
Abstract
Bacterial RNA polymerase employs extra-cytoplasmic function (ECF) σ factors to regulate context-specific gene expression programs. Despite being the most abundant and divergent σ factor class, the structural basis of ECF σ factor-mediated transcription initiation remains unknown. Here, we determine a crystal structure of Mycobacterium tuberculosis (Mtb) RNAP holoenzyme comprising an RNAP core enzyme and the ECF σ factor σH (σH-RNAP) at 2.7 Å, and solve another crystal structure of a transcription initiation complex of Mtb σH-RNAP (σH-RPo) comprising promoter DNA and an RNA primer at 2.8 Å. The two structures together reveal the interactions between σH and RNAP that are essential for σH-RNAP holoenzyme assembly as well as the interactions between σH-RNAP and promoter DNA responsible for stringent promoter recognition and for promoter unwinding. Our study establishes that ECF σ factors and primary σ factors employ distinct mechanisms for promoter recognition and for promoter unwinding.
Similar content being viewed by others

Introduction
Transcription initiation is the first and the most tightly regulated step of bacterial gene expression1,2,3. σ factors are required for transcription initiation4. After forming a complex with the RNA polymerase (RNAP) core enzyme, σ factors guide RNAP to promoter DNA, open double-stranded DNA (dsDNA) to form a transcription bubble, facilitate synthesis of initial short RNA transcripts, and later assist in promoter escape4,5,6.
Bacterial σ factors are classified into two types—σ70- and σ54-type factors based on their distinct structures and mechanisms. The σ70-type factors can be further classified into four groups according to their conserved domains4. Group-1 σ factors (or primary σ factors) contain domains σ1.1, σ1.2, σNCR, σ2, σ3.1, σ3.2, and σ4; group-2 σ factors contain all domains except σ1.1; group-3 σ factors contain σ2, σ3.1, σ3.2, and σ4; while group-4 or extra-cytoplasmic function (ECF) σ factors only contain σ2 and σ47. The genomes of a majority of bacteria harbor one primary σ factor for expression of most genes (i.e., group-1 σ factor; σ70 in Escherichia coli and σA in Gram-positive bacteria; referred as σA hereafter), and multiple alternative σ factors for expression of genes with cellular- or environmental-context-dependent functions8,9. The ECF σ factors are the largest family of alternative σ factors. On average, bacterial genomes encode six ECF σ factors; the given number for a particular bacterium will vary according to its genome size and environmental complexity9,10. ECF σ factors enable bacteria to rapidly respond to a variety of stresses9,11,12 and are known to be essential for the pathogenicity of several disease-causing bacteria13,14. Mycobacterium tuberculosis has 10 ECF σ factors (σC, σD, σE, σG, σH, σI, σJ, σK, σL, and σM); deletion of ECF σ factors from M. tuberculosis results in attenuated disease progression (e.g., sigC and sigD) or in alleviated virulence (e.g., sigE and sigH)15,16.
The σA is capable of recognizing at least five conserved functional elements in the DNA sequences of gene promoters, including the “−35 element” (TTGACA)17, the “Z element”18, the “extended −10 element” (TG)19, the “−10 element” (TATAAT)17, and the “discriminator element” (GGG)20. Distinct domains of σA are responsible for interacting with these DNA elements: the domain σ4 forms sequence-specific interactions with exposed bases in the major groove of the −35 dsDNA21; the σ2.5 and σ3.1 domains reach into the major groove of the extended −10 element and make base-specific contacts22,23; and the σ2 and σ1.2 domains recognize and then unwind the −10 element dsDNA3,24.
During the process of promoter unwinding, a tryptophan dyad of σ2 (W256/W257 in Thermus aquaticus σA or W433/W434 in E. coli σ70) forms a chair-like structure that functions as a wedge to separate the dsDNA at the (−12)/(−11) junction22,23. The group-2 σ factors use the same set of residues to unwind promoter DNA; but the melting residues of group-3 σ factors are not conserved25. Subsequently, the base moieties of the unwound nucleotides at position −11 and −7 of the nontemplate strand—A(−11)(nt) and T(−7)(nt)—are flipped out and inserted into pre-formed pockets by σ2 and σ1.23,24. Domain σ1.2 also recognizes the discriminator element by flipping out the guanine base of G(−6)(nt) and inserting it into a pocket24. Although σ3.2 does not read the promoter sequence directly, it is essential for transcription initiation. Domain σ3.2 reaches into the RNAP active site cleft and “pre-organizes” template single-stranded DNA (ssDNA)24. Domain σ3.2 also blocks the path of the extending RNA chain (>5 nt)26,27 thereby contributing to both initial transcription pausing28 and promoter escape29,30.
Each category of known ECF σ factors recognizes promoters bearing a unique sequence signature at the −35 and the −10 elements10,31. In contrast to the high tolerance to sequence variation at the −35 and the −10 promoter elements exhibited by the primary σ factor, the ECF σ factors have stringent requirements for sequence identity in the −35 and the −10 elements and for spacer length between these two elements through an unknown mechanism8,32. Although both the primary and ECF σ factors recognize the −35 element via σ4 and recognize the −10 element via σ2, the protein sequences of these two domains are not well conserved, and the consensus sequences of the two corresponding DNA elements vary. Crystal structures of individual σ2 or σ4 domains of ECF σ factors complexed with cognate DNA have suggested that these ECF domains bind the −35 and the −10 elements differently than does the primary σ factor, implicating a unique means of promoter recognition by ECF σ factors33,34. Another striking difference was revealed by a sequence analysis showing the surprising fact that ECF σ factors do not contain σ3 domains (σ3.1 and σ3.2), but instead contains a linker—highly variable in both length and sequence—to connect the σ2 and σ4 domains4. This fact immediately raises the question of how these σ factors perform multiple steps of transcription initiation that the σ3 domain performs in the primary σ factor.
A recent crystal structure of E. coli σE2/−10 ssDNA binary complex suggests that bacterial ECF σ factors probably recognize and unwind promoters through a unique mechanism. Specifically, E. coli σE employs a flexible “specificity loop” to recognize a flipped master nucleotide of the −10 element and probably unwinds at a distinct position compared with that of σ70 by using non-conserved melting residues34. In contrast to the large collection of structural information of primary σ factor, no structure of bacterial RNAP complex with ECF σ factor is available. Therefore, it is largely unknown how ECF σ factors form a holoenzyme with RNAP and how ECF σ factors work alongside RNAP to recognize and to unwind promoter DNA. Here we report the crystal structure of an ECF σ factor-RNAP holoenzyme comprising M. tuberculosis RNAP and σH at 2.70 Å resolution. We also report the crystal structure of an ECF σ factor-RNAP transcription initiation complex comprising M. tuberculosis σH-RNAP holoenzyme, a full transcription bubble of promoter DNA, and an RNA primer at 2.80 Å resolution. The crystal structures present detailed interactions among RNAP, ECF σ factors, and promoter DNA. The structures together with data from biochemical assays collectively establish the structural basis of RNAP holoenzyme assembly, promoter recognition, and promoter unwinding by the ECF σ factors.
Results
The crystal structure of M. tuberculosis σH-RNAP holoenzyme
The crystals of M. tuberculosis σH-RNAP holoenzyme were unexpectedly obtained during an initial attempt to crystallize M. tuberculosis σH-RPo (Supplementary Figs. 1a, c–f). The crystal structure of M. tuberculosis σH-RNAP holoenzyme at 2.7 Å resolution was determined by molecular replacement using a Mycobacterium smegmatis RNAP core enzyme (PDB: 5TW1) [https://www.rcsb.org/structure/5TW1] as the searching model35. The Fo–Fc map shows unambiguous density for σH residues 22–195 (Table 1; Supplementary Fig. 2a) and the anomalous difference map shows clear density for 4 out of 5 Se atoms, validating the σH model (Supplementary Fig. 2a).
σH2 (residues 22–99) and σH4 (residues 140–195) fold into independent helical domains (Fig. 1a, c). Lacking the σ1.1, σ1.2, and σNCR domains of σA, the σH2 domain is very compact, containing only four α helices (Fig. 1c). The “specificity loop” (residues 72–79 in σH2; Supplementary Fig. 2a) known to be essential for recognition of the −10 element is disordered (no electron density), in contrast to the pre-organized specificity loop in the σA-RNAP holoenzyme (Fig. 1b, d and Supplementary Fig. 2b–c). Lacking σ1.2, the domain that forms extensive interactions with the specificity loop of σA, probably accounts for the disordered conformation of the specificity loop in σH (Fig. 1c, d). As occurs in σA2, the σH2 domain resides in a cleft between the RNAP-β lobe and the RNAP-β′ coiled-coil (β′CC) and makes extensive electrostatic interactions with the latter (Fig. 1e). Notably, the residues contacting β′CC of both σA and ECF σ factors are conserved (Supplementary Figs. 2d, e and 3), suggesting that β′CC probably serves as an anchor point for the σ2 domain of the majority of bacterial σ factors.

The crystal structure of Mtb σH-RNAP holoenzyme. a Schematic diagram of Mtb σH. Ordered regions in the structure are indicated by dashes. b Schematic diagram of T. thermophilus σA. c σH in the crystal structure of Mtb σH-RNAP holoenzyme. The disordered specificity loop is shown by blue dashes. d σA in the crystal structure of T. thermophilus σA-RNAP (PDB: 1IW7) [https://www.rcsb.org/structure/1iw7] was used for comparison due to no available structure of Mtb σA-RNAP holoenzyme. e Front and top views of Mtb σH-RNAP holoenzyme. βFTH, the flap-tip helix on RNAP-β subunit; βCTH, the C-terminal helix on RNAP-β subunit; β′CC, the coiled-coil on RNAP-β′ subunit. σA1.1, purple; σA1.2, pink; σANCR, cyan; σ2, blue; σA3.1, orange; σ3.2, green; σ4, red. RNAP-α subunits, light orange; RNAP-β subunit, black; RNAP-β′ subunit, gray; RNAP-ω subunit, light cyan
The σH4 domain enfolds the flap-tip helix of the RNAP-β subunit (βFTH; Figs. 1e and 2a). The hydrophobic residues contacting the βFTH are conserved between the σA and the ECF σ factors (Supplementary Figs. 2f–g and 3). Surprisingly, we discovered another anchor point for the σH4 domain on RNAP—a C-terminal helix of the RNAP-β subunit (βCTH; residues 1145–1157; Figs. 1e and 2a, and Supplementary Fig. 2h and m). The interaction with βCTH was not observed in any of the previously reported bacterial σA-RNAP structures22,24,36,37. To explore the contribution of such interaction to the transcription activity of σH-RNAP, we performed in vitro transcription experiments using wild-type or βCTH-deleted Mtb σH-RNAP holoenzyme and pClpB promoter variants with −35/−10 spacer lengths ranging from 15 to 19 bp. The wild-type σH-RNAP was most transcriptionally active with a promoter of 17-bp spacer (Fig. 2b), consistent with a study reporting that most σH-regulated promoters have a 17-bp spacer38. The βCTH-deletion variant caused impaired transcription activity from promoter with the optimal spacer length (17 bp) but showed little effect on promoter with sub-optimal spacer lengths (16 and 18 bp) (Fig. 2b), suggesting that the interactions between βCTH and σH are important for the transcription activity of σH. Intriguingly, deletion of βCTH caused a general increase of σA-dependent transcription activity from promoter with spacer lengths 15–19 bp (Supplementary Fig. 2i).

The interaction between Mtb RNAP core enzyme and σH. a Both βFTH and βCTH interact with σH4. Colors are as in previous figure. b The in vitro transcription activity of σH-RNAP(WT) (green bars) and σH-RNAP(∆βCTH) (gray bars) from pClpB promoter variants with −35/−10 spacer length of 15–19 base pairs. “122 nt” indicates length of run-off RNA products. c The interaction between σH2/σH4 linker (σH3.2) and RNAP core enzyme. RNAP-β subunit was omitted for clarity. The location of catalytic Mg2+ is shown by a dashed purple circle. BH, bridge helix. RNAP-β′ subunit, gray. σH2, σH3.2, and σH4, blue, green, and red respectively. d The in vitro transcription activity from pClpB promoter of RNAP holoenzymes comprising σH derivatives. H2-H3.2-H4, wild-type σH; H2---H4, two individual domains of σH2 and σH4; H2-DL-H4, a chimeric σH with σH3.2 replaced by a disordered loop with an equivalent residue number; H2-E3.2-H4, a chimeric σH with σH3.2 replaced by Mtb σE3.2; H2-L3.2-H4, a chimeric σH with σH3.2 replaced by Mtb σL3.2; H2-M3.2-H4, a chimeric σH with σH3.2 replaced by Mtb σM3.2. The experiments were repeated in triplicate, and the data are presented as mean ± S.E.M. Source data of b and c are provided as a Source Data file
The most surprising finding in the structure of σH-RNAP is the interaction between RNAP and the linker region connecting σH2 and σH4. The linker is the least conserved region among the ECF σ factors and shares no sequence similarity with the linker of σA (Supplementary Fig. 3). Our structure shows that the linker region of σH dives into the active site cleft and emerges out from the RNA exit channel of RNAP (Figs. 1e and 2c, and Supplementary Fig. 4a–b). This interaction creates an entry channel for template ssDNA loading into the active site cleft during RPo formation, but blocks the exit pathway of extending RNA during subsequent transcription initiation events. The path of the σH2/σH4 linker in RNAP is similar to that of σA3.2 (Supplementary Fig. 4c–d), so we designated the σH2/σH4 linker as σH3.2.
To examine the significance of the interaction between σH3.2 and RNAP, we tested the in vitro transcription activity of RNAP holoenzyme comprising σH variants with the σH3.2 domain either deleted or swapped. Deleting σH3.2 or replacing σH3.2 with a protein sequence known to be disordered completely abolished the transcription activity of σH (lanes II and III in Fig. 2d), indicating that σH3.2 does not simply serve as a σH2/σH4 linker; rather, the interaction between σH3.2 and the active site cleft of RNAP is essential for transcription. Interestingly, replacing σH3.2 with the σ2/σ4 linker from other ECF σ factor partially recovered the transcription activity (lanes IV, V, and VI in Fig. 2d and Supplementary Fig. 4e). Based on these results, we infer that other ECF σ factors probably have a functional σ3.2 domain that, while divergent in sequence, likely binds RNAP in a way somehow analogous to σH3.2.
The overall structure of Mtb σH-RPo
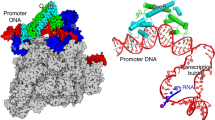
To understand how σH-RNAP holoenzyme recognizes promoter DNA and initiates transcription, we sought to determine a crystal structure of σH-RPo. We assembled the complex by incubating the RNAP core enzyme, σH, and a synthetic nucleic-acid scaffold (Fig. 3d, and Supplementary Fig. 1b, 1g–j). The synthetic scaffold comprises an upstream DNA duplex (−34 to −10 with respect to transcription start site at +1) with a consensus −35 element (GGAACA), a non-complimentary transcription bubble (−9 to +2) with a consensus −10 element (GTT), a 7-nt RNA primer complimentary to template DNA (−6 to +1), and a downstream DNA duplex (+3 to +13). We determined the crystal structure of σH-RPo at 2.8 Å resolution by molecular replacement using the crystal structure of Mtb σH-RNAP holoenzyme as a search model (Table 1). The Fo–Fc map contoured at 2.5 σ shows clear density for all nucleotides of nontemplate ssDNA, template ssDNA, and RNA primer of the transcription bubble, as well as for all the nucleotides of the upstream and downstream DNA duplexes (Fig. 3e).

The crystal structure of Mtb σH-RPo. a Front and top views of σH-RPo overall structure. The α, β, β′, and ω subunits of RNA polymerase core enzyme are shown as ribbon and colored in light orange, gray, black, and light cyan respectively. The σH2, σH3.2, and σH4 are shown as ribbon and colored as in above figures. The nontemplate DNA, template strand DNA, and RNA strands are shown in surface and colored in orange, yellow, and cyan, respectively, except the −35 element (green), the −10 element (purple), and the CRE (black). The location of the catalytic Mg2+ is indicated by a dashed circle. b Both Mtb σH-RPo (violet) and Mtb σA-RPo (light gray; PDB: 5UHA) [https://www.rcsb.org/structure/5uha] show closed clamp conformation. c The comparison of upstream double-stranded DNA (dsDNA), transcription bubble, and downstream dsDNA in Mtb σH-RPo (colored as in a) and Taq σA-RPo (light gray; PDB: 4XLN) [https://www.rcsb.org/structure/4XLN]. d Summary of protein–nucleic acid interactions. Solid line, van del Waals interactions; dashed line, polar interactions. Colors are as in above. Red box, interactions with the −35 element (details in Fig. 4a); gray box, interactions with the −35/−10 spacer (details in Fig. 4b); blue box, interactions of the single-stranded DNA (ssDNA) in transcription bubble (details in Figs. 4e and 5a–d); green box, interactions with the DNA/RNA hybrid (details in Fig. 5e). The numbers in parenthesis are corresponding positions in σA-regulated promoters. e The simulated-annealing omit Fo–Fc electron density map (nucleic acids removed; green; contoured at 2.5 σ) and model for nucleic acids
In the σH-RPo structure, σH makes the same interactions with RNAP as in the structure of σH-RNAP holoenzyme. The RNAP clamp adopts a closed conformation as in σA-RPo24,39, consistent with previous single-molecule fluorescence resonance energy transfer results40 and supporting the idea that clamp closure is also an obligatory step of RPo formation in ECF σ-mediated transcription initiation (Fig. 3b). The DNA/RNA hybrid resides in the active site cleft in a post-translocation state, and the downstream DNA duplex is accommodated in the main channel. The conformations of the DNA/RNA hybrid and downstream DNA duplex in σH-RPo and σA-RPo are similar (Fig. 3c)24.
The σH-RPo structure revealed multiple interactions responsible for promoter recognition and promoter unwinding by σH-RNAP that we will describe in detail in each of the subsequent sections of our manuscript. These include: (1) σH4 inserts into the major groove and reads the sequence of the −35 element (Figs. 3a and 4a, and Supplementary Fig. 6a); (2) the RNAP-β′ subunit stabilizes the upstream DNA duplex by contacting the phosphate backbone of nucleotides at positions −23, −20, and −19 (Figs. 3a and 4b); (3) σH2 unwinds dsDNA using an apparently distinct mechanism (Figs. 3a and 4e); (4) σH2 and the RNAP-β subunit recognize sequences at four positions in the −10 element via interactions with nontemplate ssDNA (Figs. 3a and 5a–c); (5) the RNAP-β subunit recognizes the “CRE element” DNA sequence via interactions with nontemplate ssDNA (Figs. 3a and 5d, and Supplementary Fig. 7a); and (6) σH3.2 guides the template ssDNA into the RNAP active center and forms interactions with the DNA/RNA hybrid in the active site cleft (Figs. 3a and 5e, and Supplementary Fig. 7g).

The interaction between upstream promoter DNA and RNAP in the Mtb σH-RPo structure. a The interaction between σH4 and the −35 element. σH4, red ribbon. The residues making base-specific interactions are presented as ribbon and half-transparent spheres; the C, O, N, and S atoms of the residues are colored in white, red, blue, and yellow respectively. The C, O, N, and P atoms of the −35 DNA are colored in green, red, blue, and orange, respectively. H-bond, blue dash. b The interaction between RNAP and −35/−10 spacer region of upstream dsDNA. The residues making polar interactions with DNA are shown and colored as above. The C atoms of the −35/−10 spacer DNA are colored in yellow (template DNA) or orange (nontemplate DNA), and the rest of atoms are colored as above. c The in vitro transcription activity of σH derivatives comprising alanine substitution on σH4. d M181A changes the sequence specificity for positions −30 and −31 of the −35 element. e The promoter melting by σH. The numbers in parenthesis correspond to positions in σA-RPo. The −10 element is colored in purple. The residues involved in promoter melting are shown in stick and colored in white (C atoms), red (O atoms), and blue (N atoms). f The in vitro transcription activity of σH2 derivatives with alanine or tryptophan substitutions. The experiments were repeated in triplicate, and the data are presented as mean ± S.E.M. Source data of c, d, and f are provided as a Source Data file

The interaction between transcription bubble and RNAP in the Mtb σH-RPo structure. a Recognition of the “G−11T−10T−9” sequence in the −10 element by σH2. b Stacking of −7(nt) and −6(nt) nucleotides by RNAP-β subunit and σH2. c The recognition of G−5(nt) by RNAP-β subunit and σH2. d The interaction between RNA polymerase (RNAP) and CRE element. Colors are as above. e The interaction between RNAP and DNA/RNA hybrid. f The in vitro transcription activity of RNAP derivatives comprising alanine substitutions of DNA-contacting residues on RNAP-β subunit and σH. The experiments were repeated in triplicate, and the data are presented as mean ± S.E.M. Source data of f are provided as a Source Data file
The interactions of σH 4 with the −35 element
σH-regulated promoters have a distinct consensus sequence at their −35 elements (5′-GGAAYA-3′; from −34 to −29; Supplementary Fig. 5a)38,40,41. Alternation of DNA sequences at each of the positions from −34 to −29 resulted in substantial loss of transcription activity (Supplementary Fig. 5b). In the structure, σH4 binds to the major groove of dsDNA of the −35 element and makes base-specific polar interactions with nucleotides at three (−34, −33, and −31) out of six positions (Figs. 3d and 4a, and Supplementary Fig. 6a). The G−34(nt) makes two H-bonds with R186 through its O6 and N7 atoms; the G−33(nt) makes one H-bond with S182 through its O6 atom; and the A−31(nt) makes one H-bond with M181. Moreover, M181 forms extensive van der Waals interactions with nucleotides at positions −31 and −30 of the template strand. The interactions are important, as alanine substitutions of R186 or S182 resulted in substantial loss of transcription activity (Fig. 4c). Interestingly, the M181A mutant had increased transcription activity, but this came at the apparent expense of relaxing its sequence stringency for the positions −31 and −30 (Fig. 4d), suggesting that M181 partially accounts for sequence specificity of the two positions.
The rest of the −35 element (−32, −30, and −29) makes no base-specific interactions. Previous crystal structure of E. coli σE4/−35 dsDNA and a structural model of Streptomyces coelicolor σR4/−35 dsDNA reported a local DNA shape readout (straight helix with a narrow minor groove) at this region33,42. We observed a similar DNA conformation (Supplementary Fig. 6b) in our crystal structure, suggesting a general mode of promoter recognition for the ECF σ factors. Such DNA structure might assist the binding of −35 dsDNA to the σH4 surface perhaps by making favorable interactions through its phosphate backbones with polar residues of σH4 including Y166, K167, T179, R183, H185, and R188 (Fig. 3d and Supplementary Fig. 6a). Consistently with this, losing any of these interactions causes a substantial loss of transcription activity (Fig. 4c).
The interactions of σH-RNAP with the −35/−10 spacer
In the crystal structure of Mtb σH-RPo, σH-RNAP contacts phosphate backbones of the spacer region between the −35 and the −10 elements at three positions (Fig. 3d): (1) R77 of the RNAP-β′ zinc-binding domain contacts the nucleotide at position −23; (2) R37 of the RNAP-β′ zipper domain contacts the nucleotides at positions −20 and −19; and (3) K96 and R99 of σH2 contact the nucleotide at position −14 (Fig. 4b). These interactions probably stabilize the conformation of the upstream DNA duplex, likely promoting the engagement of the upstream duplex with σH4 and σH2 for subsequent promoter unwinding. Mutating K96 and R99 causes a mild loss of transcription activity, suggesting the importance of these interactions (Fig. 4f).
The promoter DNA unwinding function of σH
The electron density map unambiguously shows that the T:A base pair at position −10 (corresponding to position −12 of promoters for σA) is unwound, despite the fact that the −10 nucleotides in the synthetic nucleotide scaffold were designed to be complimentary (Figs. 3d and 4e). This observation strongly suggests that σH2 unwinds promoter DNA at the −11/−10 junction (corresponding to (−13)/(−12) of promoters targeted by σA; Fig. 4e and Supplementary Fig. 6c, d). This clearly confirms the hypothesis that the ECF σ factors unwind promoter DNA starting from a distinct position as compared to σA32,34. In the structure, N88 blocks the pathway of upstream dsDNA and serves as a wedge to disrupt the stacking of base pairs at the positions −11 and −10. The base pair at position −10 is subsequently forced open by σN88 via a competitive H-bond between the Watson-Crick atom of the T−10(nt) and N88 (Fig. 4e). Two unwound nucleotides on the nontemplate strand DNA (T−10(nt) and T−9(nt)) are stabilized by two adjacent pockets of σH2; these pockets are where the sequence identities are “read” (Supplementary Fig. 6F). Moreover, two unwound nucleotides on the template strand DNA (A−10(t) and T−9(t)) are also trapped in a cleft created by the RNAP-β lobe and σH2. Specifically, the base moieties of A−10(t) and T−9(t) form a stack with βR395, σY90, and σY94, and the phosphate moieties are stabilized by βK428, βN419, and σQ98. The functional importance of these residues for promoter DNA unwinding was underscored by our finding that their substitution with alanine resulted in defects in transcription (I85G, I85A, and N88A; Fig. 4f).
Structure superimposition between σH-RPo and σA-RPo shows that the promoter DNA position at which unwinding is initiated differs between σA and σH by one base pair; σH unwinds promoter DNA at a position 1 bp upstream of the position at which σA unwinds its promoter DNA (the (−12)/(−11) junction for σA; the −11/−10 junction for σH corresponding to the (−13)/(−12) junction for σA; Supplementary Fig. 6c–e). A tryptophan dyad (W433/W434 in E. coli or W256/W257 in T. aquaticus) is essential for promoter unwinding at the (−12)/(−11) junction by σA 22,23,43,44, but the residues at the corresponding positions of σH (R84/I85) are not conserved (Supplementary Fig. 3). Mutating R84 and I85 in σH to tryptophan (I85W, R84W, or I85W/R84W) resulted in substantial loss of transcription activity, confirming that σH opens promoter through a different mechanism than σA and supporting the mechanism proposed for E. coli σE (Fig. 4f)34.
The interactions of σH-RNAP with the −10 element
σH-regulated promoters contain a “G−11T−10T−9” consensus sequence at the −10 element (Supplementary Fig. 5a)38,40,41. Alteration of the DNA sequence at any of these positions resulted in complete loss of promoter activity (Supplementary Fig. 5b), helping explain the reported finding from previous bioinformatic studies that the −10 element is the most conserved region among ECF σ factors10,31. Our crystal structure shows that the base moiety of C−11(t) of the −11 G:C pair makes one H-bond with N92 and extensive Van der Waal interactions with I91, alanine substitution of N92 or I91 resulted in modest or substantial decrease of transcription, respectively (Fig. 5a, f), providing a structural explanation for sequence recognition at this position. Our crystal structure further reveals that σH recognizes the nucleotides of the next two positions via two protein pockets on the surface of σH2 (Fig. 5a).
T−10(nt) is accommodated by a shallow protein pocket on σH2 wherein I85 forms a stack with the base of T−10(nt) at the pocket bottom and W81 supports the sugar moiety of T−10(nt) on one side of the pocket (Fig. 5a). N88 on the other side of the pocket makes a H-bond with the base moiety of T−10(nt), likely contributing to the sequence specificity known to occur for this position. Alanine substitution of I85 or W81 causes severe defects in transcription activity (Figs. 4f and 5f), emphasizing their importance. Sequence alignment of the 10 Mtb ECF σ factors shows that the I85 and W81 are highly conserved (Supplementary Fig. 3), suggesting the −10(nt) pocket probably exists on other ECF σ factors. Alanine substitution of N88 causes defects in transcription activity (Fig. 4f), and the sequence alignment shows that N88 is the most frequent residue at this position. However, other polar residues occur at this position (e.g., a histidine for σC and σD, and an arginine for σI, σJ, and σK) (Supplementary Fig. 3), suggesting that this position may help determine sequence specificity for position −10 of the promoter DNA.
T−9(nt) is flipped out and inserted into a protein pocket formed by the “specificity loop” of σH234. The thymine base of T−9(nt) stacked on top of W81 makes one H-bond with the main-chain atom of σH residue R73 and two H-bonds with the side-chain atoms of σ residues T76 and N77 on the specificity loop (Fig. 3d and Supplementary Fig. 6f). F72 and R73 contact the thymine base via van der Waals interactions. Mutating T−9(nt) completely abolished promoter activity (Supplementary Fig. 5b), and alanine substitution of σH residues contacting T−9(nt) (F72A, T76A, N77A, and W81A) severely decreased transcription activity from the consensus promoter (Fig. 5f), verifying the requirement for the T−9(nt)/σH2 interaction for transcription. The fact that both the primary and the ECF σ factors use the specificity loop to read the sequence identity (position −9 of σH-regulated promoters corresponding to position (−11) of σA-regulated promoters; Supplementary Fig. 6f–h) implies the central importance of this position in promoter DNA3,24,34. Outside of these crucial positions, σH2 forms fewer interactions with nontemplate nucleotides (positions −8, −7, and −6). In the structure, the base moiety of nucleotide at position −8 is disordered (no electron density) and the base moieties of −7 and −6 nucleotides are sandwiched between residue T405 of the RNAP-β subunit and residue Y83 of σH2 (Figs. 3d and 5b, and Supplementary Fig. 6i).
A surprising finding in the σH-RPo crystal structure is that σH2 flips the guanine base of G−5(nt) (corresponding to the (−7) position of σA-regulated promoters) and inserts into a shallow pocket created by σH2 and the RNAP-β gate loop (Fig. 5c). In this pocket, G−5(nt) forms a stacking interaction with R282, two H-bonds with E285 on the RNAP-β gate loop, and has van der Waals interactions with D38 and Q39 of σH2. Mutations of the RNAP-β gate loop (βR282A or βE285A) cause severely reduced transcription activity (Fig. 5f). As the RNAP-β gate loop makes base-specific contacts to G−5(nt), we tested whether this position exhibits sequence preference. Results of in vitro transcription assays showed that promoters with C or G at this position have much higher transcription activity compared with T or A (Supplementary Fig. 5b), suggesting a sequence preference of C~G > T~A at such position. Our results therefore show that in σH-regulated promoters, the consensus sequence of the −10 elements is extended to “G−11T−10T−9N−8N−7N−6S−5”. It is worth noting that σA also accommodates the T(−7)(nt) nucleotide in a protein pocket3,24; however, the protein pocket is mainly formed by residues from σA1.2 and σA224, and the structural features that determine sequence specificity are located on σA (Supplementary Fig. 6i–k).
Interactions of σH-RNAP with the CRE
The guanine base of G+2(nt) is inserted into the “G” pocket in σH-RPo (Fig. 5d) and makes essentially the same interaction with residues in the “G” pocket as the G(+2)(nt) does in the σA-RPo complex (Figs. 3d and 5d, and Supplementary Fig. 7d–f)24. However, in contrast to the σA-RPo, in which the nontemplate T(+1)(nt) forms a stacking interaction with βW211, the nontemplate T+1(nt) in σH-RPo was pushed out from the base-stacking. Instead, the nucleotide immediately upstream of +1 makes the stacking interaction with βW211 in σH-RPo (Fig. 5d and Supplementary Fig. 7a–c). The base moieties of the remaining nucleotides (−4 to −1) between the CRE and −10 elements are stacked in a row between L45 on σH2 and W211 on the RNAP-β subunit (Fig. 5d and Supplementary Fig. 7a).
Interactions of the template ssDNA
In the σH-RPo structure, σH and the RNAP-β subunit form an “T-ssDNA entry channel” that guides the template ssDNA into the RNAP active center cleft (Supplementary Fig. 4a). Along the channel, σH and RNAP form extensive interactions with the template ssDNA (σM47, σR49, σY90, σY94, σQ98, σY04, βR395, βN419, βP422, βK428, and β′R334; Fig. 3d). Compared to σA, which forms extensive interactions between the σA3.2 finger with the ssDNA template nucleotides the active center cleft24, σH forms fewer interactions (Fig. 3d and Supplementary Fig. 7g–i). The 5′ terminus of the 7-nt RNA is positioned very closely to the tip of σH3.2; extending the RNA by even one additional nucleotide would likely result in steric hindrance (Fig. 5e and Supplementary Fig. 7g). Since σH3.2 occupies the RNA exit channel and must be displaced by the nascent RNA chain, such hindrance may be the trigger for the release of σH and promoter DNA during promoter escape.
Discussion
The structural basis of transcription initiation by the primary σ factor has been studied extensively, but little is known about how the ECF σ factors—the largest and most diverse group of σ70 family factors—initiate transcription. In this study, we present high-resolution crystal structures of M. tuberculosis σH-RNAP holoenzyme and σH-RPo complexes along with comprehensive mutational analyses. Our study demonstrates the structural basis for RNAP holoenzyme formation and transcription initiation by the ECF σ factors.
Our structures show that σH binds to RNAP in a similar way to σA, in which σ2 and σ4 stay on the surface of RNAP, and σ3.2 inserts into the active center. The interactions of σH2 and σH4 with RNAP were thus anticipated and supported a structure model of E. coli σE-RNAP holoenzyme45, as the residues contacting the βFTH and β′CC domains are conserved between the ECF and primary σ factors. However, the interactions of σH3.2 with RNAP are unexpected, showing similarity in neither sequence nor secondary structure between the σ3.2 regions of σECF and σA (Supplementary Fig. 3). In vitro transcription experiments show that σH3.2 is essential for the transcription activity of σH-RNAP; removing or replacing the linker with an unrelated sequence completely abolished its transcription activity (Fig. 2d). It is worth noting that the B-reader loop of TFIIB reaches into the active site cleft of yeast pol II in a similar way to σ3.246. Considering that this general mode of interaction appears to be conserved between prokaryotic RNAP and eukaryotic pol II, it is reasonable to propose that the σ2/σ4 linker of other bacterial ECF σ factors very likely also inserts into the active site cleft of RNAP. Our transcription assays show that chimeric ECF σ factors with swapped σ3.2 domains retain function in transcription, albeit with reduced activity (Fig. 2d and Supplementary Fig. 4e), supporting this idea. An intriguing question to be answered is how RNAP uses the same channels to accommodate different σ3.2 domains.
σA-RPo crystal structures show that σA3.2 contacts nucleotides at the template strand of ssDNA and pre-organizes it into an A-form helical conformation in a manner compatible with pairing of initial nucleotide triphosphates (NTPs)24,26. These interactions provide explanations for the effects of σA3.2 on de novo RNA synthesis26,47,48. The structure predicts that the σA3.2 finger has to be displaced by an RNA molecule of >4-nt in length and that the σA3.2 loop in the RNA exit channel has to be cleared during the promoter escape process. The interactions observed in the σA-RPo structure underscore the key role of the σA3.2 on abortive production, pausing, and promoter escape in transcription initiation28,29,49,50. In our crystal structures of σH-RPo, we show that σH3.2 guides the template ssDNA into the active site cleft and forms interactions with template ssDNA (Figs. 3d and 5e). We propose that σH3.2 probably functions similarly to σA3.2 during transcription initiation: by stabilizing the template ssDNA and facilitating binding of initial NTPs. The crystal structure of σH-RPo also indicates that σH3.2 should collide with RNA molecules >7 nt in length and that the σH3.2 domain must dissociate from the RNAP RNA exit channel during promoter escape (Fig. 5e), raising the possibility that the σ3.2 of the ECF factors functions like σA3.2 during abortive production, pausing, and promoter escape in transcription initiation.
Our structure of σH-RPo suggests that substantial differences exist between how individual domains σH-RNAP and σA-RNAP interact with their cognate promoter DNA. Both σH4 and σA4 use the same α-helix to bind the −35 element, but the positions of DNA on the α-helix differ by one α-helical turn, resulting a ~ 4 Å difference in the position of the −35 element on the σ4 surface (Supplementary Fig. 6B). A previous crystal structure of the E. coli σE4/−35 element binary complex is superimposable on our σH-RPo (Supplementary Fig. 6b)33, suggesting that the distinct mode of interaction that we observed with the −35 element is likely used by other ECF σ factors.
σH-RNAP “reads” the sequence of the −10 element differently than does σA-RNAP. In our crystal structure of σH-RPo, we discovered that base moieties of three nucleotides—T−10(nt), T−9(nt), and G−5(nt) (corresponding to the positions (−12), (−11) and (−7) of σA-regulated promoters)—were flipped out and inserted into three respective protein pockets on σH (Figs. 3d and 5a–c), in contrast to the two protein pockets known for base moieties of A(−11)(nt) and T(−7)(nt) on σA3,24. This extra pocket for T−10(nt) on σH was also suggested in a previous structure of a E. coli σE2/−10 element binary complex (Supplementary Fig. 6g)34. Sequence alignment of multiple ECF σ factors and σA revealed that residues forming the T−10(nt) pocket are generally conserved between ECF σ factors but are distinct from σA, suggesting that other ECF σ factors likely also recognize the nucleotide at this position using similar protein pockets (Supplementary Fig. 3).
σH-RNAP uses different protein regions to accommodate the flipped guanine base of G−5(nt) (corresponding to position (−7) of σA-regulated promoters) than does σA-RNAP for T(−7)(nt) (Supplementary Fig. 6i–k). The guanine base of G-5(nt) is sandwiched between σH2 and the RNAP-β gate loop, while the thymine base of T(−7)(nt) resides in a pocket on σA1.23,24. Our mutation study of the G−5(nt) pocket residues demonstrated that the RNAP-β gate loop functions to recognize this particular nucleotide (Fig. 5f), thus raising the possibility that other σECF-RNAP holoenzymes may also bind and read a nucleotide in the nontemplate ssDNA in a manner analogous to σH-RNAP.
σH-RNAP also engages the −10 element differently than does σA-RNAP. We found that the protein pockets for T−9(nt) and G−5(nt) on σH -RNAP do not exist in the absence of promoter DNA (Fig. 6a, b). In the crystal structure of σH-RNAP, the specificity loop, which recognizes the T−9(nt) is disordered; and the RNAP-β gate loop is too far away from the σH2 to form the G−5(nt) pocket (Fig. 6a, b). Such conformational differences support an “induced-fit” model of interaction between σH-RNAP and nontemplate ssDNA, in contrast to the accepted “lock-and-key model” for the interaction between σA-RNAP and nontemplate ssDNA (Fig. 6c, d)3,24,51.

The induced-fit mechanism of promoter recognition by Mtb σH-RNAP. a The T−9(nt) pocket does not exist in σH-RNAP holoenzyme (left) but is induced by DNA binding in σH-RPo (right) structures. b The G−5(nt) pocket does not exist in σH-RNAP holoenzyme (left) but is induced by DNA binding in σH-RPo (right) structures. The pockets are presented in cartoon (top) and surface (bottom). c The schematic of induce-fit model of promoter recognition by σH-RNAP. d The schematic of lock-and-key model of promoter recognition by σA-RNAP
σH-RNAP recognizes the G+2(nt) of CRE in a same way as does σA-RNAP (Supplementary Fig. 7d–f). As the residues that form the “G” pocket are solely from the RNAP core enzyme, it is possible that other ECF σ-RNAP holoenzymes are probably able to read the sequence identity of nucleotide at position +2 of the promoter DNA. However, whether the sequence content at this position affects other events (transcription start site selection, slippage synthesis, etc.) during transcription initiation by ECF σ-RNAP as σA-RNAP remains to be determined52.
Our crystal structures suggest that σH employs a distinct mechanism to unwind promoter DNA compared to σA (Supplementary Figure 6C-E): (1) σH and σA use residues with positions that differ by one α-helical turn on the σ2.3 α-helix (N88 for Mtb σH vs. W433/W434 for Ec σA, or W256/W257 for Taq σA) to unwind promoter DNA; (2) σH and σA unwind promoter DNA at positions differing by one base pair ((−13)/(−12) junction for σH vs. −(12)/(−11) junction for σA); and (3) σH traps and reads two unwound nucleotides (T(−12)(nt) and T(−11)(nt) immediately after the unwinding points), whereas σA only traps and reads one unwound nucleotide (A(−11)(nt)). Although it is unclear whether trapping of the flipped nucleotides initiates or facilitates the event of promoter unwinding, such interactions play crucial roles during RPo formation.
Campagne et al. recently identified a similar protein pocket on E. coli σE2 for the T(−12)(nt) in a crystal structure of E. coli σE bound to the −10 element ssDNA, and predicted that E. coli σE unwinds promoter dsDNA at the (−13)/(−12) junction34. Our crystal structure of σH-RPo clearly confirms the unwinding position proposed in the study by Campagne et al.. Sequence alignment of multiple ECF σ factors and σA revealed that most of the ECF σ factors do not contain the tryptophan dyad of σA at corresponding positions, but instead share a conserved (−12) pocket (Supplementary Figure 3). Therefore, it is possible that the ECF σ factors share the same unwinding mechanism as Mtb σH and Ec σE.
Our mutation study of the σH-regulated promoter showed that substitution of the consensus sequence at almost every position on the −35 element and −10 element abolished transcription activity (Supplementary Figure 5B). Moreover, extending or shortening the spacer of −35/−10 elements substantially reduced promoter activity (Fig. 2b). These results confirmed previously reported observations that ECF σ factors require a consensus sequence at the −35/−10 elements as well as a rigid spacer on promoter DNA to efficiently initiate transcription8,32. Our structures and results from biochemical experiments provide explanations for the promoter stringency of σH. We show that the interactions among σH and RNAP, the unwinding mechanism, and the induced-fit mode of promoter recognition work in concert to collectively confer the high specificity exhibited by σH and probably by other ECF σ factors as well.
We have shown that σH employs residues different from σA to unwind promoter DNA. The well-conserved tryptophan dyad of σA functions very efficiently for promoter unwinding; substitutions of the tryptophan dyad in σA resulted in severely reduced transcription activity43,44; and sequence variations at corresponding positions account for inferior DNA unwinding capacity of other alternative σ factors25. Given that ECF σ factors lack the tryptophan dyad at corresponding positions, we infer σH (and probably other ECF σ factors) unwinds promoter DNA less efficiently than does σA. This putative sub-optimal unwinding efficiency could be compensated by employing a very-high-affinity consensus sequence of promoter DNA to facilitate its loading4,8,25. Our proposed induced-fit mode of nontemplate ssDNA binding by σH-RNAP at the position immediate downstream of unwinding—i.e. T−9(nt)—also require the consensus promoter sequence to induce formation of correct conformation of the “specificity loop” (Fig. 6c); RNAP is not able to efficiently propagate promoter unwinding downstream without firmly anchoring the “master” nucleotide—A−11(nt) for E. coli σ70 corresponding to T−9(nt) for Mtb σH—as demonstrated in the case of E. coli σ70-RNAP53,54,55.
In conclusion, we demonstrate the structural basis of RNAP holoenzyme formation and transcription initiation by the ECF σ factors, thereby deepening our understanding the basic mechanisms of transcription initiation used by the largest and most diverse group of bacterial initiation factors. Our work will facilitate the rational design of orthogonal transcription units based on ECF σ factors and should help computational chemistry and other efforts to design selective antibacterial agents through the inhibition ECF σ factor-mediated transcription initiation.
Methods
Plasmid construction
The plasmids used in this study are listed in Supplementary 1. For construction of the expression plasmid pTolo-EX5-MtbσH, the M. tuberculosis σH gene amplified from M. tubercolusis genomic DNA (see Supplementary Data 1 for primer information) was cloned into the pTolo-EX5 plasmid (Tolo Biotech.) using NcoI and XhoI restriction sites. The pTolo-EX5-MtbσH derivatives bearing single or double mutations were generated through site-directed mutagenesis (Transgen biotech).
The pTolo-EX5-MtbσH derivatives encoding chimeric σH were generated by replacing the DNA fragment encoding Mtb σH3.2 (aa 96–144) with DNA fragments encoding Ec σA (aa 164–212; disordered acidic loop of the non-conserved region), Mtb σE3.2 (aa 150–189), Mtb σL3.2 (aa 78–122), or Mtb σM3.2 (aa 98–137) in pTolo-EX5-MtbσH (Tolo Biotech).
The pACYCDuet-Mtb-rpoA-rpoZ plasmid was constructed by replacing Mtb rpoD with Mtb rpoZ in parent plasmid pACYCDuet-Mtb-rpoA-sigA plasmid using KpnI and NdeI (Supplementary Table 1). The pETduet-Mtb-rpoB-rpoC derivatives bearing single mutations were generated through site-directed mutagenesis (Transgen Biotech; Supplementary Table 1 and Supplementary Data 1).
For construction of plasmids for in vitro transcription assays of Mtb σH, the promoter region (−50 to +51) of ClpB gene amplified from M. tuberculosis genomic DNA was cloned into pEASY-Blunt simple vectors, resulting in pEASY-Blunt-pClpB (Transgen Biotech; Supplementary Table 1 and Supplementary Data 1). The derivatives of pEASY-Blunt-pClpB with varied −35/−10 spacer lengths were obtained by site-directed mutagenesis (Supplementary Figure 2J). The promoter region (−50 to +51) of Rv2466c gene amplified from M. tuberculosis genomic DNA was cloned into pEASY-Blunt simple vectors, resulting in pEASY-Blunt-pRv2466c (Supplementary Table 1 and Supplementary Data 1; Supplementary Figure 2L).
The derivatives of pARTaq-N25–100-TR2 for in vitro transcription assays of Mtb σA with varied −35/−10 spacer lengths were obtained by site-directed mutagenesis (Supplementary Figure 2K).
Protein preparation
For preparation of M. tuberculosis σH, E. coli BL21(DE3) cells (NovoProtein) carrying pTolo-EX5-MtbσH were cultured in Luria-Bertani broth (LB) at 37 °C, and the expression of N-terminal sumo-tagged Mtb σH was induced at 18 °C for 14 h with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at OD600 of 0.8. Cells were harvested by centrifugation (8000 × g, 4 °C), re-suspended in lysis buffer (20 mM Tris-HCl (pH 8.0), 0.5 M NaCl, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, and protease inhibitor cocktail (bimake.cn)) and lysed using an Avestin EmulsiFlex-C3 cell disrupter (Avestin, Inc.). The lysate was centrifuged (16,000 × g; 45 min, 4 °C) and the supernatant was loaded on to a 2 mL column packed with Ni-NTA agarose (SMART, Inc.). The protein was washed by lysis buffer containing 20 mM imidazole and eluted with lysis buffer containing 250 mM imidazole. The eluted fraction was digested by tobacco etch virus protease and dialyzed overnight in dialysis buffer (20 mM Tris-HCl (pH 8.0), 0.2 M NaCl, 1% (v/v) glycerol, and 0.5 mM β-mercaptoethanol). The sample was loaded onto a second Ni-NTA column and the cleaved protein was retrieved from the flow-through fraction. The sample was diluted to the dialysis buffer with 0.05 M NaCl and further purified through a Heparin column (HiTrap Heparin HP 5 mL column, GE Healthcare Life Sciences) with buffer A (20 mM Tris-HCl (pH 8.0), 0.05 M NaCl, 1% (v/v) glycerol, and 1 mM dithiothreitol (DTT)) and buffer B (20 mM Tris-HCl (pH 8.0), 1 M NaCl, 1% (v/v) glycerol, and 1 mM DTT). Fractions containing M. tuberculosis σH was concentrated to 5 mg/mL and stored at −80 °C. The M. tuberculosis σH derivatives were prepared by the same procedure.
For preparation of selenomethionines (SeMet)-labeled M. tuberculosis σH, BL21 (DE3) strains carrying pTolo-EX5-MtbσH were cultured in SelenoMet base medium supplemented with nutrient mix (Molecular Dimensions) at 37 °C. The amino-acid mixture containing selemethionine was added into the culture at OD600 of 0.4 and the protein expression was induced with 0.5 mM IPTG at OD600 of 0.8 for 14 h at 18 °C. The SeMet-labeled Mtb σH was purified as described above.
The M. tuberculosis RNAP core enzyme was expressed and purified from E. coli BL21(DE3) carrying pETDuet-Mtb-rpoA-rpoZ and pACYCDuet-Mtb-rpoB-rpoC as described56. The protein sample was concentrated to 5 mg/mL and stored at −80 °C.
Nucleic acid scaffolds
Nucleic acid scaffolds for assembly of σH-RPo* for crystallization of σH-RNAP holoenzyme was prepared from synthetic oligos (nontemplate DNA: 5′-GTTGTGCTGGGCGTCACGGATGCA-3′; template DNA: 5′-TGCATCCGTGAGTCGGT-3′, Sangon Biotech, Supplementary Figure 1A) by an annealing procedure (95 °C, 5 min followed by 2 °C-step cooling to 25 °C) in annealing buffer (5 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 10 mM MgCl2).
Nucleic acid scaffolds for crystallization of σH-RPo was prepared from synthetic oligos (nontemplate DNA: 5′-CGGAACAGTTGCGACTTAGACGTGGTTGTGGGAGCTGCTATACTCTCC-3′; template DNA: 5′-GGAGAGTATAGGTCGAGGGTGTACCACGTCTAAGTCGCAACTGTTCC-3′, Sangon Biotech; and RNA: 5′-CCCUCGA-3′, Genepharma; Fig. 3c) by an annealing procedure (95 °C, 5 min followed by 2 °C-step cooling to 25 °C) in annealing buffer (5 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 10 mM MgCl2).
M. tuberculosis σH-RPo complex reconstitution
The M. tuberculosis σH-RPo and σH-RPo* were reconstituted from M. tuberculosis RNAP core enzyme, σH (or SeMet-σH), and nucleic acid scaffolds. The RNAP core enzyme, σH, and nucleic acid scaffolds were mixed at a 1:4:1.2 molar ratio and incubate at 4 °C overnight. The mixture was loaded on a HiLoad 16/60 Superdex S200 column (GE Healthcare, Inc.) equilibrated in 20 mM Tris-HCl (pH 8.0), 0.1 M NaCl, 1%(v/v) glycerol, and 1 mM DTT. Fractions containing Mtb σH-RPo were collected, concentrated to 7.5 mg/mL, and stored at −80 °C.
Structure determination of M. tuberculosis σH-RNAP holoenzyme
The structure of σH-RNAP holoenzyme was obtained during an attempt for obtaining the σH-RPo* with the fork transcription bubble DNA scaffold (no RNA oligo in the scaffold). The initial screen was performed by a sitting-drop vapor diffusion technique. Crystals grown from optimized reservoir solution A (1 μL 0.2 M NaAc, 0.1 M sodium citrate (pH 5.5), and 10% PEG4000 mixed with 1 μL 7.5 mg/mL protein complex) for 3 days at 22 °C were harvested for X-ray diffraction data collection. Crystals were soaked in stepwise fashion to reservoir solution A containing 18%(v/v) (2R, 3R)-(−)-2,3-butanediol (Sigma-Aldrich) and cooled in liquid nitrogen. The crystals of σH-RNAP derivative containing SeMet-labeled σH were obtained by analogous procedure.
Data were collected at Shanghai Synchrotron Radiation Facility (SSRF) beamlines 17U and 19U1, processed using HKL200057. The structure was solved by molecular replacement with Phaser MR58 using the structure of M. smegmatis core enzyme in a M. smegmatis transcription initiation complex (PDB: 5TW1)35 [https://www.rcsb.org/structure/5TW1] as the search model. Only one molecule of RNAP core enzyme was found in one asymmetric unit. The electron density maps show clear signal for σH. Cycles of iterative model building and refinement were performed in Coot59 and Phenix60. Residues of σH were built into the model at the last stage of refinement. No density of nucleic acid was observed in all stages of refinements, suggesting that the nucleic acids dissociated during crystallization resulting in a crystal of σH-RNAP holoenzyme. The final model of Mtb σH-RNAP holoenzyme was refined to Rwork and Rfree of 0.218 and 0.258, respectively. Analogous procedures were used to refine the structures of σH-RNAP holoenzyme with SeMet-labeled σH.
Structure determination of M. tuberculosis σH-RPo
The initial screen of σH-RPo was performed by a sitting-drop vapor diffusion technique. Crystals grown from reservoir solution B (1 μL 2% Tacsimate pH 5.0, 0.1 M Sodium citrate pH 5.6, 16 % PEG3350 mixed with 1 μL 7.5 mg/mL protein complex) for 15 days at 22 °C were harvested for X-ray diffraction data collection. Crystals were soaked in stepwise fashion to the reservoir solution B containing 18%(v/v) (2R, 3R)-(−)-2,3-butanediol (Sigma-Aldrich) and cooled in liquid nitrogen. Data were collected at SSRF beamlines 17U and 19U1, processed using HKL200057. The structure was solved by molecular replacement with Phaser MR58 using the structure of M. tuberculosis σH-RNAP as a search model. Only one molecule of σH-RNAP was found in one asymmetric unit. The electron density map showed clear signals for nucleotides in transcription bubble and downstream DNA duplex after initial rigid-body refinement, and clear signals for nucleotides in upstream DNA duplex after iterative cycles of model building and refinements in Coot59 and Phenix60. The nucleotides were built into the model at the last stage, and the final model of Mtb σH-RPo was refined to Rwork and Rfree of 0.220 and 0.255, respectively.
In vitro transcription assay
Transcription assays with M. tuberculosis RNAP σH-holoenzyme were performed as follows: reaction mixtures contained (20 μL): 80 nM M. tuberculosis RNAP core enzyme; 1 μM M. tuberculosis σH; 40 mM Tris-HCl, pH 7.9; 75 mM KCl; 5 mM MgCl2; 2.5 mM DTT; and 12.5% glycerol. Reaction mixtures were incubated for 10 min at 37 °C, and then supplemented with 2 μL promoter DNA (1 μM; amplified from pEASY-Blunt-pClpB; Supplementary Data 1), and further incubated for 10 min at 37 °C. The reaction was initiated by adding 0.7 μL NTP mixture (3 mM [α-32P]UTP (0.04 Bq/fmol), 3 mM ATP, 3 mM GTP, and 3 mM CTP), and RNA synthesis was allowed to proceed for 10 min at 37 °C. Reactions were terminated by adding 8 μL loading buffer (10 mM EDTA, 0.02% bromophenol blue, 0.02% xylene cyanol, and 98% formamide), boiled for 2 min, and stored in ice for 5 min. Reaction mixtures were applied to 15% urea-polyacrylamide slab gels (19:1 acrylamide/bisacrylamide), electrophoresed in 90 mM Tris-borate (pH 8.0) and 0.2 mM EDTA, and analyzed by storage-phosphor scanning (Typhoon; GE Healthcare, Inc.).
Transcription assays with M. tuberculosis RNAP σA-holoenzyme were performed essentially as above except that σA instead of σH were added and N25 promoter DNA were used (amplified from pARTaq-N25-100-TR2; Supplementary Table 1 and Supplementary Data 1).
Transcription assays using M. tuberculosis RNAP σH-holoenzyme and pRv2466c promoters were also performed essentially as above with subtle modifications. The reaction mixtures (20 μL) containing 160 nM M. tuberculosis RNAP core enzyme, 1 μM M. tuberculosis σH, 40 mM Tris-HCl, pH 7.9, 75 mM KCl, 5 mM MgCl2, 2.5 mM DTT, and 12.5% glycerol were incubated for 10 min at 37 °C, and then supplemented with 2 μL promoter DNA (1 μM, amplified from pEASY-pRv2466c; Supplementary Table 1 and Supplementary Data 1), and further incubated for 10 min at 37 °C. The reactions were initiated by adding 4 μL NTP mixture (0.1 mM ATP, 0.1 mM GTP, 0.1 mM CTP, and 7 μM [α-32P]UTP (5.6 Bq/fmol)) and were allowed to proceed for 20 min at 37 °C. The reactions were terminated and the transcripts were separated and visualized as above.
Quantification and statistical analysis
All biochemical assays were performed at least three times independently. Data were analyzed with SigmaPlot 10.0 (Systat Software Inc.).
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The accession numbers for the coordinates and structure factors for M. tuberculosis σH-RNAP and σH-RPo in this paper are PDB: 5ZX3 and 5ZX2, respectively. The source data underlying Figs. 2b, d, 4c, d, f, and 5f, and Supplementary Figs. 2i, 4e, and 5b are provided as a Source Data file. Other data are available from the corresponding author upon reasonable request.
References
Browning, D. F. & Busby, S. J. Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 14, 638–650 (2016).
Lee, D. J., Minchin, S. D. & Busby, S. J. Activating transcription in bacteria. Annu. Rev. Microbiol. 66, 125–152 (2012).
Feklistov, A. & Darst, S. A. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase sigma subunit. Cell 147, 1257–1269 (2011).
Feklistov, A., Sharon, B. D., Darst, S. A. & Gross, C. A. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu. Rev. Microbiol. 68, 357–376 (2014).
Ruff, E. F., Record, M. T. Jr. & Artsimovitch, I. Initial events in bacterial transcription initiation. Biomolecules 5, 1035–1062 (2015).
Saecker, R. M., Record, M. T. Jr. & Dehaseth, P. L. Mechanism of bacterial transcription initiation: RNA polymerase-promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 412, 754–771 (2011).
Paget, M. S. Bacterial sigma factors and anti-sigma factors: structure, function and distribution. Biomolecules 5, 1245–1265 (2015).
Gruber, T. M. & Gross, C. A. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57, 441–466 (2003).
Mascher, T. Signaling diversity and evolution of extracytoplasmic function (ECF) sigma factors. Curr. Opin. Microbiol. 16, 148–155 (2013).
Staron, A. et al. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol. Microbiol. 74, 557–581 (2009).
Helmann, J. D. Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Curr. Opin. Microbiol. 30, 122–132 (2016).
Fiebig, A., Herrou, J., Willett, J. & Crosson, S. General stress signaling in the alphaproteobacteria. Annu. Rev. Genet. 49, 603–625 (2015).
Woods, E. C. & McBride, S. M. Regulation of antimicrobial resistance by extracytoplasmic function (ECF) sigma factors. Microbes Infect. 19, 238–248 (2017).
Flentie, K., Garner, A. L. & Stallings, C. L. Mycobacterium tuberculosis transcription machinery: ready to respond to host attacks. J. Bacteriol. 198, 1360–1373 (2016).
Rodrigue, S., Provvedi, R., Jacques, P. E., Gaudreau, L. & Manganelli, R. The sigma factors of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 30, 926–941 (2006).
Sachdeva, P., Misra, R., Tyagi, A. K. & Singh, Y. The sigma factors of Mycobacterium tuberculosis: regulation of the regulators. FEBS J. 277, 605–626 (2010).
Shultzaberger, R. K., Chen, Z., Lewis, K. A. & Schneider, T. D. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res. 35, 771–788 (2007).
Yuzenkova, Y., Tadigotla, V. R., Severinov, K. & Zenkin, N. A new basal promoter element recognized by RNA polymerase core enzyme. EMBO J. 30, 3766–3775 (2011).
Barne, K. A., Bown, J. A., Busby, S. J. & Minchin, S. D. Region 2.5 of the Escherichia coli RNA polymerase sigma70 subunit is responsible for the recognition of the ‘extended-10’ motif at promoters. EMBO J. 16, 4034–4040 (1997).
Feklistov, A. et al. A basal promoter element recognized by free RNA polymerase sigma subunit determines promoter recognition by RNA polymerase holoenzyme. Mol. Cell 23, 97–107 (2006).
Campbell, E. A. et al. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol. Cell 9, 527–539 (2002).
Bae B., Feklistov A., Lass-Napiorkowska A., Landick R., Darst S. A. Structure of a bacterial RNA polymerase holoenzyme open promoter complex. ELife 4, e08504 (2015).
Feng, Y., Zhang, Y. & Ebright, R. H. Structural basis of transcription activation. Science 352, 1330–1333 (2016).
Zhang, Y. et al. Structural basis of transcription initiation. Science 338, 1076–1080 (2012).
Koo, B. M., Rhodius, V. A., Nonaka, G., deHaseth, P. L. & Gross, C. A. Reduced capacity of alternative sigmas to melt promoters ensures stringent promoter recognition. Genes Dev. 23, 2426–2436 (2009).
Basu, R. S. et al. Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J. Biol. Chem. 289, 24549–24559 (2014).
Zuo, Y. & Steitz, T. A. Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol. Cell 58, 534–540 (2015).
Duchi, D. et al. RNA polymerase pausing during initial transcription. Mol. Cell 63, 939–950 (2016).
Petushkov, I. et al. Interplay between sigma region 3.2 and secondary channel factors during promoter escape by bacterial RNA polymerase. Biochem. J. 474, 4053–4064 (2017).
Samanta, S. & Martin, C. T. Insights into the mechanism of initial transcription in Escherichia coli RNA polymerase. J. Biol. Chem. 288, 31993–32003 (2013).
Rhodius, V. A. et al. Design of orthogonal genetic switches based on a crosstalk map of sigmas, anti-sigmas, and promoters. Mol. Syst. Biol. 9, 702 (2013).
Campagne, S., Allain, F. H. & Vorholt, J. A. Extra cytoplasmic function sigma factors, recent structural insights into promoter recognition and regulation. Curr. Opin. Struct. Biol. 30, 71–78 (2015).
Lane, W. J. & Darst, S. A. The structural basis for promoter −35 element recognition by the group IV sigma factors. PLoS Biol. 4, e269 (2006).
Campagne, S., Marsh, M. E., Capitani, G., Vorholt, J. A. & Allain, F. H. Structural basis for −10 promoter element melting by environmentally induced sigma factors. Nat. Struct. Mol. Biol. 21, 269–276 (2014).
Hubin, E. A. et al. Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. ELife 6, e22520 (2017).
Lin, W. et al. Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol. Cell 66, 169–179 e168 (2017).
Degen, D. et al. Transcription inhibition by the depsipeptide antibiotic salinamide A. ELife 3, e02451 (2014).
Manganelli, R. et al. Role of the extracytoplasmic-function sigma factor sigma(H) in Mycobacterium tuberculosis global gene expression. Mol. Microbiol. 45, 365–374 (2002).
Chakraborty, A. et al. Opening and closing of the bacterial RNA polymerase clamp. Science 337, 591–595 (2012).
Raman, S. et al. The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J. Bacteriol. 183, 6119–6125 (2001).
Song, T., Song, S. E., Raman, S., Anaya, M. & Husson, R. N. Critical role of a single position in the −35 element for promoter recognition by Mycobacterium tuberculosis SigE and SigH. J. Bacteriol. 190, 2227–2230 (2008).
Kim, K. Y., Park, J. K. & Park, S. In Streptomyces coelicolor SigR, methionine at the −35 element interacting region 4 confers the −31’-adenine base selectivity. Biochem. Biophys. Res. Commun. 470, 257–262 (2016).
Juang, Y. L. & Helmann, J. D. A promoter melting region in the primary sigma factor of Bacillus subtilis. Identification of functionally important aromatic amino acids. J. Mol. Biol. 235, 1470–1488 (1994).
Fenton, M. S., Lee, S. J. & Gralla, J. D. Escherichia coli promoter opening and −10 recognition: mutational analysis of sigma70. EMBO J. 19, 1130–1137 (2000).
Campbell, E. A. et al. Crystal structure of Escherichia coli sigmaE with the cytoplasmic domain of its anti-sigma RseA. Mol. Cell 11, 1067–1078 (2003).
Sainsbury, S., Niesser, J. & Cramer, P. Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature 493, 437–440 (2013).
Murakami, K. S., Masuda, S. & Darst, S. A. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 A resolution. Science 296, 1280–1284 (2002).
Kulbachinskiy, A. & Mustaev, A. Region 3.2 of the sigma subunit contributes to the binding of the 3’-initiating nucleotide in the RNA polymerase active center and facilitates promoter clearance during initiation. J. Biol. Chem. 281, 18273–18276 (2006).
Pupov, D., Kuzin, I., Bass, I. & Kulbachinskiy, A. Distinct functions of the RNA polymerase sigma subunit region 3.2 in RNA priming and promoter escape. Nucleic Acids Res. 42, 4494–4504 (2014).
Lerner, E. et al. Backtracked and paused transcription initiation intermediate of Escherichia coli RNA polymerase. Proc. Natl Acad. Sci. USA 113, E6562–E6571 (2016).
Liu, X., Bushnell, D. A. & Kornberg, R. D. Lock and key to transcription: sigma-DNA interaction. Cell 147, 1218–1219 (2011).
Vvedenskaya, I. O. et al. Massively systematic transcript end readout, “MASTER”: transcription start site selection, transcriptional slippage, and transcript yields. Mol. Cell 60, 953–965 (2015).
Heyduk, E., Kuznedelov, K., Severinov, K. & Heyduk, T. A consensus adenine at position -11 of the nontemplate strand of bacterial promoter is important for nucleation of promoter melting. J. Biol. Chem. 281, 12362–12369 (2006).
Lim, H. M., Lee, H. J., Roy, S. & Adhya, S. A. “master” in base unpairing during isomerization of a promoter upon RNA polymerase binding. Proc. Natl Acad. Sci. USA 98, 14849–14852 (2001).
Tomsic, M. et al. Different roles for basic and aromatic amino acids in conserved region 2 of Escherichia coli sigma(70) in the nucleation and maintenance of the single-stranded DNA bubble in open RNA polymerase-promoter complexes. J. Biol. Chem. 276, 31891–31896 (2001).
Banerjee, R., Rudra, P., Prajapati, R. K., Sengupta, S. & Mukhopadhyay, J. Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis (Edinb.) 94, 397–404 (2014).
Otwinowski, Z. & Minor, W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Acknowledgements
Work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29020000), the National Natural Science Foundation of China (31670067 and 31822001), and the Leading Science Key Research Program of the Chinese Academy of Sciences (QYZDB-SSW-SMC005). We thank Prof. Richard Ebright for generous gifts of pACYCDuet-Mtb-rpoA-rpoD, pETDuet-Mtb-rpoB-rpoC, and pARTaq-N25–100-TR2 constructs; Prof. Xiaoming Zhang for generous gift of M. tuberculosis genomic DNA; and Tolo Biotechnology for generous gift of pTolo-EX vectors. We thank the staff at beamline BL18U1/BL19U1 of National Center for Protein Science Shanghai (NCPSS), and at beamline BL17U1 of Shanghai Synchrotron Radiation Facility for assistance during data collection.
Author information
Authors and Affiliations
Contributions
L.L. solved the structures. C.F. performed in vitro transcription assays. N.Z. assisted in structure determination. T.W. prepared pACYCDuet-Mtb-rpoA-rpoZ and purified proteins. Y.Z. designed experiments, analyzed data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal peer review information: Nature Communications thanks Xiaodong Zhang and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Fang, C., Zhuang, N. et al. Structural basis for transcription initiation by bacterial ECF σ factors. Nat Commun 10, 1153 (2019). https://doi.org/10.1038/s41467-019-09096-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-09096-y
This article is cited by
-
Antiviral signalling by a cyclic nucleotide activated CRISPR protease
Nature (2023)
-
Structure of the transcription open complex of distinct σI factors
Nature Communications (2023)
-
Structural basis of template strand deoxyuridine promoter recognition by a viral RNA polymerase
Nature Communications (2022)
-
Structural basis of ribosomal RNA transcription regulation
Nature Communications (2021)
-
Transcriptional regulation by σ factor phosphorylation in bacteria
Nature Microbiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
