF2) was generally considered as a bioisostere for a hydroxy goup (-OH)41. Replacement of a hydroxy group of a drug molecule with a difluoromethyl group may result in a Me-too or Me-better drug molecule. Consequently, a difluoromethylated compound 6, which is a mimic of histamine H3 receptor42, was synthesized in 71% overall yield and 90:10 e.r. after four steps.
Discussion
Inspired by the mechanistic studies disclosed that a highly reactive zincate [Ph2ZnBr]Li would facilitate the transmetalation step of the nickel-catalyzed cross-coupling reaction, we successfully developed a highly enantioselective nickel-catalyzed coupling of easily available α-bromobenzyl fluooalkanes with a variety of lithium aryl boronates in the presence of stochiometric amount of ZnBr2. Thus, the protocol may serve as a versatile, efficient, and convenient approach for the rapid access of chiral benzhydryl fluoroalkane derivatives. The application of the high-reactive lithium aryl zincate [Ar2ZnBr]Li in other transition-metal-catalyzed cross-coupling reactions are undergoing currently in our laboratory.
Methods
Coupling of trifluoromethylated secondary benzyl bromides
In a glove box, phenylpinacolboronate ester (5.1 g, 25 mmol) was weighted into a 100 mL Schlenk tube, and 40 mL of anhydrous THF was added. The mixture was taken out from the glove box and cooled at −20 °C. n-BuLi (25 mmol, 10 mL, 2.5 M in Hexanes) was added. The mixture was stirred at −20 °C for 2 h. Then the Schlenk tube was taken into the glove box, the solvents were removed under vacuum to give lithium phenyl pinacol boronate.
In an argon-filled glove box, lithium organoborate (371 mg, 0.900 mmol, 3.00 equiv.), ligand L2 (26.8 mg, 0.0750 mmol, 0.250 equiv.), ZnBr2 (67.5 mg, 0.300 mmol, 1.00 equiv.), and NiBr2.DME (18.5 mg, 0.0600 mmol, 0.200 equiv) were placed into a 25 mL Schlenk tube. To this vial was added 5.0 mL of anhydrous DME/diglyme(v/v = 1:1). The Schlenk tube was taken out from the glove box and cooled at −15 °C. α-Bromo-4-methoxycarbonylbenzyl trifluoromethyl 1a (89.1 mg, 0.300 mmol) was added and the mixture was stirred at -15 °C for 12 h. The mixture was quenched by addition of water (5.0 mL) and extracted with Et2O (10.0 mL × 3). The organic layer was combined, dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by column chromatography on silica gel with pentane/ethyl acetate as the eluent to give (S)-methyl 4-(2,2,2-trifluoro-1-phenylethyl)benzoate 3a as a yellow liquid (77% yield, 96:4 e.r.).
Data availability
Experimental procedures and characterization data are available within this article and its Supplementary Information. Data are also available from the corresponding author on request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers 1868035 and 1898246. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
References
Glorius, F. Asymmetric cross-coupling of non-activated secondary alkyl halides. Angew. Chem. Int. Ed. 47, 8347 (2008).
Iwasaki, T., Kambe, N. & Ni-Catalyzed, C. -C. Coupling using alkyl electrophiles. Top. Curr. Chem. (Z.) 373, 66 (2016).
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692 (2017).
Choi, J. & Fu, G. C. Transition metal–catalyzed alkyl-alkyl bond formation: another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017).
Fisher, C. & Fu, G. C. Asymmetric nickel-catalyzed negishi cross-couplings of secondary α-bromo amides with organozinc reagents. J. Am. Chem. Soc. 127, 4594 (2005).
Lou, S. & Fu, G. C. Nickel/bis(oxazoline)-catalyzed asymmetric kumada reactions of alkyl electrophiles: cross-couplings of racemic α-bromoketones. J. Am. Chem. Soc. 132, 1264 (2010).
Arp, F. O. & Fu, G. C. Catalytic enantioselective negishi reactions of racemic secondary benzylic halides. J. Am. Chem. Soc. 127, 10482 (2005).
Binder, J. T., Cordier, C. J. & Fu, G. C. Catalytic enantioselective cross-couplings of secondary alkyl electrophiles with secondary alkylmetal nucleophiles: negishi reactions of racemic benzylic bromides with achiral alkylzinc reagents. J. Am. Chem. Soc. 134, 17003 (2012).
Song, S. & Fu, G. C. Nickel-catalyzed asymmetric negishi cross-couplings of secondary allylic chlorides with alkylzincs. J. Am. Chem. Soc. 130, 2756 (2008).
Jiang, X. -J. & Gandelman, M. Enantioselective suzuki cross-couplings of unactivated 1‐fluoro-1- haloalkanes: synthesis of chiral β‐, γ‐, δ‐, and ε‐fluoroalkanes. J. Am. Chem. Soc. 137, 2542 (2015).
Owston, N. A. & Fu, G. C. Asymmetric alkyl-alkyl cross-couplings of unactivated secondary alkyl electrophiles: stereoconvergent suzuki reactions of racemic acylated halohydrins. J. Am. Chem. Soc. 132, 11908 (2010).
Lu, Z., Wilsily, A. & Fu, G. C. Stereoconvergent amine-directed alkyl-alkyl suzuki reactions of unactivated secondary alkyl chlorides. J. Am. Chem. Soc. 133, 8154 (2011).
Schmidt, J., Choi, J., Liu, A. T., Slusarczyk, M. & Fu, G. C. A general, modular method for the catalytic asymmetric synthesis of alkylboronate esters. Science 354, 1265 (2016).
Lou, S. & Fu, G. C. Enantioselective alkenylation via nickel-catalyzed cross-coupling with organozirconium reagents. J. Am. Chem. Soc. 132, 5010 (2010).
Saito, B. & Fu, G. C. Enantioselective alkyl-alkyl suzuki cross-couplings of unactivated homobenzylic halides. J. Am. Chem. Soc. 130, 6694 (2008).
Dai, X., Strotman, N. A. & Fu, G. C. Catalytic asymmetric hiyama cross-couplings of racemic α-bromo esters. J. Am. Chem. Soc. 130, 3302 (2008).
Caeiro, J., Sestelo, J. P. & Sarandeses, L. A. Enantioselective nickel-catalyzed cross-coupling reactionsof trialkynylindium reagents with racemic secondary benzyl bromides. Chem. Eur. J. 14, 741 (2008).
Fang, H. et al. Transmetal-catalyzed enantioselective cross-coupling reaction of racemic secondary benzylic bromides with organoaluminum reagents. Org. Lett. 18, 6022 (2016).
Varenikov, A. & Gandelman, M. Synthesis of chiral α-trifluoromethyl alcohols and ethers via enantioselective hiyama cross-couplings of bisfunctionalized electrophiles. Nat. Commun. 9, 3566 (2018).
Suzuki, A. Organoboron compounds in new synthetic reactions. ACC Chem. Res. 15, 178 (1982).
Hall, D. G. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine. (Wiley-VCH, Weinheim, 2005).
Fyfe, J. W. B. & Watson, A. J. B. Recent developments in organoboron chemistry: old dogs, new tricks. Chem 3, 31 (2017).
Zhou, J. & Fu, G. C. Suzuki cross-couplings of unactivated secondary alkyl bromides and iodides. J. Am. Chem. Soc. 126, 1340 (2004).
Huang, W. -C., Wan, X. -L. & Shen, Q. Enantioselective construction of trifluoromethoxylated stereogenic centers by a nickel-catalyzed asymmetric suzuki–miyaura coupling of secondary benzyl bromides. Angew. Chem. Int. Ed. 56, 11986 (2017).
Hagmann, W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 51, 4359 (2008).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320 (2008).
Zhou, Y. et al. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II−III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem. Rev. 116, 422 (2016).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822 (2018).
Brambilla, M. & Tredwell, M. Palladium-catalyzed Suzuki–Miyaura cross-coupling of secondary a-(trifluoromethyl)benzyl tosylates. Angew. Chem. Int. Ed. 56, 11981 (2017).
Budnikova, Y. H., Vicic, D. A. & Klein, A. Exploring mechanisms in Ni terpyridine catalyzed C–C cross-coupling reactions—a review. Inorganics 6, 18 (2018).
Procter, R. J. et al. A zinc catalyzed C(sp3)-C(sp2) Suzuki–Miyaura cross-coupling reaction mediated by aryl-zincates. Chem. Eur. J. 23, 15889 (2017).
Anderson, T. J. & Vicic, D. A. Direct observation of noninnocent reactivity of ZnBr2 with alkyl halide complexes of nickel. Organometallics 23, 623 (2004).
Ma, J. -A. & Cahard, D. Asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 104, 6119 (2004).
Ma, J. -A. & Cahard, D. Update 1 of: asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 108, PR1 (2008).
Nie, J., Guo, H. -C., Cahard, D. & Ma, J. -A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 111, 455 (2011).
Yang, X. Y., Wu, T., Phipps, R. J. & Toste, F. D. Advances in catalytic enantioselective fluorination, Mono-, Di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 105, 826 (2015).
Liang, Y. & Fu, G. C. Stereoconvergent negishi arylations of racemic secondary alkyl electrophiles: differentiating between a CF3 and an alkyl group. J. Am. Chem. Soc. 137, 9523 (2015).
Banik, S. M., Medley, J. W. & Jacobsen, E. N. Catalytic, asymmetri difluorination of alkenes to generate difluoromethylated stereocenters. Science 353, 51 (2016).
Bos, M. et al. Catalytic enantioselective synthesis of highly functionalized difluoromethylated cycloprapanes. Angew. Chem. Int. Ed. 56, 13319 (2017).
Gehling, V. S. et al. Discovery, design and synthesis of indole-based EZH2 inhibitors. Bioorg. Med. Chem. Lett. 25, 3644 (2015).
Sessler, C. D. et al. J. CF2H, a hydrogen bond donor. J. Am. Soc. Chem. 139, 9325 (2017).
Allison, B. D., Carruthers, N. I., Letavic, M. A., Santillan, J. A. & Shan, C. R. Substituted benzamide modulators of the histamine H3 receptor. WO2008002816A1. 2008-01-03 (2008).
Acknowledgements
This work was supported by National Natural Science Foundation of China (21625206, 21632009, 21572258, 21421002) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000).
Author information
Authors and Affiliations
Contributions
W.C.H. performed the experiments. M.H. conducted the experiments related monofluoromethylated substrates. X.L.W. participated in the determination of enantioselectivity. Q.L.S. supervised the project. Q.L.S. and W.C.H. co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, W., Hu, M., Wan, X. et al. Facilitating the transmetalation step with aryl-zincates in nickel-catalyzed enantioselective arylation of secondary benzylic halides. Nat Commun 10, 2963 (2019). https://doi.org/10.1038/s41467-019-10851-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-10851-4
This article is cited by
-
Asymmetric construction of allylicstereogenic carbon center featuring atrifluoromethyl group via enantioselective reductive fluoroalkylation
Nature Communications (2022)
-
Organometallic catalysis under visible light activation: benefits and preliminary rationales
Photochemical & Photobiological Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
