
Abstract
Anaplastic thyroid cancer (ATC) and advanced differentiated thyroid cancers (DTCs) show fatal outcomes, unlike DTCs. Here, we demonstrate mutational landscape of 27 ATCs and 86 advanced DTCs by massively-parallel DNA sequencing, and transcriptome of 13 ATCs and 12 advanced DTCs were profiled by RNA sequencing. TERT, AKT1, PIK3CA, and EIF1AX were frequently co-mutated with driver genes (BRAFV600E and RAS) in advanced DTCs as well as ATC, but tumor suppressors (e.g., TP53 and CDKN2A) were predominantly altered in ATC. CDKN2A loss was significantly associated with poor disease-specific survival in patients with ATC or advanced DTCs, and up-regulation of CD274 (PD-L1) and PDCD1LG2 (PD-L2). Transcriptome analysis revealed a fourth molecular subtype of thyroid cancer (TC), ATC-like, which hardly reflects the molecular signatures in DTC. Furthermore, the activation of JAK-STAT signaling pathway could be a potential druggable target in RAS-positive ATC. Our findings provide insights for precision medicine in patients with advanced TCs.
Similar content being viewed by others


Introduction
The molecular understating of differentiated thyroid cancer (DTC) was expanded by recent comprehensive studies1,2. The Cancer Genome Atlas (TCGA) proposed two molecular subtypes of papillary thyroid cancer (PTC), BRAFV600E-like and RAS-like, based on transcriptome analysis1. Moreover, our group demonstrated a third molecular subtype, Non-BRAF-Non-RAS (NBNR), which is closely associated with follicular-patterned thyroid tumors, including follicular adenoma (FA) and minimally invasive follicular thyroid cancer (miFTC)2. According to the aforementioned studies, the molecular classification of thyroid cancer (TC) better explains its underlying characteristics than a histological classification.
Anaplastic TC (ATC), which accounts for 2% or fewer of TC cases, is one of the most aggressive human malignancies3. The median survival of ATC patients is about 3–5 months after diagnosis4. In addition, advanced DTCs, such as poorly differentiated, metastatic, or widely invasive types, also show poor outcomes5,6,7. Nonetheless, there is no effective therapy to prolong the survival of patients with those forms of TC. Although the molecular characteristics of DTC have been analyzed, the underlying mechanism of its progression to advanced DTC and ATC has not been fully elucidated. Several studies have reported that multiple mutational hits in tumor suppressor genes (TSGs) or oncogenes were involved in the development of ATC, but the majority of those reports were confined to genomic alterations8,9. Thus, the need for further transcriptomic analysis of ATC and advanced DTCs is increased to discover molecular mechanisms potentially involved in tumor progression and targets for treatment.
In this study, we apply various types of massively parallel sequencing technology to 113 advanced TCs, including 27 ATCs and 86 advanced DTCs, to reveal their genomic and transcriptomic characteristics. We expect that this work will broaden the current molecular understanding of advanced TCs and lead to more efficient diagnostic and therapeutic strategies for them.
Results
Mutational landscape of ATC and advanced DTCs
We have preliminarily analyzed 13 ATCs, 3 focal ATC/poorly differentiated TCs (PDTCs), and 9 widely invasive follicular TCs (wiFTCs) by whole-genome sequencing (WGS) or whole-exome sequencing (WES), and extended the dataset with 88 additional samples using targeted sequencing. In total, 113 advanced TCs, including 27 ATCs, 15 PDTCs, 28 focal ATC/PDTCs, 12 wiFTCs, and 31 metastatic papillary TCs (PTCs) were investigated for mutational profiling. Targeted sequencing was also performed on 13 ATCs and 3 focal ATC/PDTCs which were analyzed by WGS and the concordance rate between two methods was 91.89% (Supplementary Data 1). We collected tissues from primary (76/113), distant metastatic (19/113), and locally recurred or residual sites (18/113), respectively. The clinicopathological characteristics of the patients according to histology are shown in Table 1.
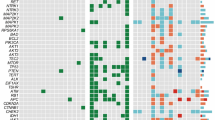
The mutational landscape of 113 advanced TCs is illustrated in Fig. 1. BRAFV600E and RAS (40.74% and 44.44%, respectively) were recognized as major driver genes in ATC, whereas no fusion gene was identified. RET fusions (CCDC6-RET and NCOA4-RET) were discovered in PDTC, focal ATC/PDTC, and metastatic PTC. We also found NFE2L2 mutation which is frequently altered in lung squamous cell carcinoma and recently identified in TC as fusion driver10,11. In PDTC, BRAFV600E and RAS mutations were found with the same frequency (26.67%), but most focal ATC/PDTCs had a BRAFV600E mutation (82.14%). wiFTC and metastatic PTC showed frequencies of BRAFV600E (0% and 64.52%, respectively) and RAS mutations (66.67% and 22.58%, respectively) similar to those found in FA/miFTC and PTC, respectively1,2. In metastatic PTC, we also discovered SPOPP94R which is repeatedly reported in various types of TC2,12,13.

The mutational landscape of ATC and advanced DTCs. Each column represents an individual tumor. Only genes harboring mutations confirmed to be somatic in at least one tumor were displayed. Genes were sorted by their functions as oncogenes, tumor suppressors, splicing machinery gene, RET fusion, and telomere lengthening genes. The right bar chart represents the frequencies of gene alterations across 113 advanced TCs. FU, LN, NA, PT, LRT, and DMT indicate follow-up, lymph node, not available, primary tumor, locally recurred or residual tumor, and distant metastatic tumor, respectively
TERT was the most recurrently co-mutated gene in ATC, with a frequency of 55.56% (Fig. 2a), as previously reported9 (56.12%; Supplementary Fig. 1a). It was also frequently altered in advanced DTCs (46.67%, 39.29%, and 47.39% in PDTCs, focal ATC/PDTCs, and metastatic PTCs, respectively). Notably, wiFTC showed an extremely high frequency of TERT alterations (91.67%), even more frequent than in ATC, and other alterations were barely discovered in this subtype. We also identified ATRX mutations in ATC, focal ATC/PDTC, wiFTC, and metastatic PTC.

Recurrently altered genes in ATC and advanced DTCs. The frequencies of recurrent gene alterations among diverse types of TC were represented by radar charts. a TERT, b TSGs, c TP53, and d the frequencies of AKT1/PIK3CA and EIF1AX co-mutations with BRAFV600E or RAS in diverse types of TC (blue). The mutational frequencies without BRAFV600E and RAS were also displayed (orange). e The distribution of recurrent oncogene mutations across 113 TCs. f The functional domains of PDE8B–TERT fusion. g The expression level of TERT at each exon with PDE8B-TERT fusion. h The expression level of TERT by alteration type. P-values from DESeq2 were represented. i A schematic illustration of TERT upstream translocation. The breakpoints were pointed out by arrows
Furthermore, 74.07% of ATCs harbored mutations in TSGs such as TP53, CDKN2A, PTEN, LATS1, LATS2, CTNNA2, TET1, and BRCA1 (Fig. 2b). Among them, TP53 was the most commonly altered TSG (48.15%; Fig. 2c). Notably, we confirmed two types of germline TP53 mutation (E11Q and R49H) in ATC (11.11%), wiFTC (8.33%), and metastatic PTC (3.23%; Supplementary Fig. 2). TSG mutations were also found in advanced DTCs (26.67%, 21.43%, 25.81%, and 16.67% in PDTCs, focal ATC/PDTCs, metastatic PTCs, and wiFTCs, respectively), and these frequencies were much higher than those of DTC1,2 (1.41% in PTCs and 7.41% in FA/miFTCs).
We also identified AKT1/PIK3CA and EIF1AX co-mutations in ATC (Fig. 2d), as in previous reports8,9. Co-mutations of these genes were frequent in ATCs with BRAFV600E (36.36%) and RAS (66.67%) mutations, respectively, and they were mutually exclusive with each other (Fig. 3e). AKT1/PIK3CA and EIF1AX mutations also showed increased incidences with the aggressiveness. Additionally, a somatic mutation in U2AF1 (S34F), which is a spliceosomal machinery gene known to be involved in the progression of acute myeloid leukemia14, was identified in one focal ATC/PDTC (Fig. 1).

CDKN2A loss in ATC and advanced DTCs. a Detection of CDKN2A loss in ATC using WGS. b Detection of CDKN2A loss in ATC using targeted sequencing. c The frequency of CDKN2A loss among diverse types of TC were represented by a radar chart. d The distribution of TP53 and CDKN2A alterations across 113 advanced TCs. e The effect of CDKN2A loss on thyroid differentiation score (TDS) in ATC. P-values from two-tailed Mann–Whitney U-test were represented. f The effect of CDKN2A loss on disease-specific survival in patients with ATC and advanced DTCs. del and 2n indicate CDKN2A copy number loss and neutral, respectively. g The effect of p16 expression on disease-specific survival in patients with ATC and advanced DTCs. P-values from Log-rank test were represented
The additional mutated genes, such as KMT2D, ATM, CHEK2, ATM, CHEK2, NF1, NF2, and MEN1, that are putatively involved in the progression of TC were also discovered (Supplementary Figs. 1b and 3). However, it is possible that they might be rare germline mutations as matched normal DNA of the targeted sequenced tumors were not analyzed.
Somatic TERT rearrangements in wiFTC
As we described earlier, TERT alteration was most frequently identified in wiFTC (91.67%). In this subtype, two structural rearrangements within or adjacent to TERT, as well as promoter mutations, were found. We identified TERT fusion gene, PDE8B-TERT, which has not been described in TC (Fig. 2f). After the breakpoint, elevated TERT expression was found which is a hallmark of fusion gene15 (Fig. 2g). PDE8B is known to be involved in thyroid function and actively expressed in the thyroid gland16,17. Although TCGA group did not describe TERT fusion in TC, another group identified MTMR12-TERT fusion in PTC (TCGA-BJ-A4O9-01) from TCGA cohort (see URLs). We found remarkably elevated TERT expression in wiFTC with PDE8B-TERT and PTC with MTMR12-TERT from TCGA (Fig. 2h and Supplementary Fig. 4).
Meanwhile, one wiFTC showed increased expression of TERT without fusion or promoter mutations, hence we performed WGS to identify a structural rearrangement adjacent to TERT as previous reports18,19. As a result, an inter-chromosomal translocation, t(2;5)(2q;5p), at 21 kilobases upstream from TERT was discovered (Supplementary Fig. 5). The huge up-regulation of TERT in tumors with intergenic rearrangements is known to be induced by super-enhancer hijacking18,19. We also pinpointed typical-enhancers and super-enhancer in the partner region of upstream translocation (Fig. 2i). The distance between hijacked super-enhancer and TERT (179 kb) was closer than that of the original target gene of the super-enhancer, RGPD4 (337 kb). Moreover, we found the unusual intergenic expression by RNA sequencing (RNA-seq) alignment after the breakpoint, which suggests the consequential effect of this translocation (Supplementary Fig. 6). Two TERT rearrangements were confirmed by polymerase chain reaction (PCR) and Sanger sequencing (Supplementary Fig. 7). Additionally, the expression levels of TERT, induced by promoter mutations, were significantly higher in ATC than in DTC (P = 0.006; Fig. 2h). This may be due to the expansion of sub-clones with TERT promoter mutations in ATC, as previously described8.
Other genomic characteristics of ATC and advanced DTCs
Tumor mutational burden (TMB) of ATC was higher than that of other types of TC (P < 0.001 for each; Supplementary Fig. 8). However, wiFTC did not show a higher TMB than FA/miFTC (P = 0.40). Using whole-genome sequenced tumors, we assessed the mutational signature, which suggests the particular mutagenesis processes in the cancer genome20. We found that most tumors (15/16) had signature 5, the most common signature in many cancers with unknown etiology (Supplementary Figs. 9 and 10). Meanwhile, one focal ATC/PDTC presented signature 2, which is associated with the activation of the AID/APOBEC family of cytidine deaminases20.
The arm-level somatic copy number alterations (SCNA) burden was higher in ATC relative to DTCs (Supplementary Fig. 11a). In particular, BRAFV600E-positive ATC showed a dramatic increment of arm-level SCNA compared with BRAFV600E-positive PTC21 (P < 0.001; Supplementary Fig. 11b). RAS-positive ATC also had a higher burden of arm-level SCNA than FA/miFTC12 (P = 0.01; Supplementary Fig. 11c). wiFTC showed a similar level of arm-level SCNA as ATC (P = 0.59).
Prognostic significance of CDKN2A loss for disease-specific mortality
Furthermore, we discovered the recurrent copy number altered regions in ATC (Supplementary Table 1). Among them, 9p21.3 was the only region that covers cancer-related genes, CDKN2A and CDKN2B (Fig. 3a). It was the most significant copy number loss region, and we also successfully identified CDKN2A loss using targeted sequencing approach (Fig. 3b). CDKN2A was the second most frequently altered TSG in ATC, with a frequency of 22.22% (Fig. 3c), consistent with a previous study9 (23.47%; Supplementary Fig. 1). Most CDKN2A alterations (72.73%) occurred without TP53 mutations (Fig. 3d). CDKN2A loss was also identified in advanced DTCs (6.67%, 3.56%, and 6.45% in PDTCs, focal ATC/PDTCs, and metastatic PTCs, respectively), but not in FA/miFTC and PTC1,12. We also found one missense mutation (T79I) in focal ATC/PDTC.
Intriguingly, thyroid differentiation score (TDS) in ATCs with CDKN2A loss was significantly lower than those with wild-type (P < 0.001; Fig. 3e). We also found that CDKN2A loss was significantly associated with increased disease-specific mortality in patients with ATC and advanced DTCs (P = 0.03 and P < 0.001 for each; Fig. 3f). The hazard ratios (HRs) were 6.67 (95% confidence interval [CI], 1.34–33.12) and 9.88 (95% CI, 1.97–49.57), respectively, after adjustments for the age at surgery, sex, distant metastasis, and tumor origin (Supplementary Table 2). However, other recurrently mutated genes were not closely associated with patient survival (Supplementary Figs. 12 and 13).
We validated the relationship between CDKN2A loss and patient outcome by p16 immunohistochemistry (IHC) using tissue microarray (TMA) of 57 ATC and advanced DTC samples, of which 42 were sequenced samples (Supplementary Fig. 14a). All tissues (5/5) with CDKN2A loss, and 43.2% of CDKN2A wild-type (16/37) were p16-negative (Supplementary Fig. 14b). p16 expression displayed the association between poor disease-specific survival in patients with ATC and advanced DTCs (P = 0.03 for both; Fig. 3g). In ATC, p16-negative status increased the risk of disease-specific mortality (HR, 35.25; 95% CI, 1.38–898.79) after adjusting the age at surgery and sex, although the statistical significance was lost without adjustment for covariates or after additional adjustments for distant metastasis and tumor origin (Supplementary Table 3). In advanced DTCs, the hazard ratio was not calculated since p16-positive patients were all censored.
Transcriptome landscape of ATC and advanced DTCs
Transcriptome of 13 ATCs, 3 focal ATC/PDTCs, and 9 wiFTCs which were sequenced by WGS or WES were profiled by RNA-seq, then compared with data from 162 DTCs of our previous study2. Using K-means clustering via principal component analysis (PCA), the molecular subtype of each tumor was determined. The majority of the ATCs (10 of 13) showed a clear separation from the BRAFV600E-like, RAS-like, and NBNR which are three molecular subtypes of DTC, and formed a fourth cluster (Fig. 4a). We designated this molecular subtype as ATC-like, and this cluster did not show the molecular distinctions resulting from the types of driver mutations that have been demonstrated in DTC1,2. One ATC with no alteration in cancer-related genes was also classified as ATC-like, and showed similar DNA methylation patterns to those of other ATC-like tumors (Supplementary Fig. 15). The expression profiles of focal ATC/PDTCs seemed to be dominated by the preceding DTC, since they were all classified as BRAFV600E-like.

The transcriptome landscape of ATC and advanced DTCs. a The result of K-means clustering via PCA. The types of tumors and driver mutations were represented by shape and color, respectively. b The results of BRAFV600E-RAS score (BRS) analysis were represented by box plots. P-values from two-tailed Mann–Whitney U-test were represented. c TDS and ERK score were displayed on a scatter plot. d The results of TDS analysis and e ERK score analysis were represented by box plots. P-values from two-tailed Mann–Whitney U-test were represented. f The heatmap represents the expression profile of 16 genes associated with thyroid function and metabolism in TCs. The right panel represents gene expression levels in normal thyroid tissues
TCGA’s BRAFV600E-RAS score (BRS) analysis displayed a similar result to that of K-means clustering via PCA; BRAFV600E-positive and RAS-positive ATCs displayed no variance in BRS (P = 0.91; Fig. 4b), in contrast to PTC (P < 0.001). For wiFTC, one tumor was classified as ATC-like by K-means clustering via PCA, but most of them were RAS-like or NBNR (Fig. 4a). However, BRS analysis suggested that wiFTC is differentiated from FA/miFTC on the molecular level (P = 0.001; Fig. 4b). Then, we performed TDS and ERK score analyses, which illustrate thyroid differentiation and the activity of the MAPK-signaling pathway, respectively1. Lower TDS and higher ERK scores were observed in ATC than in DTC (Fig. 4c). ATC showed similar thyroid differentiation regardless of the driver mutation (P = 0.67; Fig. 4d), and MAPK-signaling pathway was even more activated in RAS-positive ATC than in BRAFV600E-positive ATC (P = 0.02; Fig. 4e). Unlike ATC, ERK score was not higher in wiFTC (P = 0.88), but TDS was significantly lower (P < 0.001) compared with FA/miFTC. In wiFTC, the expression of seven genes (SLC5A8, TPO, FOXE1, DIO2, TG, GLIS3, and SLC26A4) that are related to thyroid metabolism and function was significantly reduced (Fig. 4f). Furthermore, most of thyroid metabolism genes were repressed in ATC, except for three genes (THRA, NKX2-1, and PAX8).
Potential druggable targets in ATC
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway22 enrichment analysis using differentially expressed genes (DEGs) was performed to discover the biologically relevant pathways that are regulated during the progression of TC. The alterations of specific pathways during the progression of BRAFV600E-positive or RAS-positive ATC were demonstrated separately. We found that various pathways, such as the MAPK-signaling pathway, focal adhesion, extracellular matrix (ECM) receptor interaction, p53 signaling, and cell adhesion molecules (CAMs), which were initially increased in PTC relative to the normal thyroid, were further activated in BRAFV600E-positive ATC (Fig. 5a). Notably, the VEGF-signaling pathway and the Notch-signaling pathway, which are closely associated with angiogenesis23,24, but were not activated in PTC, were significantly elevated in BRAFV600E-positive ATC.

The potential druggable targets of ATC. a The top 15 significantly up-regulated KEGG pathways in BRAFV600E-positive and RAS-positive ATCs. The significance of these pathways were also noted in PTC, FA/miFTC, and wiFTC, as they were also found within the top 15 significantly up-regulated pathways of each tumor. b The increment levels of genes in the JAK-STAT-signaling pathway in RAS-positive ATC compared with FA/miFTC/wiFTC were represented by Log2(fold-change) values. Asterisks indicate the genes that were also up-regulated BRAFV600E-positive ATC. c Quantitative reverse transcription polymerase chain reaction measurement of expression of JAK-STAT-signaling pathway genes and d cell viabilities analyzed by cell counting kit-8 assay, in CAL72 cells following treatment with ruxolitinib (1, 10, 20, and 25 μM). Ctrl denotes control. All data were expressed as mean ± standard deviation. *P < 0.05 from two-tailed Mann–Whitney U-test (compared with controls). e The expression levels of CD274 and PDCD1LG2 in various types of TC. f The relationship between the up-regulation of two immunotherapeutic genes and CDKN2A loss. Samples were sorted by high to low TDS
In RAS-positive ATC, the MAPK-signaling pathway, focal adhesion, ECM receptor interaction, p53-signaling pathway, cell cycle, and CAMs were also further elevated compared with three types of follicular-patterned thyroid tumors (Fig. 5a). Except for the MAPK-signaling pathway, these pathways were not activated in wiFTC relative to FA/miFTC. Moreover, the JAK-STAT-signaling pathway, which was not detected in the three types of follicular-patterned tumors, was activated in RAS-positive ATC (Fig. 5b). In order to evaluate the potential ability of activated pathways in ATC as druggable targets, we performed functional in vitro experiments with ATC cell lines. In BRAFV600E-positive ATC cell lines, we were unable to demonstrate effects of inhibition of VEGF-Notch signaling. Whereas, in CAL62, the RAS-positive ATC cells, JAK inhibition with ruxolitinib decreased the expression of SOCS3, BCL2L1, and MYC which are the downstream molecules of JAK-STAT pathway (Fig. 5c), and then we confirmed reduced cellular proliferation (Fig. 5d).
We also found that the calcium-signaling pathway and various pathways related to metabolism, such as glycerolipid metabolism and fatty acid metabolism, were down-regulated in ATC compared with DTC (Supplementary Fig. 16). In addition to the aforementioned pathways, the expression levels of CD274 and PDCD1LG2, which encode PD-L1 and PD-L2, respectively, were increased in some ATCs (Fig. 5e). Interestingly, the up-regulation of these genes were found in ATCs with CDKN2A loss (Log2fold-change [FC] = 2.38 and 2.85 for each; Fig. 5f).
Discussion
In this study, we elucidated the genomic and transcriptomic landscape of ATC and advanced DTCs using diverse types of sequencing technologies. Our mutational profiling confirmed that multiple hits of genetic alterations promote the progression of TC, as described in previous reports8,9. Moreover, we were able to extend the current state of knowledge about the transcriptomic characteristics of advanced TCs.
Several studies demonstrated the association between TERT promoter mutation and aggressive clinicopathological features of TC25,26,27. In this study, TERT was the most frequently co-mutated gene in both ATC and advanced DTCs in addition to main driver genes (BRAFV600E and RAS). Especially, 91.67% of wiFTCs harbored TERT alterations, including promoter mutations and two rearrangements, but only 25.00% of them harbored the additional mutation in an oncogene or TSG. Therefore, this result again underscores the importance of TERT in metastatic, invasive, and early aggressive characteristics of DTC, rather than anaplastic transformation of TC. The activation of TERT in cancer was thought to be caused by point mutations in its promoter region28. However, structural rearrangements were also reported to trigger the extreme up-regulation of TERT and be associated with tumor aggressiveness18,19,29. Likewise, we determined the high expression level of TERT in two tumors with PDE8B-TERT fusion gene and inter-chromosomal translocation, t(2;5)(2q;5p), in TERT upstream region. Although two TERT rearrangements across 113 TCs were found, there might be more tumors with these alterations, since targeted sequencing method which was performed on most of study subjects (77.88%) did not cover intronic and intergenic regions.
We also confirmed that the prevalence of second-hit and third-hit in oncogenes and TSGs were increased in ATC. Intriguingly, co-mutations in the oncogenes, AKT1/PIK3CA or EIF1AX, were frequently discovered in ATC and advanced DTCs with BRAFV600E or RAS mutations, respectively. This suggests that the advancement of TC can be predicted by different markers according to the main driver mutation of DTC. TP53 and CDKN2A were the most frequently altered TSGs in ATC and advanced DTCs, as described in a recent report9. The prognostic significance of CDKN2A has been well described for diverse types of cancer30,31, but not yet for TC. Using TDS analysis, we found that ATC with CDKN2A loss presented the poorest thyroid differentiation. Moreover, we demonstrated an association between CDKN2A loss and increased disease-specific mortality in patients with ATC or advanced DTCs, even after adjustment for the other potential prognostic factors.
Altogether, our results exhibited the potential contribution of TERT and diverse oncogenes (AKT1/PIK3CA and EIF1AX) in the early progression of DTC, and higher relation of TSGs (e.g., TP53 and CDKN2A) in anaplastic change of TC. Notably, CDKN2A loss may be a strong prognostic factor for patients with advanced DTCs as well as ATC.
It is well established that the molecular characteristics of DTC are determined by the types of driver mutations1,2. Two molecular subtypes, BRAFV600E-like and RAS-like, were first proposed by TCGA1, and our group showed the existence of a third subtype, NBNR2. Based on molecular subtype, TC displays differential regulation of signaling pathways. Landa et al. reported that RAS-positive ATCs lost the molecular characteristics that were exhibited in RAS-positive DTC and that all ATCs characterized to be BRAFV600E-like, regardless of the driver mutation8. The remarkable transcriptomic changes in RAS-positive ATC were also confirmed in this study. Moreover, we were able to demonstrate the extended molecular perspective of ATC with the support of our previously published RNA-seq data from 162 DTCs2; both BRAF-positive and RAS-positive ATCs presented a similar transcriptome profile, irrespective of their driver mutation, but were grouped into another subtype, ATC-like, rather than BRAFV600E-like.
In addition to ATC-like, we identified potential druggable pathways in ATC. In both BRAFV600E-positive and RAS-positive ATCs, the MAPK-signaling pathway and several cell–cell communication pathways were further up-regulated compared with DTC. Furthermore, we found the activation of VEGF and Notch-signaling pathways in BRAFV600E-positive ATC and the JAK-STAT-signaling pathway in RAS-positive ATC, which were not activated in DTC. Although we demonstrated that the cell viability was regulated by inhibition of the activated JAK-STAT signaling in RAS-positive ATC cell line in vitro, inhibition of Notch signaling did not affect BRAFV600E-positive ATC cell lines. It is possible that the effect might not have been shown, since there would be other pathways that regulate VEGF-Notch signaling32. In addition to the aforementioned pathways, previous reports showed that cell lines with CDKN2A/p16 loss is linked to response to CDK4/6 inhibitors, such as palbociclib33,34,35 and to resistance to BRAFV600E-selective inhibitor, vemurafenib, in metastatic BRAFV600E PTC cells36. Therefore, our findings would give clues to choose appropriate target agents for ATC.
A putative association between the APOBEC family of cytidine deaminases and the progression of ATC was recently reported, but it was demonstrated by an analysis based on only a small number of variants (8–20)9. In this study, we clearly showed the presence of mutational signature 2 in one focal ATC/PDTC with over 10,000 variants that were discovered by WGS. Hence, targeting APOBEC mutagenesis may be an option for treatment of a few advanced TCs.
Immunotherapy is the most promising state-of-the-art for cancer therapy37. Growing evidence indicates that patients who have a tumor with a high TMB are highly responsive to immunotherapy38,39. We also found a higher TBM in ATC than in DTC, consistent with recent studies8,9. Furthermore, ATCs with CDKN2A loss displayed up-regulation of CD274 (PD-L1) and PDCD1LG2 (PD-L2), which are favorable immunotherapeutic targets for treating cancer37, in agreement with a previous report that showed the expression of PD-L1 in ATC40. The underlying reason of the increased expression of CD274 and PDCD1LG2 in ATC with CDKN2A loss is not unveiled in this study, but their potential relationship in non-small cell lung cancer were previously reported41,42. Put together, immunotherapy for ATC might be effective, considering the increased TMB and expression of PD-L1/PD-L2, especially for ATC with CDKN2A loss.
In conclusion, this study presented a comprehensive analysis of the genomic and transcriptomic alterations associated with the progression of DTC to its advanced and anaplastic types. We expect that our findings will provide more tailored diagnostic and therapeutic interventions for these fatal diseases.
Methods
Ethics statement
This study was approved by the institutional review board of Seoul National University Hospital, in accordance with the Declaration of Helsinki (approved ID: H-1307-034-501). Written informed consents were obtained from all patients.
Patients
Fresh frozen or formalin-fixed paraffin-embedded (FFPE) tissues from 113 patients with advanced TCs, including 27 ATCs, 15 PDTCs, 28 focal ATC/PDTCs, 12 wiFTCs, and 31 metastastic PTCs, who underwent thyroidectomy were analyzed using massively parallel sequencing method. The clinicopathological characteristics of 113 patients according to histology are shown in Table 1 and the clinical information of individual patient is provided in Supplementary Data 2. Median follow-up duration was 38.0 (interquartile range, 11.0–109.2) months and mean age of patients was 60.7 ± 14.1 years. Patients with ATC showed the highest mortality rate (74.1%) and the shortest disease-specific survival (median, 6.9 months), while those with PDTC and focal ATC/PDTC showed higher rates of no evidence of disease (73.3% and 67.9%, respectively) and relatively longer disease-specific survival (60.3 and 109.2 months, respectively) compared to other histologic types of advanced thyroid cancer. For transcriptomic profiling, RNA-seq data of 162 patients with DTC from our previous study2 were applied as reference dataset for newly sequenced 25 samples.
Pathological diagnosis
For the purposes of accurate diagnosis, previous pathologic specimens were re-evaluated by an experienced pathologist (J.-K.W.). ATC or PDTC was defined as a tumor in which ≥10% of its volume was occupied by undifferentiated or poorly differentiated cells, while the focal ATC/PDTC was defined as a tumor in which <10% of the tumor volume was occupied by undifferentiated or poorly differentiated cells in the background of differentiated cancer43,44. As there is no definitive established pathologic definition for this, the definitions adopted in this study were based on the experience of clinicians and pathologists. The ATC component in tumors mixed with DTC was defined based on the following features: the nuclei without the characteristic features of DTC and showing a greater ratio of nucleus/cytoplasm, nuclear pleomorphism other than the features of DTC, and a more solid growth pattern with or without p53 expression. PDTC was defined on the basis of the Turin proposal for the use of uniform diagnostic criteria45, and was confirmed if showing a solid, trabecular, or insular growth pattern with the absence of conventional nuclear features of papillary carcinoma, and the presence of at least one of the following features: tumor necrosis, mitotic count ≥3/10 high-power field, or convoluted nuclei. wiFTC was defined in the case of widespread infiltration of thyroid tissue and/or vascular invasion according to WHO criteria46. All metastatic PTC accompanied distant metastasis in other organs and 77.42% of them (24/31) also had lymph node metastasis.
Massively parallel DNA sequencing
We performed WGS using Hiseq X instrument (Illumina, San Diego, CA, USA). For WGS, fragmentation of gDNA samples (except for T75 and N75) followed standard Illumina protocols except for an additional restriction enzyme digest step at the beginning of the work flow: a 1 μg of the DNA was first cut with a single methyl-sensitive restriction endonuclease, HpaII. Once digested, DNA was washed with Qiagen’s QIAquick PCR Purification Kit and sheared to a median size of 300 bp using a Covaris S220. Tumor and matched normal samples (except for T1) were sequenced with average sequencing depth of 72.23X and 35.74X, respectively.
WES (including WGS of T75 and N75) and targeted sequencing were performed with Hiseq 2000 and 2500 instruments, respectively (Illumina, San Diego, CA, USA), according to manufacturer’s instruction. For WES, we captured whole-exome region using SureSelect Human All Exon V4 kit, and tumor and matched normal samples were sequenced with average depth of 129.27X and 138.54X, respectively. Also, we downloaded the previously published WES data regarding PTC (n = 28) and FA/FTC (n = 18) for comparative analyses12,21. Paired-end reads were aligned to GRCh37.p13 reference using BWA47. Duplicated reads were removed by Picard tools (see URLs). Insertion/deletion (indel) realignment and base quality score recalibration (BQSR) was performed by Genome Analysis Tool Kit (GATK)48.
For targeted sequencing, we designed custom DNA capture probes using the Agilent SureDesign (see URLs). This custom probes contained the exonic regions of 57 genes, 13 regions for fusion gene rearrangements, and one region for TERT promoter mutation (Supplementary Table 4). The target genes were selected by our preliminarily findings from WGS/WES and the previously reported genes in TC1,2,8,49,50. Moreover, four genes (STARD9, HUWE1, BAZ2B, and MCM6) which were discovered in our unpublished work about distant metastasis of FTC were included. The average sequencing depth of 421.54X were achieved and matched normal samples were not included.
Massively parallel RNA sequencing
We conducted RNA-seq on 13 ATCs, 3 focal ATC/PDTCs, and 9 wiFTCs which were also sequenced by WGS and WES, respectively. Read alignment and gene expression quantification were performed using STAR51 and HTSeq52, respectively. DEGs were found with DESeq253 as following criteria: (1) adjusted P < 0.05 and (2) |Log2FC| ≥ 1. For DEG analysis, we only used BRAFV600E-positive classical PTC and RAS-positive FA/miFTC as comparison groups for BRAFV600E-positive ATC (PTC origin) and RAS-positive ATC (FTC origin), respectively. Then, DEGs were subjected to KEGG pathway enrichment analysis by Molecular Signatures Database22,54. As massively parallel DNA sequencing, we also performed indel realignment and BQSR using GATK48.
Scoring analysis
We used three scoring analysis methods from TCGA study1: TDS, ERK score, and BRS. For TDS, 16 genes that are related to thyroid function and metabolism were subtracted by median across all tumors samples and their average value were used. For ERK score, 52 MAPK-signaling pathway genes subtracted by median across all tumors and their average value were used. For BRS, single sample gene set enrichment analysis55,56 from GenePattern (see URLs) was performed with 71 gene signatures from TCGA study.
Variant detection
For all sequencing methods, MuTect and GATK’s HaplotypeCaller were applied to discover single nucleotide variant (SNV) and indel, respectively48,57. For MuTect, we applied somatic detection mode when matched normal sample is available. All variants were annotated by ANNOVAR58. To determine variants that are putatively associated with the progression of TC, we only kept variants which are not commonly found in The Exome Aggregation Consortium database (minor allele frequency <0.1%)59. Furthermore, missense SNVs which were functionally predicted as deleterious by PolyPhen2 or SIFT60,61, and the loss of function variants, such as nonsense, splicing site, and frameshift variants were retained for subsequent analysis. Then, we separated genes into two groups to avoid potential bias derived from the absence/presence of matched normal samples as follows: (1) genes that were confirmed to have somatic mutations from WGS/WES analysis (Fig. 1) and (2) genes that were highly suspected to have germline mutations (Supplementary Fig. 1).
Structural variation detection
To discover SCNA, we used FACETS and EXCAVATOR2 for WGS and WES data, respectively62,63. Then, GISTIC2 deduced the arm-level SCNA and the significantly altered chromosome regions64. For targeted sequencing data, CNVKit was implemented excluding antitarget regions with parameter ‘-m haar’ in segmentation step to determine CDKN2A deletion65. We considered the segment with Log2ratio ≤−0.6 as homozygous deletion, since this threshold reproduced the consistent results compared with the results from WGS. For targeted sequencing data, we discovered fusion genes when more than five split or discordant reads were found. For RNA-seq data, we used MOJO (see URLs) to identify fusion genes.
Mutational signature analysis
To assign specific mutational signature of each cancer genome, the average hierarchical clustering analysis was performed using mutational signatures from whole-genome-sequenced samples with 30 reference signatures. The mutation type probabilities of 96 motifs were extracted by SomaticSignatures66. The reference signatures were downloaded from COSMIC (see URLs).
Enhancer prediction
We used H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) data to identify enhancers in human thyroid gland. The ChIP-seq data of thyroid gland from 54 years old male was downloaded from ENCODE project67. We defined super-enhancers as described in other study68. In short, 76 bp single-end reads were aligned to the GRCh37 reference genome using bowtie 0.1.1 using following parameters: ‘-k 2’, ‘-m 2’, ‘-n 2’, ‘-S’, and ‘–best’69. Then, MACS 1.4.2 identified the enrichment regions of H3K27ac in thyroid gland following parameters: ‘-p 1e-9’, ‘–keep-dup = auto’, ‘-w –S –space = 50’, and ‘-g hs’70. Using output of MACS, ROSE identified the super-enhancers71. The constituent enhancers were stitched together if they are within 12,500 bp. If constituent enhancers were fully contained within promoter region (window ± 2000 bp from the transcription start site), they were excluded from stitching. At last, we separated super-enhancers and typical-enhancers from each other by isolating an inflection point of H3K27ac signal versus enhancer rank.
DNA methylation analysis
HpaII, methylation-sensitive restriction enzyme, recognizes and digests CCGG sites, if second cytosine is unmethylated72. Therefore, global DNA methylation pattern could be investigated using WGS with assistance of this characteristic of HpaII. After alignment of HpaII-digested paired-end reads to GRCh37 reference genome, we extracted the mapped reads which span CCGG sites of GRCh37. Then, we calculated DNA methylation level of second cytosine based on 10 possible patterns of spanning read. For this, only reads with high mapping quality (≥20) were used. Four patterns which span CCGG site but do not contain information of DNA methylation status were excluded from calculation. At last, we analyzed only CCGG sites that were appropriately covered by mapped reds (≥5) across 16 ATC tumors.
Polymerase chain reaction analysis and Sanger sequencing
TP53 germline mutations, TERT promoter mutations, and TERT rearrangements were confirmed by PCR and Sanger sequencing. The PCR was performed by using a StepOne Plus real-time PCR system (Applied Biosystems, Foster City, CA, USA). The PCR primer sequences are listed in Supplementary Table 5. Sanger sequencing with PCR product was conducted with a BigDyeTM Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) using an ABI 3730XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). For TERT promoter mutations, Primer #1 was mainly used and when the result was not clear, we used Primer #2 to confirm the result. It was performed on all the subject in this study, as well as FAs and miFTCs which were included in our previous study2.
TMAs and immunohistochemical staining
For the IHC analysis of p16, we constructed TMAs including 17 ATCs and 40 advanced DTCs (15 PDTCs and 25 focal PD/ATCs). TMAs were constructed from 2-mm-diameter cores derived from representative tumor areas of FFPE tissue blocks. p16 IHC was performed using the DAKO Omnis autostainer (DAKO-Agilent Technologies, Santa Clara, CA, USA) with a mouse anti-p16 monoclonal antibody (1:4, F. Hoffmann-La Roche, Basel, Switzerland).
Cell cultures and in vitro inhibition assays
CAL62, a human ATC cell line harboring KRASG12R were kindly provided by Dr. Yong-Hyun Jeon (Kyungpook National University Hospital, Daegu, Republic of Korea). CAL62 cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% of fetal bovine serum and incubated at 37 °C in a humidified atmosphere containing 5% of CO2. Approximately 0.8 × 105 cells/mL were then seeded on a 12-well culture plate. After 12 h, CAL62 cells were treated with ruxolitinib (1, 10, 20, and 25 μM; Selleckchem, Houston, TX, USA), a JAK1/2 inhibitor. After 24 h of treatment, mRNAs were extracted for the analysis of expression levels of downstream genes of JAK-STAT- signaling pathway. The primers are listed in Supplementary Table 5. After 48 h of treatment, cell viability was then determined by using a CCK-8 assay (Dojindo, Kumamoto, Japan). The CCK-8 solution (50 μL) was added to each well and, after 50 min incubation, absorbance at 450 nm was measured with a microplate reader (Molecular devices, San Jose, CA, USA).
Statistical analyses
For statistical comparisons, two-tailed Mann–Whitney U-test was used by SPSS 23.0 (IBM Co., Armonk, NY, USA). Disease-specific survival curves were generated by Kaplan–Meier method and compared by Log-rank test using survfit() function of survminer in the R programming language. For Cox proportional hazard model, coxph() function was applied.
URLs
TCGA Fusion Gene Database. Agilent SureDesign. Picard tools, GenePattern, MOJO, COSMIC.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All sequencing reads were submitted the European Genome Phenome Archive (https://www.ebi.ac.uk/ega/) with accession number EGAS00001003540.
References
Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 (2014).
Yoo, S. K. et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 12, e1006239 (2016).
Keutgen, X. M., Sadowski, S. M. & Kebebew, E. Management of anaplastic thyroid cancer. Gland Surg. 4, 44–51 (2015).
Molinaro, E. et al. Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 13, 644–660 (2017).
D’Avanzo, A. et al. Follicular thyroid carcinoma: histology and prognosis. Cancer 100, 1123–1129 (2004).
Durante, C. et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 91, 2892–2899 (2006).
Ibrahimpasic, T. et al. Outcomes in patients with poorly differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 99, 1245–1252 (2014).
Landa, I. et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Invest. 126, 1052–1066 (2016).
Pozdeyev, N. et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 24, 3059–3068 (2018).
Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Alaei-Mahabadi, B., Bhadury, J., Karlsson, J. W., Nilsson, J. A. & Larsson, E. Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers. Proc. Natl Acad. Sci. USA 113, 13768–13773 (2016).
Jung, S. H. et al. Mutational burdens and evolutionary ages of thyroid follicular adenoma are comparable to those of follicular carcinoma. Oncotarget 7, 69638–69648 (2016).
Ye, L. et al. The genetic landscape of benign thyroid nodules revealed by whole exome and transcriptome sequencing. Nat. Commun. 8, 15533 (2017).
Graubert, T. A. et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 44, 53–57 (2011).
Ju, Y. S. et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 22, 436–445 (2012).
Taylor, P. N. et al. Whole-genome sequence-based analysis of thyroid function. Nat. Commun. 6, 5681 (2015).
Hayashi, M. et al. Molecular cloning and characterization of human PDE8B, a novel thyroid-specific isozyme of 3’,5’-cyclic nucleotide phosphodiesterase. Biochem. Biophys. Res. Commun. 250, 751–756 (1998).
Valentijn, L. J. et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 47, 1411–1414 (2015).
Peifer, M. et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Rubinstein, J. C. et al. Shifting patterns of genomic variation in the somatic evolution of papillary thyroid carcinoma. BMC Cancer 16, 646 (2016).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999).
Kofler, N. M. et al. Notch signaling in developmental and tumor angiogenesis. Genes Cancer 2, 1106–1116 (2011).
Koch, S. & Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2, a006502 (2012).
Song, Y. S. et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 122, 1370–1379 (2016).
Liu, R. & Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 23, R143–R155 (2016).
Yin, D. T. et al. Clinicopathological significance of TERT promoter mutation in papillary thyroid carcinomas: a systematic review and meta-analysis. Clin. Endocrinol. 85, 299–305 (2016).
Bell, R. J. et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348, 1036–1039 (2015).
Davis, C. F. et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26, 319–330 (2014).
Endo, M. et al. Prognostic significance of p14ARF, p15INK4b, and p16INK4a inactivation in malignant peripheral nerve sheath tumors. Clin. Cancer Res. 17, 3771–3782 (2011).
Aesif, S. W. et al. Loss of p16(INK4A) expression and homozygous CDKN2A deletion are associated with worse outcome and younger age in thymic carcinomas. J. Thorac. Oncol. 12, 860–871 (2017).
Song, Y. S. et al. Aberrant thyroid-stimulating hormone receptor signaling increases VEGF-A and CXCL8 secretion of thyroid cancer cells, contributing to angiogenesis and tumor growth. Clin. Cancer Res. 25, 414–425 (2019).
Elvin, J. A. et al. Clinical benefit in response to palbociclib treatment in refractory uterine leiomyosarcomas with a common CDKN2A alteration. Oncologist 22, 416–421 (2017).
Konecny, G. E. et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin. Cancer Res. 17, 1591–1602 (2011).
Wiedemeyer, W. R. et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc. Natl Acad. Sci. USA 107, 11501–11506 (2010).
Duquette, M. et al. Metastasis-associated MCL1 and P16 copy number alterations dictate resistance to vemurafenib in a BRAFV600E patient-derived papillary thyroid carcinoma preclinical model. Oncotarget 6, 42445–42467 (2015).
Farkona, S., Diamandis, E. P. & Blasutig, I. M. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 14, 73 (2016).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Rizvi, H. et al. Molecular determinants of response to anti-Programmed Cell Death (PD)-1 and anti-Programmed Death-Ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 36, 633–641 (2018).
Chintakuntlawar, A. V. et al. Expression of PD-1 and PD-L1 in anaplastic thyroid cancer patients treated with multimodal therapy: results from a retrospective study. J. Clin. Endocrinol. Metab. 102, 1943–1950 (2017).
Zhang, Y., Marin-Acevedo, J. A., Azzouqa, A. G., Manochakian, R. & Lou, Y. Association of CDKN2A gene alteration with high expression of PD-L1. J. Clin. Oncol. 36, 9102–9102 (2018).
Sterlacci, W., Fiegl, M., Droeser, R. A. & Tzankov, A. Expression of PD-L1 identifies a subgroup of more aggressive non-small cell carcinomas of the lung. Pathobiology 83, 267–275 (2016).
Lee, D. Y. et al. Recurrence and survival after gross total removal of resectable undifferentiated or poorly differentiated thyroid carcinoma. Thyroid 26, 1259–1268 (2016).
Lee, D. Y. et al. Changes of clinicopathologic characteristics and survival outcomes of anaplastic and poorly differentiated thyroid carcinoma. Thyroid 26, 404–413 (2016).
Volante, M. et al. Poorly differentiated thyroid carcinoma: the Turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am. J. Surg. Pathol. 31, 1256–1264 (2007).
DeLellis, R. A., Organització Mundial de la, S, International Agency for Research on C. Pathology and Genetics of Tumours of Endocrine Organs, 3rd edn (IARC Press, 2004).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Stransky, N., Cerami, E., Schalm, S., Kim, J. L. & Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846 (2014).
Costa, V. et al. New somatic mutations and WNK1-B4GALNT3 gene fusion in papillary thyroid carcinoma. Oncotarget 6, 11242–11251 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7, Unit 7, 20 (2013).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
D’Aurizio, R. et al. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 44, e154 (2016).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 12, e1004873 (2016).
Gehring, J. S., Fischer, B., Lawrence, M. & Huber, W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics 31, 3673–3675 (2015).
Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Loven, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Reyna-Lopez, G. E., Simpson, J. & Ruiz-Herrera, J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 253, 703–710 (1997).
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Science, ICT & Future Planning (grant number: NRF-2016R1A2B4012417).
Author information
Authors and Affiliations
Contributions
J.-S.S. and Y.J.P. conceived and designed study. S.-K.Y. and Y.S.S. wrote the manuscript. S.-K.Y. and J.H. performed bioinformatic analyses. H.H.K. and G.J. performed TERT promoter sequencing and PCR. J.-K.W. contributed to the histopathological review. J.-Y.S. performed massively parallel sequencing experiments. E.K.L., Y.A.K., S.-j.K., S.W.C., E.-J.C., K.E.L. and J.-I.K. contributed to data interpretation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks Cristina Ronchi and other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoo, SK., Song, Y.S., Lee, E.K. et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat Commun 10, 2764 (2019). https://doi.org/10.1038/s41467-019-10680-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-10680-5
This article is cited by
-
Dual targeting of MAPK and PI3K pathways unlocks redifferentiation of Braf-mutated thyroid cancer organoids
Oncogene (2024)
-
Genomic alterations in thyroid cancer: biological and clinical insights
Nature Reviews Endocrinology (2024)
-
Ruxolitinib induces apoptosis and pyroptosis of anaplastic thyroid cancer via the transcriptional inhibition of DRP1-mediated mitochondrial fission
Cell Death & Disease (2024)
-
Emerging roles of circular RNAs in regulating the hallmarks of thyroid cancer
Cancer Gene Therapy (2024)
-
Current evidences in poorly differentiated thyroid carcinoma: a systematic review and subsection meta-analysis for clinical decision making
Endocrine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
