
Abstract
Currently, only a few 18F-radiolabeling methods were conducted in aqueous media, with non-macroelement fluoride acceptors and stringent conditions required. Herein, we describe a one-step non-solvent-biased, room-temperature-driven 18F-radiolabeling methodology based on organophosphine fluoride acceptors. The high water tolerance for this isotope-exchange-based 18F-labeling method is attributed to the kinetic and thermodynamic preference of F/F over the OH/F substitution based on computational calculations and experimental validation. Compact [18/19F]di-tert-butyl-organofluorophosphine and its derivatives used as 18F-labeling synthons exhibit excellent stability in vivo. The synthons are further conjugated to several biomolecular ligands such as c(RGDyk) and human serum albumin. The one-step labeled biomolecular tracers demonstrate intrinsic target imaging ability and negligible defluorination in vivo. The current method thus offers a facile and efficient 18F-radiolabeling pathway, enabling further widespread application of 18F.
Similar content being viewed by others
Introduction
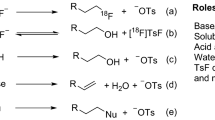
Positron emission tomography (PET) is a highly sensitive, quantitative, noninvasive imaging technique, and therefore is one of the most popular imaging modalities for disease diagnosis and monitoring of therapeutic effects1. The 18F-radionuclide contributes greatly to the success of this imaging method and is often preferred for PET imaging due to its wide availability, favorable half-life (109.8 min), high isotopic purity, and low positron energy (640 keV), allowing sequential acquisition of PET images with a relatively high resolution2. However, to synthesize 18F-labeled PET tracers, traditional labeling methods are mostly based on carbon-fluorine bond formation, which usually demands multiple steps and/or harsh reaction conditions such as heating at high temperatures in organic media3. Moreover, most 18F-radiolabeling reactions require strict anhydrous conditions due to the limited reactivity of the fluoride anion as a nucleophile in aqueous solution4 (Fig. 1a). Feasible and efficient 18F-labeling methods in physiological media remain highly desirable for the introduction of 18F into useful biomolecules, such as water-soluble peptides and antibodies that bind to overexpressed receptors in various inflammatory tissues and tumors2.

Overview of 18F-labeling methods for solvent/heat-sensitive biomolecules. a Commonly used multistep synthetic route beginning with drying of aqueous [18F]F−. b One-step 18F-labeling of biomolecules via an organophosphine fluoride acceptor that allows for efficient labeling with good RCYs (>50%) under mild conditions within 5–15 min
A few studies5,6,7,8 reported that fluorinase enzyme can catalyze the incorporation of 18F into PET radiotracers under aqueous conditions. However, the high substrate specificity severely limits the scope of application for this enzyme method7. Several direct aqueous 18F-labeling approaches have been reported, using organofluorosilicons2, 9,10,11,12,13,14,15, aluminum complexes16,17,18,19 or trifluoroborates20,21,22,23,24,25,26,27,28 as non-carbon fluoride acceptors to afford one-step labeling of peptides. These non-macroelement fluoride acceptor-based methods exploit their Lewis acid characteristics. Lewis acid promoters, such as SnCl4 and AlCl3, are sometimes required to activate the fluorine-element bond to facilitate [18F]F¯ incorporation in these systems29. The high bond enthalpy and low activation energy for the binding of Si, Al, and B with F− lead to the formation of stable bonds with 18/19F− instead of being attacked by and bonded with OH−30.
However, these methods still pose obvious drawbacks: (i) limited stability and high lipophilicity of the organosilicon-based 18F-labeling blocks; (ii) specific pH requirement for trifluoroborate-based 18F-labeling; (iii) steric effect of bulky Al-18F-based chelate synthons; and iv) potential biosafety issue due to possible metal contamination in the final product. Recently, a bulky [GaF3(BnMe2-tacn)], another metal chelate system, was labeled in aqueous solutions by 18F/19F isotopic exchange but showing slight defluorination of the 18F-labeled product in plasma29. Collectively, non-solvent-biased 18F-labeling methods with macroelement-composed fluoride acceptors remain unexplored.
The phosphorus (P) element is essential for life and phosphines are components of deoxyribonucleic acid, ribonucleic acid, adenosine triphosphate, and phospholipids31. The formation heat of phosphine fluoride radicals is −12.5 ± 5 kcal mol−1 while those of silicon fluoride, boron fluoride, and fluorocarbon radicals are −4.8 ± 3, −27.7 ± 3.3, and 61.0 ± 2 kcal mol−1, respectively32. Therefore, phosphorus, boron, and silicon readily form more stable products with fluorine (compared to fluorocarbon). Phosphorus is strongly fluorophilic based on previous reports33, 34. For example, the affinity of F-P in PF5 (380 kJ mol−1) is even higher than that in F-B in BF3 (346 kJ mol−1). Preparation of compounds containing the P-18F bond was demonstrated by 18F-labeling of phospho-pesticide35 and NCH-PF533 in organic solvents under heating condition.
Here we rationally design and develop organophosphine precursors that allow for easy synthesis and efficient radiolabeling with high radiochemical yields (RCYs) and molar activities under mild conditions. In this way, 18F-labeled biomolecules are successfully obtained in either organic solvents or aqueous media with high RCY up to 100% within 15 min, as shown in Fig. 1b. Proof-of-concept of 18F-labeling with heat/solvent-sensitive biomolecules in one step is demonstrated by biomolecular tracers include 18F-labeled human serum albumin (HSA) for blood pool imaging and 18F-labeled c(RGDyk) which targets the integrin αvβ3 receptor of the U87MG cell (glioblastoma). Thus, the method reported here permits non-specialist-manageable rapid 18F-labeling of extensive biomolecules and their use in PET imaging.
Results
Mechanism of rapid H2O-resistant 18F/19F exchange
We first performed computational density functional theory (DFT) calculations to simulate the F/F isotope exchange processes on the organophosphine fluoride acceptors with substitutions of varying steric hindrance. Using the B3LYP method36,37,38,39,40, two-step addition-elimination pathways for both F/F isotope exchange and the competing OH/F substitution (i.e., hydrolysis) were identified for five representative organophosphine fluorides (1–5). Detailed reaction mechanisms and free energy profiles are shown in Fig. 2. Such a mechanistic path suggests an inversion of the tetravalent phosphorus after a single F/F exchange. Hence, the overall reactions are not strictly free energy neutral but negligible. The OH/F exchange processes share similar reaction pathways with the exception that [F…H2O]− binds with the phosphorus center using its oxygen atom in the first addition step.

Mechanism of H2O-resistant 18F/19F exchange interpreted by free energy parameters. Geometries are optimized at the B3LYP/6-31+G* level of theory, and single point calculations are performed at the CAM-B3LYP/6-311++G** level of theory. Two substituent groups on the phosphorus center are simplified as two large spheres in the molecular structures for clarity. a Reaction pathways and free energy (kcal mol−1) profiles for the F/F isotope exchange process of five selected reactant systems. b Reaction pathways and free energy (kcal mol−1) profiles for the OH/F exchange process of the same five selected reactant systems
The calculated free energy profiles show that the pentacoordinate intermediates are unstable compared to the initial materials for both F/F isotope exchange and OH/F substitution. A comparison of the relative stabilities of TS1-X and TS2-X (X = 1–5) to those of TSO1-X and TSO2-X shows that the free energy barriers for the F/F exchange processes were distinctly lower than the corresponding values for the OH/F substitution. The differences in relative free energy values between TS1-X and TSO1-X for the 1–5 reaction systems were −3.6, −4.1, −5.1, −7.2, and −4.7 kcal mol−1, respectively, while those between TS2-X and TSO2-X for the five reaction systems were −5.9, −7.8, −7.1, −8.1, and −4.4 kcal mol−1, respectively. These results strongly indicate that the F/F isotope exchange is kinetically more favorable than the corresponding OH/F substitution for all of the reactant systems. According to the calculated relative free energies of the transition states, the predicted rates for F/F isotope exchange follow the order of 1 ≈ 2 > 5 > 4 > 3. In addition, the OH/F substitution processes exhibit endothermic free energy values (e.g., ∆G = 2.3–5.1 kcal mol−1), suggesting that these processes are neither spontaneous nor thermodynamically favorable. Overall, the DFT calculations demonstrate that the 18F/19F isotope exchange occurs automatically rather than the competing OH/F substitution in terms of both kinetics and thermodynamics (Table 1).
Synthesis of organofluorophosphine fluoride acceptors
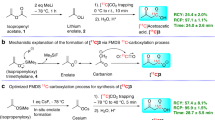
Next, the designed organofluorophosphine fluoride acceptors were prepared via oxidative coupling between various hydrophosphine oxide and F−. Specifically, dibutylphosphinic fluoride (1), diphenylphosphinic fluoride (2), tert-butyl(phenyl)phosphinic fluoride (3) and benzyl 2-(tert-butylfluorophosphoryl)-2-methylpropanoate (4) were synthesized from phosphine oxides using the Reformatsky-type reaction41, followed by copper-promoted fluorination of phosphine42. Compound 1 was found to be difficult to obtain and only 19F and 31P NMR spectra were obtained because of its volatility and low yield.
Radiochemistry
For radiolabeling tests, [18F]F− solution unloaded from a cyclotron target was diluted and added to reaction vials pre-charged with the organofluorophosphine fluoride acceptors. Rapid 18F-labeling was achieved in high RCYs by 18F/19F isotope exchange in aqueous or organic solvents without heating. The RCYs were further measured by changing H2O ratio from 0 to 95% at graded precursor concentrations and reaction temperatures. For the first try, one-step 18F-labeling in >95% aqueous solutions was achieved with >50% RCY under mild conditions31. As the precursor concentrations and reaction temperatures increased, the RCYs improved significantly. Under the same labeling conditions, [18F]3 and [18F]4 were more rapidly labeled with higher yields than [18F]2. [18F]3 and [18F]4 achieved a 100% labeling yield in dimethyl sulfoxide (DMSO) at 75 °C, as shown in Table 2. The molar activity of manually labeled [18F]4 was measured to be 0.01–0.05 Ci μmol−1 (depending on the amount of starting radioactivity, decay-uncorrected) at the end of synthesis. Detailed radiolabeling procedures and calculations are described in the supporting information.
Stability of 18F-labeled organofluorophosphine
In vitro stabilities of [18F]2, [18F]3, and [18F]4 in human serum, saline, and ethanol, respectively, are shown in Supplementary Figure 5. Among these 18F-labeled fluorophosphines, [18F]4 exhibited the highest in vivo stability (Table 1). During a metabolic stability examination, [18F]4 remained 100% intact at 2 h post-injection and was more stable than [18F]3, which is in good agreement with the predictions from the free energy barrier calculations (Table 1). MicroPET/CT imaging with [18F]4 in healthy ICR mice was then performed to investigate the in vivo metabolic process. No increase in bone uptake was observed up to 120 min post-injection. The radioactivity was primarily concentrated in the bladder and gallbladder over time (see Supplementary Figure 7). 2,3,5,6-Tetrafluorophenyl 2-(tert-butylfluorophosphoryl)-2-methylpropanoate (DBPOF-COOC6HF4, 5), a derivative with an activated ester, was synthesized and conjugated to several peptide ligands of clinical interest43, 44 to obtain one-step-labeling precursors.
Radiosynthesis of 18F-DBPOF-c(RGDyk) and PET imaging
Using a manual labeling procedure, 18F-DBPOF-c(RGDyk) as a peptide ligand was prepared at an average RCY (the amount of activity in the final product expressed as the percentage of starting activity) of >30% in 25 min with >98% purity after purification with a C-18 solid-phase extraction cartridge. In order to obtain cGMP compliant 18F-DBPOF-c(RGDyk), we established an automated procedure using a commercial multifunction radiosynthesis module PET-MF-2V-IT-I (Beijing PET Technology, China). The schematic layout of the automated synthesis module is shown in Supplementary Figure 4.
Quality control analysis protocols were established according to the United States Pharmacopeia guidelines for PET radiopharmaceuticals. Average RCY (the amount of activity in the final product expressed as the percentage of starting activity) was 15–25% for the automated procedure. The radiochemical purity of 18F-DBPOF-c(RGDyk) was confirmed to be >95% by radio-HPLC analysis. The molar activity was measured to be 0.06–0.13 Ci μmol−1 for the product prepared using the automated module, compared to 0.006–0.01 Ci μmol−1 for the manually prepared product. MicroPET/CT imaging data, shown in Fig. 3a, indicated an uptake of 18F-DBPOF-c(RGDyk) with standardized uptake value (SUV) of 0.76 in U87MG gliomablastoma, and a target to non-target ratio of 2.4. No obvious signal was detected in the tumor as illustrated in the inset image of Fig. 3a, demonstrating that the specific uptake was blocked by DBPOF-c(RGDyk).

MicroPET/CT imaging with 18F-DBPOF-c(RGDyk) and 18F-DBPOF-HSA. a A PET image from a dynamic scan with 18F-DBPOF-c(RGDyk) in U87MG nude mice bearing gliomablastoma, reconstructed at 23 min post-injection with the tumor (T), live (L), and kidney (K) indicated by white arrows. The inset image in the right corner showed no specific tumor uptake indicated by a white arrow. b Time-activity curves of 18F-DBPOF-c(RGDyk) in the tumor, bone, blood, kidney and muscle. Uptake values are presented by means ± standard deviations (n = 3). c A PET image from a 60 min dynamic scan with 18F-DBPOF-HAS in healthy female Wistar rats, reconstructed at 60 min post-injection with the heart (H) and artery (A) indicated by white arrows. d Time-activity curves of 18F-DBPOF-HSA in the heart, liver, bone and muscle. Uptake values are presented by means ± standard deviations (n = 3). Source data are provided as a Source Data file
Radiosynthesis of 18F-DBPOF-HAS and PET imaging
Human serum albumin (HSA, 66.5 kDa) is a heat-sensitive globular protein that exhibits high solubility and stability with a plasma half-life of 16–18 h45. DBPOF-HSA was prepared by simple conjugation of DBPOF-COOC6HF4 to HAS within 30 min. By manual 18F-labeling, the RCY (the amount of activity in the final product expressed as the percentage of starting activity) of 18F-DBPOF-HSA was >5% with molar activity of ~0.03 Ci μmol−1 in shorter synthesis time than other radiofluorination strategies.
PET imaging of healthy female Wistar rats was conducted 1 h after intravenous injection of 18F-DBPOF-HSA. A whole-body PET/CT of a healthy rat 1 h after 18F-DBPOF-HSA injection through tail vein is demonstrated in Fig. 3c. The heart ventricles the and the peripheral blood vessels are clearly revealed. Other organs can be also observed but with lower activity concentrations than that in the central vessels. Analysis of 18F-DBPOF-HSA biodistribution in rats showed high retention in blood with SUVs of 2.4 ± 0.6, 1.4 ± 0.7, and 1.0 ± 0.1 at 0.2, 0.5, and 1 h, respectively. The tissue uptake from the microPET/CT experiments agreed well with the biodistribution results at matched time points. These results indicated that the introduction of DBPOF synthon into HSA and subsequent radiolabeling did not affect the structural or functional integrity of HSA.
Discussion
Biomolecules, such as proteins, carbohydrates, and nucleic acids, are excellent vectors for monitoring physiological and biochemical lesions via PET imaging due to their high affinity and high specificity for many pathogenic targets46 as well as superior biocompatibility. Most of these vectors are soluble and remain intact in water alone or mixed aqueous solvents rather than in organic solvents, while anionic [18F]F−, with its high solvation energy, becomes inactive in the same solvents. Consequently, the direct incorporation of 18F is challenging, especially in the case of temperature-, pH- and solvent-sensitive macromolecules. Fortunately, efficient aqueous labeling approaches are theoretically feasible by employing highly fluorophilic fluoride acceptors with very low activation energy and high hydrolysis energy for selective fluoro-substitution. Indeed, fluorophosphine fluoride acceptors have been shown to undergo rapid radiofluorination reaction in at least partially aqueous media10.
Theoretical DFT calculations confirmed that the F/F exchange of organophosphines is more favorable both kinetically and thermodynamically than the OH/F exchange, which is consistent with our experimental findings. The detailed reaction mechanisms and free energy variations were illustrated for the organophosphine labels with five representative reactants. In our calculations, the 1:1 hydrogen-bond complex of H2O with F− (F−·H2O) was modeled as a nucleophile to account for the solvation effect of water. Nucleophilic substitution at the tetravalent phosphorus center may occur via either a concerted pathway or a stepwise addition-elimination pathway. The latter method involves a pentavalent adduct intermediate owing to the addition of a nucleophile without the loss of a leaving group.
In addition to the spontaneity and aqueous tolerance of these radiolabeling reactions, the compact prothesis configuration and favorable in vivo behavior of the resulting PET tracers are also important. [18F]2 was found to be unstable at pH 7.4–7.6 in human serum, saline, and ethanol because the steric hindrance of benzene is relatively small. In contrast, [18F]3 and [18F]4 exhibited remarkable thermal and hydrolytic stability. The stabilities of 18F-labeled fluorodiphenylphosphines are consistent with the steric parameters of these compounds, i.e., greater steric hindrance leads to better stability. By virtue of their small size, zero net charge, and negligible decomposition, the physicochemical properties of the 18F-labeled tracers prepared via the organophosphine fluoride acceptors are similar to those of the unlabeled analogs.
Phosphorus compounds are used in many modern homogeneous catalyst systems because the P-X bonds are thermally robust in general and unlikely to break down by transition metals at the catalysis conditions47. Fluorophosphines with bulky or electron-withdrawing substituents, such as (C6F5)2PF, (CF3)2PF, and tBu2PF, are also found to be thermally stable48,49,50,51. Synthesis and screening of more fluorophosphine fluoride acceptors with various leaving groups are continuing in our laboratories.
In summary, we have developed a procedure for the radiolabeling of a series of organofluorophosphine biomolecules using 18F-fluoride. Low activation energy is required for the formation of these P-F bonds in comparison to P-OH bonds. As a result, 18F-labeling can be performed in aqueous media and high radiochemical yields achieved even at room temperature, and acceptable molar activities that are critical for targeted imaging52. This inexpensive, rapid, one-step 18F-radiolabeling procedure holds great promise for widespread applications in radiopharmaceutical industry to produce a variety of biomolecules as potential PET tracers.
Methods
Automated radiosynthesis of 18F-DBPOF-c(RGDyk)
No-carrier-added [18F]F− was obtained by bombardment of a H218O target with 11-MeV protons in a RDS 111 cyclotron (Siemens, Germany) and delivered to the PET-MF-2V-IT-I synthesis module. The aqueous [18F]F−solution (600–1200 mCi) was mixed with a solution of the unlabeled precursor DBPOF-c(RGDyk) (0.8–2.4 mg, 1–3 µmol) in ~0.5 mL of DMSO/H2O (1:1, v/v, pH = 7.4) in the reaction vial. The vial was kept at room temperature for 15 min without stirring. The reaction mixture was diluted with 10.0 mL of H2O and loaded onto a C18 light cartridge (Waters, USA). The cartridge was flushed twice with 10.0 mL of pure water each to remove the unreacted [18F]F−, then eluted with 1.0 mL ethanol to recover 90–150 mCi of radiochemically pure product collected in a glass vial. This solution was then formulated in 0.9% sodium chloride (9.0 mL) and filtered through a 0.22-µm sterile Millex® GV (vented filter, Millipore, Billerica, MA, USA) into a sterile dose vial (Mallinckrodt, Hazelwood, MO, USA) fitted with a vent needle (Sartorius Stedim Biotech GmbH, Göttingen, Germany). A small sample was removed for quality control analysis. The radiolabeling procedure was repeated at least three times. Detailed HPLC profiles and procedure for molar activity measurements are presented in the Supplementary Information.
Radiosynthesis of 18F-DBPOF-HAS
DBPOF-HAS (2–3 mg) was dissolved in 150 μL of 0.01 M phosphate buffered saline (pH = 7.36) in a glass vial. Aqueous [18F]F− solution (50 μL, 30–50 mCi) unloaded from the cyclotron target was added to the reaction vial. The vial was gently shaken at room temperature for 15 min. With the reaction impurities (i.e., free [18F]F−) removed by a radio-HPLC equipped with a Xtimate SEC-300 column (Welch, China), 1.5–2.8 mCi of the desired product was collected. A small sample was removed for quality control analysis. Radiochemical purity was measured by a radio-HPLC with a Xtimate SEC-300 column. The radiolabeling was repeated at least three times. Detailed analytical HPLC procedures are presented in the Supplementary Information.
MicroPET/CT imaging
All animal studies were performed under the animal use and care regulations approved by Center of Animal Care and Use Committee, Xiamen University. Dynamic microPET-CT scans were acquired by an Inveon microPET/CT (Siemens, Germany). Reconstruction of PET images was performed with 3D OPMAP algorithm.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information Files or from the corresponding author on reasonable request.
References
Wahl, R. L. & Buchanan, J. W. Principles and Practice of Positron Emission Tomography. (Lippincott Williams and Wilkins, Philadelphia, 2002).
Wängler, C. et al. Silicon-[18F]Fluorine radiochemistry: basics, applications and challenges. Appl. Sci. 2, 277–302 (2012).
Li, K. P. et al. Improved and optimized one-pot method for N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) synthesis using microwaves. Appl. Radiat. Isot. 94, 113–117 (2014).
Zhan, C. G. & Dixon, D. A. Hydration of the fluoride anion: structures and absolute hydration free energy from first-principles electronic structure calculations. J. Phys. Chem. 108, 2020–2029 (2004).
Onega, M. et al. An enzymatic route to 5-deoxy-5-[18F]fluoro-D-ribose, a [18F]-fluorinated sugar for PET imaging. Chem. Commun. 46, 139–141 (2010).
Pascali, G., Watts, P. & Salvadori, P. A. Microfluidics in radiopharmaceutical chemistry. Nucl. Med. Biol. 40, 776–787 (2013).
Thompson, S. et al. A localized tolerance in the substrate specificity of the fluorinase enzyme enables “last-step” 18F fluorination of a RGD peptide under ambient aqueous conditions. Angew. Chem. Int. Ed. 53, 8913–8918 (2014).
Thompson, S. et al. A two-step fluorinase enzyme mediated 18F labelling of an RGD peptide for positron emission tomography. Chem. Commun. 51, 13542–13545 (2015).
Li, Y., Schaffer, P. & Perrin, D. M. 18F-labeling techniques for positron emission tomography. Sci. Chi. Chem. 56, 1682–1692 (2013).
Thorp-Greenwood, F. L. & Coogan, M. P. Multimodal radio-(PET/SPECT) and fluorescence imaging agents based on metallo-radioisotopes: current applications and prospects for development of new agents. Dalton. Trans. 40, 6129–6143 (2011).
Wangler, B. et al. Chelating agents and their use in radiopharmaceutical sciences. Mini-Rev. Med. Chem. 11, 968–983 (2011).
Wangler, C. et al. One-step 18F-labeling of peptides for positron emission tomography imaging using the SiFA methodology. Nat. Protoc. 7, 1946–1955 (2012).
Kostikov, A. P. et al. Oxalic acid supported Si-18F-radiofluorination: one-step radiosynthesis of N-succinimidyl 3-(di-tert-butyl[18F]fluorosilyl)benzoate ([18F]SiFB) for protein labeling. Bioconjug. Chem. 23, 106–114 (2012).
Dijkgraaf, I. et al. PET of tumors expressing gastrin-releasing peptide receptor with an 18F-labeled bombesin analog. J. Nucl. Med. 53, 947–952 (2012).
Schirrmacher, R., Wangler, C. & Schirrmacher, E. Recent developments and trends in 18f-radiochemistry: syntheses and applications. Mini-Rev. Org. Chem. 4, 317–329 (2007).
McBride, W. J. et al. New lyophilized kit for rapid radiofluorination of peptides. Bioconjug. Chem. 23, 538–547 (2012).
Gelovani, J. G. et al. Quantitative analysis and comparison study of [18F]AlF-NOTA-PRGD2, [18F]FPPRGD2 and [68Ga]Ga-NOTA-PRGD2 using a reference tissue model. PLoS. ONE. 7, e337506 (2012).
D’Souza, C. A. et al. High-yielding aqueous 18F-labeling of peptides via Al18F chelation. Bioconjug. Chem. 22, 1793–1803 (2011).
McBride, W. J. et al. Improved 18F labeling of peptides with a fluoride-aluminum-chelate complex. Bioconjug. Chem. 21, 1331–1340 (2010).
Ting, R. et al. Arylfluoroborates and alkylfluorosilicates as potential PET Imaging agents: high-yielding aqueous biomolecular 18F-labeling. J. Am. Chem. Soc. 127, 13094–13095 (2005).
Ting, R. et al. Toward [18F]-labeled aryltrifluoroborate radiotracers: in vivo positron emission tomography imaging of stable aryltrifluoroborate clearance in mice. J. Am. Chem. Soc. 130, 12045–12055 (2008).
Ting, R. et al. Fast 18F labeling of a near-infrared fluorophore enables positron emission tomography and optical imaging of sentinel lymph nodes. Bioconjug. Chem. 21, 1811–1819 (2010).
Li, Y. et al. One-step and one-pot-two-step radiosynthesis of cyclo-RGD-18F-aryltrifluoroborate conjugates for functional imaging. Am. J. Nucl. Med. Mol. Imaging 3, 44–56 (2013).
Liu, S. et al. “Kit like” 18F labeling method for synthesis of RGD peptide-based PET probes. Am. J. Nucl. Med. Mol. Imaging 3, 97–101 (2013).
Li, Y. et al. 18F-click labeling of a bombesin antagonist with an alkyne-18F-ArBF3 -: in vivo PET imaging of tumors expressing the GRP-receptor. Am. J. Nucl. Med. Mol. Imaging 3, 57–70 (2013).
Liu, Z. et al. Stoichiometric leverage: rapid 18F-aryltrifluoroborate radiosynthesis at high specific activity for click conjugation. Angew. Chem. Int. Ed. 52, 2303–2307 (2013).
Li, Y., Schaffer, P. & Perrin, D. M. Dual isotope labeling: conjugation of 32P-oligonucleotides with 18F-aryltrifluoroborate via copper(I) catalyzed cycloaddition. Bioorg. Med. Chem. Lett. 23, 6313–6316 (2013).
Liu, Z. et al. An organotrifluoroborate for broadly applicable one-step 18F-labeling. Angew. Chem. Int. Ed. 53, 11876–11880 (2014).
Monzittu, F. M. et al. Rapid aqueous late-stage radiolabelling of [GaF3(BnMe2-tacn)] by 18F/19F isotopic exchange—towards new PET Imaging Probes. Angew. Chem. Int. Ed. 57, 6658–6661 (2018).
Burke, B. P., Clemente, G. S. & Archibald, S. J. Boron-18F containing positron emission tomography radiotracers: advances and opportunities. Contrast Media Mol. Imaging 10, 96–110 (2015).
Kamat, S. S., Williams, H. J. & Raushel, F. M. Intermediates in the transformation of phosphonates to phosphate by bacteria. Nature 480, 570–573 (2011).
Luo, Y. R. Comprehensive handbook of chemical bond energies (CRC Press, Boca Raton, 2007).
Vabre, B. et al. Radiofluorination of a NHC-PF5 adduct: toward new probes for 18F PET imaging. Chem. Commun. 53, 8657–8659 (2017).
Bayne, J. M. & Stephan, D. W. Phosphorus Lewis acids: emerging reactivity and applications in catalysis. Chem. Soc. Rev. 45, 765–774 (2016).
Studenov, A. R. et al. New radiolabelling chemistry: synthesis of phosphorus-[18F]fluorine compounds. J. Label. Compd. Radiopharm. 48, 497–500 (2005).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Yanai, T., Tew, D. P. & Handy, N. C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004).
Scalmani, G. & Frisch, M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 132, 114110 (2010).
Plata, R. E. & Singleton, D. A. A case study of the mechanism of alcohol-mediated Morita Baylis-Hillman reactions. The importance of experimental observations. J. Am. Chem. Soc. 137, 3811–3826 (2015).
Zhang, L. et al. Meta-selective C-H alkylation of 2-phenylpyridine catalyzed by ruthenium: DFT study on the mechanism and regioselectivity. Eur. J. Org. Chem. 2018, 5268–5277 (2018).
Kielbasinski, P. & Mikolajczyk, M. Novel-approach to the synthesis of alkoxycarbonylmethylphosphine and bis(alkoxycarbonylmethyl)phosphine pxides based on a Reformatsky-type reaction. Synth.-Stuttg. 2, 144–146 (1995).
Liu, N. et al. Copper-promoted oxidative-fluorination of arylphosphine under mild conditions. Chem. Commun. 50, 10879–10882 (2014).
Cai, H. & Conti, P. S. RGD-based PET tracers for imaging receptor integrin alphav beta3 expression. J. Label. Compd. Radiopharm. 56, 264–279 (2013).
Li, Z. et al. Rapid aqueous [18F]-labeling of a bodipy dye for positron emission tomography/fluorescence dual modality imaging. Chem. Commun. 47, 9324–9326 (2011).
Hoffend, J. et al. Gallium-68-DOTA-albumin as a PET blood-pool marker: experimental evaluation in vivo. Nucl. Med. Biol. 32, 287–292 (2005).
Zeng, J. L., Wang, J. & Ma, J. A. New strategies for rapid 18F-radiolabeling of biomolecules for radionuclide-based in vivo imaging. Bioconjug. Chem. 26, 1000–1003 (2015).
Fey, N. et al. Stable fluorophosphines: predicted and realized ligands for catalysis. Angew. Chem. Int. Ed. 51, 118–122 (2012).
Seel, V. F. & Rudolph, K. Uber Dimethylfluorphosphin and Dimethyldifluor-phosphran. Anorg. Allg. Chem. 363, 233–244 (1968).
Brown, C., Murray M. & Schmutzler R. Fluorodiphenylphosphine. J. Chem. Soc. C. 878–881 (1970).
Pabel, M., Willis, A. C. & Wild, S. B. First resolution of a free fluorophosphine chiral at phosphorus. Resolution and reactions of free and coordinate (+/−)− Fluorophenylisopropylphosphine. Inorg. Chem. 35, 1244–1249 (1996).
Haenel, J. et al. Die dimerisierung yon di(n-buty1)fluorphosphan und seine reaktion mit benzaldehyd. Z. Anorg. Allg. Chem. 607, 161–163 (1992).
Coenen, H. H. et al. Consensus nomenclature rules for radiopharmaceutical chemistry-setting the record straight. Nucl. Med. Biol. 55, v–xi (2017).
Acknowledgements
This study was supported by the National Science Foundation of China (81501534&91859113), the Excellent Youth Foundation of Fujian Scientific Committee (2018J06024), Fundamental Research Funds for the Central Universities (20720180050&20720170065), Fourth Round Fujian Health Education Joint Research Projects (WKJ2016-2-08), Scientific Research Foundation of State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics (2016ZY002), and Natural Science Foundation of Fujian Province, China (2016J05200).
Author information
Authors and Affiliations
Contributions
Z.L. and L.N. conceived the study and coordinated all the research. H.H. synthesized, characterized and radiolabeled all organophosphine fluoride acceptors and performed microPET/CT imaging. Z.L., H.H., and L.N. analyzed the results and wrote the manuscript. Mechanism calculation was carried out by L.Z., F.X., D.J., R.Z., H.Y., H.L., and J.L. helped in biological and imaging experiments. D.J. and X.Z. helped in discussions of the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal peer review information: Nature Communications thanks Jan Passchier and the other anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, H., Zhang, L., Xie, F. et al. Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors. Nat Commun 10, 989 (2019). https://doi.org/10.1038/s41467-019-08953-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-08953-0
This article is cited by
-
Cu(II)-Mediated direct 18F-dehydrofluorination of phosphine oxides in high molar activity
EJNMMI Radiopharmacy and Chemistry (2024)
-
Synthesis and preliminary evaluation of novel 11C-labeled GluN2B-selective NMDA receptor negative allosteric modulators
Acta Pharmacologica Sinica (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
