
Abstract
The influence of hydration water on the vibrational energy relaxation in a protein holds the key to understand ultrafast protein dynamics, but its detection is a major challenge. Here, we report measurements on the ultrafast vibrational dynamics of amide I vibrations of proteins at the lipid membrane/H2O interface using femtosecond time-resolved sum frequency generation vibrational spectroscopy. We find that the relaxation time of the amide I mode shows a very strong dependence on the H2O exposure, but not on the D2O exposure. This observation indicates that the exposure of amide I bond to H2O opens up a resonant relaxation channel and facilitates direct resonant vibrational energy transfer from the amide I mode to the H2O bending mode. The protein backbone motions can thus be energetically coupled with protein-bound water molecules. Our findings highlight the influence of H2O on the ultrafast structure dynamics of proteins.
Similar content being viewed by others

Introduction
Protein–water interactions play critical roles in the structure, dynamics, and function of proteins1,2,3. Strong hydrogen bonding between proteins and water results in an intimate coupling of water thermal motion to proteins2,3,4. Although the internal motions of proteins have been proposed to be slaved to the dynamics of the surrounding water molecules in the hydration shell, few direct experimental measurements are capable of capturing these interactions on the femtosecond to picosecond timescale in which hydrogen-bonding fluctuations of water proceed5,6,7. The study of vibrational energy dissipation between proteins and their surrounding water provides a powerful approach to unravel the nature of this dynamical coupling3,8 and analyze protein structures or solvent accessibility9,10. Particularly, ultrafast infrared (IR) spectroscopy such as two-dimensional (2D) IR serves as a structure-sensitive tool with fs–ps temporal resolution and has been widely used to explore the vibrational dynamics of amide I and amide A bands11,12,13,14,15. Here the amide I bands arise primarily from C=O stretching of protein backbone and are very sensitive to the hydrogen bonding between peptide bond and hydration water16. Because the frequency of amide I mode is strongly overlapped with the H2O bending mode absorption, the 2D IR experiments are generally carried out in D2O or organic solvents11,12,13,14,15. It is found that vibrational relaxation of the amide I mode occurs in ~1.2 ps (Supplementary Table 2 and Supplementary Figure 3)11,12,13,14,15. In light of this fact, the population relaxation of the amide I mode is considered as an intrinsic property of the peptide group itself and is relatively unaffected by the surrounding environment of the peptide bond and side chains11,12,13,14,15. In addition, molecular dynamics simulation of a green fluorescent protein suggested that low-frequency vibrations in proteins (collective motions of proteins) are strongly coupled with water, whereas high- and intermediate-frequency vibrations (protein backbone motions) are essentially decoupled with water8. It was therefore concluded that water serves as a thermal bath to enhance the intramolecular relaxation. It can be seen that, although the picture of vibrational dynamics of amide I mode in D2O and organic solvents has been extracted for the model compounds of peptide group and some globular proteins, the scenario of proteins in H2O is yet to be investigated, to the best of our knowledge. Consequently, the interaction mechanisms between proteins and H2O are not well understood and contradictory views have been reported17,18,19. Moreover, many major issues on this matter have not been addressed3, for example, what are the roles of resonant vibrational energy transfer in proteins; is there a “shortcut” for energy to dissipate into the solvent? Because H2O is significantly different from D2O as solvents12, the ultrafast protein dynamics in the two solvents could be quite different. Here we investigate the ultrafast vibrational dynamics of amide I mode of proteins at the H2O interface using a surface-sensitive pump–probe set-up in which a femtosecond IR pump is followed by a sum frequency generation vibrational spectroscopy (SFG-VS) probe.
SFG-VS is a powerful and versatile optical technique for characterizing the interfacial molecular structures and dynamics of various molecules including peptides and proteins in situ and in real time20,21,22,23,24,25. Recently, the IR pump–SFG probe technique has been employed to study the ultrafast vibrational dynamics of interfacial water and proteins by several groups26,27,28,29,30. Theoretically, the SFG signals from the water bending mode are extremely weak or not observable at all compared to amide I mode31. SFG-VS can therefore eliminate the interference of the H2O bending mode in the amide I region that IR spectroscopy has suffered. Indeed, many reports have confirmed that SFG spectra of amide I band are not contaminated by the contribution of H2O bending mode32,33,34,35. It can be naturally anticipated that the IR pump–SFG probe technique offers an effective optical method to investigate the vibrational dynamics of amide I band of interfacial proteins in H2O environments. However, to the best of our knowledge, such method has not been applied on the energy relaxation between amide I band and H2O. In this study, we report measurements on the ultrafast vibrational dynamics of amide I vibrations of proteins at the lipid membrane/H2O interface using several peptides, including melittin, LKα14, mastoparan (MP), influenza M2 proteins (AM2 and BM2), and KALP23 as the models. It is known that the exposure of these peptides to water can be tuned by formation of the channel or pore in lipid membrane36,37,38,39.
Results
Exposure amount of membrane-bound peptides to water
We first determine the relative amount of peptide bonds that are exposed to H2O by measuring the ratio of the hydrogen–deuterium exchange (HDX, Supplementary Note 4) of the amide proton. Previous studies indicated that the residues exposed to H2O can undergo rapid amide proton HDX as the sample is exposed to deuterium, but the part in the core of lipid bilayer (not exposed to H2O) does not exchange in several days30,40,41. Figure 1a shows the amide A spectra of peptides before and after 5-h HDX. Because the interaction between peptides and membrane can cause dehydration of the membrane surface and thus suppress the SFG signals from the interfacial water molecules, the contribution from the OH signal to the signals in the amide A region is negligible30,42. It is evident that the ssp (denoting s-, s-, and p-polarized sum-frequency output, visible input, and infrared input, respectively) SFG spectra of melittin and LKα14 in the N-H region are almost disappeared after 5-h HDX while KALP23 changes insignificantly (Fig. 1a), illustrating that melittin and LKα14 are exposed to H2O in lipid membrane because of formation of toroidal pores, but KALP23 mainly lies in the hydrophobic core of the lipid bilayer36. In addition, the amide I frequency of melittin (1651 cm−1) and LKα14 (1652 cm−1) is observed to be lower than that of KALP23 (1665 cm−1) (Fig. 1b). It has been reported that the amide I frequency of α-helices shifts from 1665 to 1648 cm−1 upon hydration because the carbonyl groups in the peptide bond form a bifurcated hydrogen bonding with water molecules (Fig. 1c)43. Formation of a hydrogen bond with the amide oxygen can soften the vibrational potential and thereby lower the frequency44,45. To qualitatively determine the 5-h HDX ratio and the amide I frequency change of these peptides, we fitted the spectra using a standard procedure, Supplementary Eq. 1. We plot the fitting amplitude ratio of the ssp SFG spectra in the N-H region after 5-h HDX and before HDX (\(\chi _{{\mathrm{5h}}\,{\mathrm{HDX}}}^{(2)}/\chi _{{\mathrm{before}}\,{\mathrm{HDX}}}^{(2)}\), Supplementary Table 3) against the amide I frequency (Supplementary Table 3). A linear correlation between the \(\chi _{{\mathrm{5h}}\,{\mathrm{HDX}}}^{(2)}/\chi _{{\mathrm{before}}\,{\mathrm{HDX}}}^{(2)}\) ratio and the amide I frequency is clearly observed (Fig. 1d). Therefore, the amide I frequency can be used as an optical marker for the exposure amount of membrane-bound peptides to water.

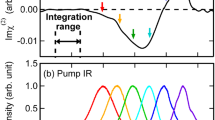
Sum frequency generation (SFG) spectra. The ssp (denoting s-, s-, and p-polarized sum-frequency output, visible input, and infrared input, respectively) SFG spectra of the peptides of amide A and amide I modes, sketch for the hydration of amide I mode, and the relationship between hydrogen–deuterium exchange (HDX) ratio and amide I frequency. a The ssp SFG spectra of the peptides in the N-H region. The spectra in black curves are measured at lipid bilayer/H2O interface while the ones in red curves are measured at lipid bilayer/D2O interface after 5-h HDX experiments. b The ssp amide I SFG spectra of peptides at lipid/H2O interface. The solid curves in a, b are the fitting curves using Supplementary Eq. 1. c Sketch for the carbonyl groups forming a bifurcated H bonding to water molecules upon hydration. d Fitting amplitude ratio \(\left( {\chi _{{\mathrm{5h}}\,{\mathrm{HDX}}}^{(2)}/\chi _{{\mathrm{before}}\,{\mathrm{HDX}}}^{(2)}} \right)\) of the ssp SFG spectra in the N-H region after 5-h HDX and before HDX against the amide I frequency. The vertical error bars represent experimental error in determining the amplitude ratio from the fitting procedure (see Supplementary Note 2). The horizontal error bars indicate the standard error of the mean of the fitting center frequency from five independent spectra
Vibrational dynamics of amide I band
We then investigate the vibrational dynamics of amide I band at the lipid bilayer/D2O and H2O interface. Figure 2a (top panel) shows the intensity decay of the amide I band of KALP23 at the 1,2-dipalmitoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt) (DPPG) lipid bilayer/D2O interface with νpump = νprobe = 1660 cm−1. Three-level vibrational model (Supplementary Figure 4 and Supplementary Note 5) has been used to extract the vibrational lifetime (T1) and thermalization time constant (Tth) of the vibrational dynamics in many condensed matters, including water46,47,48 and peptides49,50,51,52. In this model, the amide I modes are excited from ν = 0 to ν = 1 state and it takes T1 time to relax to a “hot” ground state (ν = 0*), and then the intensity of the amide I band gradually recovers. The decay of the ν = 0* state with a vibrational cooling time (Tth) leads to a full thermalization of the system. The vibrational cooling time (Tth) of the amide bond has been determined to be ~10 ps49 and is used in our fittings. It is noted that the amide I signal starts to decrease at a delay time of ~−1.2 ps in which the probe pulse significantly precedes the arrival of the pump pulse (Fig. 2a). The signal reaches a minimum at 0.1 ps and finally recovers at the positive delay time. The signal at the negative delay time is generated by the perturbed free induction decay (FID) effect53 and can be fitted by an exponential function of 1 − A0 exp(t/T0). FID process does not influence the data at the positive delay time53,54. Fit of the experimental result in Fig. 2a (top panel) for positive delay time (t ≥ 0.1 ps) using Supplementary Eq. 16 yields a relaxation time of T1 = 1.15 (±0.05) ps, confirming the same vibrational relaxation time (~1.2 ps) discovered in D2O or organic solvents by 2D IR studies. However, at the lipid bilayer/H2O interface (Fig. 2a, bottom panel), the relaxation time is found to be 1.0 (±0.05) ps, a little smaller than the value at the lipid bilayer/D2O interface.

Intensity decay of amide I band and relationship between relaxation time and amide I frequency. a The intensity decay of the amide I band of KALP23 at lipid membrane/water interface with νpump = νprobe = 1660 cm−1. The red lines are the fitting curves. The data for delay time at t ≤ 0.1 ps was fitted using the function of 1 − A0 exp(t/T0) while the data for delay time at t ≥ 0.1 ps were fitted using Supplementary Eq. 16. b Relaxation time of amide I mode against the amide I frequency. The vertical error bars represent the estimated accuracy of the fit based on the time resolution of the laser pulse width. The horizontal error bars indicate the standard error of the mean of the fitting center frequency from five independent spectra
Dependence of relaxation time on H2O exposure
Using the same experimental procedures, we investigate the vibrational dynamics of amide I band of melittin, LKα14, MP, BM2, and AM2 at the lipid membrane/H2O interface. The “pump on” and “pump off” spectra at some typical delay times and the intensity decay are shown in Supplementary Figure 5, Supplementary Figure 6, and Supplementary Note 6. Controlled experiments on the excitation of water bending mode at CaF2/H2O interface and DPPG bilayer/H2O interface indicated that the effect of the excitation of water bending mode on the vibrational dynamics of amide I is very small, at least beyond the experimental errors (Supplementary Figures 7 and 8, Supplementary Note 7). The relaxation time is determined to be 0.51 (±0.05) for melittin, 0.56 (±0.05) for LKα14, 0.64 (±0.05) for MP, 0.77 (±0.05) for BM2, which are about half of the value in D2O. For the AM2 samples, its relaxation time depends on the pH conditions, which are 0.88 (±0.05) at pH = 4, 0.93(±0.05) at pH = 6.2, and 0.97(±0.05) at pH = 8.5. All these results depict that the vibrational relaxation in H2O occurs faster than that in D2O. Because the peptides of LKα14, MP, KALP23, BM2, and AM2 all adopt a nearly pure α-helical structure in lipid membrane25,55, the difference in lifetime is not contributed by the secondary structures. Summarizing all of these results, it is found that the relaxation time of amide I mode in H2O environment linearly correlates with the amide I frequency (Fig. 2b). It is worth noticing that M2 opens its channel to form a water pore at acid environment and is therefore more exposed to water39. The results in Fig. 2b, taking into account the relationship between the amide I frequency and the exposure amount of membrane-bound peptides to water shown in Fig. 1d, reveal that the peptides that are more exposed to H2O have a shorter relaxation time. Apparently, the relaxation time is dictated by H2O exposure, which is opposite to what has been reported for peptides in D2O or organic solvents (Supplementary Figure 3, Supplementary Note 3). The hydrogen bonding mechanism could not explain this result because H2O forms weaker hydrogen bonds than D2O56.
The relaxation of amide I mode has been assumed to be the result of intramolecular vibrational redistribution (IVR) in protein secondary structures because such fast timescale cannot be explained by direct energy transfer between the amide I mode and the surrounding solvents of the peptide group11,13. However, the IVR mechanism alone cannot interpret our observation that H2O can induce a faster vibrational relaxation than D2O, which manifests that the vibrational dynamics of amide I mode in H2O must involve other relaxation pathway through additional coupling induced by H2O. The correlation between the relaxation time of amide I mode and the exposure amount of membrane-bound peptides to H2O provides experimental evidences for the transition–dipole coupling between amide I mode and the water bending mode (Fig. 3a). The vibrational mixing between them could open a resonant relaxation channel that facilitates direct vibrational energy transfer from the amide I mode to the H2O bending mode. This pathway is only present in H2O but absent in D2O because the D2O bending mode shifts to ~1220 cm−1 and is no longer resonant with the amide I mode (Fig. 3b). Indeed, the notion of vibrational mixing in the peptide unit of N-methylacetamide and dipeptides has been theoretically predicted57,58,59 and experimentally supported by the Raman band shape of amide I, which depends on H2O density60,61,62,63. In addition, such Förster-like resonant energy transfer mechanism has been observed in the case of the cyanide ion64, cyano65, and metal carbonyl complex66, in which the relaxation time in H2O is faster than that in D2O.

Coupling scheme and energy diagram. a Interaction between amide carbonyl and water through hydrogen bonding (HB) and transition–dipole coupling. b Energy-level depiction of the vibrational relaxation of the amide I mode to the water bending mode
On the other hand, according to Fig. 1c, the amide I groups can actually be considered as two components: the one coupled to water and the other not coupled to water. Therefore, the intensity decay of the amide I band shown in Supplementary Figure 6 would be analyzed using two energy relaxation processes (Supplementary Note 8): one for the amide modes coupled to water with relaxation time of 0.4 ps (a value approximate to the lifetime of water bending modes67,68) and one for the amide modes not coupled to water with relaxation time of 1.15 ps (the lifetime of amide I mode in D2O). The ratio of the component of 1.15 ps is plotted in Supplementary Figure 9. It is evident that the ratio of the component of 1.15 ps in H2O environment linearly correlates with the amide I frequency and matches well with the exposure amount of membrane-bound peptides to the water determined by HDX method (Fig. 1d). This result suggests that the relaxation time measurements combining with the site-specific labeling technique developed by Zanni et al.35,40 will offer a unique and effective optical marker to determine the hydrophobicity of specific sites.
Discussion
To conclude, we applied a time-resolved IR pump–SFG probe to investigate the ultrafast vibrational dynamics of amide I mode of proteins at the lipid membrane/H2O interface. We found that the relaxation of amide I mode strongly depends on the H2O exposure, in stark contrast to what was observed in the case of D2O exposure. This implies that the relaxation dynamics of amide I mode in H2O environment is not only controlled by the intrinsic property of the peptide group but also by a direct resonant channel that is energetically coupled with protein-bound water molecules. In other words, while D2O only serves as a thermal bath to enhance the intramolecular relaxation, H2O can also provide a “shortcut” through direct resonant channel to dissipate energy into the solvent. It highlights the limitation of using D2O to study the ultrafast structure dynamics of proteins. Our findings may be important for correctly understanding the roles of resonant vibrational energy transfer through dipole coupling in protein and hydrogen bonds between H2O and the biomolecule on the entire energy transfer pathways.
Methods
Materials
The peptides used in this study include melittin (a bee venom toxin, sequence: GIGAVLKVLTTGLPALISWIKRKRQQ), LKα14 (an amphiphilic peptide, sequence: LKKLLKLLKKLLKL), MP (sequence: INLKALAALAKKIL), influenza B M2 transmembrane domain (BM2, sequence: MLEPFQILSICSFILSALHFMAWTIGHLNQIKR), influenza A M2 transmembrane domain (AM2, sequence: SSDPLVVAASIIGILHLILWILDRL), and KALP23 (sequence: GKKLALALALALALALALALKKA). These peptides have a purity of >98% and were purchased from Shanghai Apeptide Co., Ltd. The lipids of 1-palmitoyl-2-oleoyl- sn-glycero- 3-phospho- (1’-rac-glycerol) (sodium salt) (POPG), 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt) (DMPG), DPPG, and 1,2-dimyristoyl-d54-sn-glycero-3-phosphocholine-1,1,2,2-d4-N,N,N-trimethyl-d9 (d-DMPC) were purchased from Avanti Polar Lipids (Alabaster, AL). All the peptides were dissolved in methanol (purchased from Sinopharm Chemical Reagent Co., Ltd.) with different concentrations (supplementary Table 1) and stored at −20 °C. The d-DMPC phospholipid was dissolved in chloroform (purchased from Sinopharm Chemical Reagent Co., Ltd.). DPPG, DMPG, and POPG solutions (with the concentration of 1.3 mg/mL) were prepared in mixed solvents of chloroform and methanol (with a volume ratio of 2:1) (purchased from Sinopharm Chemical Reagent Co., Ltd.). All the lipid solutions were kept at −20 °C. Right-angle CaF2 prisms were purchased from Chengdu Ya Si Optoelectronics Co., Ltd (Chengdu, China). All the chemicals were used as received. The cleaning of CaF2 prisms and preparation of lipid bilayer were performed using a standard procedure given in Supplementary Information.
Experiments and data analysis
The details about SFG-VS experiments and data analysis can be found in Supplementary Information (Supplementary Note 1, Supplementary Note 2, Supplementary Note 5, Supplementary Note 6, Supplementary Figure 1, and Supplementary Figure 2).
Data availability
The data that support the finding in current study are available from the corresponding author upon reasonable request.
References
Feng, C. J. & Tokmakoff, A. The dynamics of peptide-water interactions in dialanine: an ultrafast amide I 2D IR and computational spectroscopy study. J. Chem. Phys. 147, 085101 (2017).
Yang, J., Wang, Y. F., Wang, L. J. & Zhong, D. P. Mapping hydration dynamics around a β-barrel protein. J. Am. Chem. Soc. 139, 4399–4408 (2017).
Laage, D., Elsaesser, T. & Hynes, J. T. Water dynamics in the hydration shells of biomolecules. Chem. Rev. 117, 10694–10725 (2017).
Fenwick, R. B., Oyen, D., Dyson, H. J. & Wright, P. E. Slow dynamics of tryptophan-water networks in proteins. J. Am. Chem. Soc. 140, 675–682 (2018).
Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Ultrafast dynamics at water interfaces studied by vibrational sum frequency generation spectroscopy. Chem. Rev. 117, 10665–10693 (2017).
King, J. T. & Kubarych, K. J. Site-specific coupling of hydration water and protein flexibility studied in solution with ultrafast 2D-IR spectroscopy. J. Am. Chem. Soc. 134, 18705–18712 (2012).
Schiro, G. et al. Translational diffusion of hydration water correlates with functional motions in folded and intrinsically disordered proteins. Nat. Commun. 6, 6490 (2015).
Shenogina, N., Keblinski, P. & Garde, S. Strong frequency dependence of dynamical coupling between protein and water. J. Chem. Phys. 129, 155105 (2008).
Middleton, C. T., Buchanan, L. E., Dunkelberger, E. B. & Zanni, M. T. Utilizing lifetimes to suppress random coil features in 2D IR spectra of peptides. J. Phys. Chem. Lett. 2, 2357–2361 (2011).
Woutersen, S., Mu, Y. G., Stock, G. & Hamm, P. Subpicosecond conformational dynamics of small peptides probed by two-dimensional vibrational spectroscopy. Proc. Natl Acad. Sci. USA 98, 11254–11258 (2001).
Hamm, P., Lim, M. & Hochstrasser, R. M. Structure of the amide I band of peptides measured by femtosecond nonlinear-infrared spectroscopy. J. Phys. Chem. B 102, 6123–6138 (1998).
Ghosh, A. & Hochstrasser, R. M. A peptide’s perspective of water dynamics. Chem. Phys. 390, 1–13 (2011).
Candelaresi, M. et al. Conformational analysis of Gly-Ala-NHMe in D2O and DMSO solutions: a two-dimensional infrared spectroscopy study. J. Phys. Chem. B 117, 14226–14237 (2013).
Hamm, P., Lim, M., DeGrado, W. F. & Hochstrasser, R. M. Pump/probe self heterodyned 2D spectroscopy of vibrational transitions of a small globular peptide. J. Chem. Phys. 112, 1907–1916 (2000).
Mukherjee, P., Kass, I., Arkin, I. T. & Zanni, M. T. Picosecond dynamics of a membrane protein revealed by 2D IR. Proc. Natl Acad. Sci. USA 103, 3528–3533 (2006).
Barth, A. & Zscherp, C. What vibrations tell us about proteins. Q. Rev. Biophys. 35, 369–430 (2002).
Qin, Y. Z., Wang, L. J. & Zhong, D. P. Dynamics and mechanism of ultrafast water-protein interactions. Proc. Natl Acad. Sci. USA 113, 8424–8429 (2016).
Qin, Y. Z. et al. Molecular origin of ultrafast water-protein coupled interactions. J. Phys. Chem. Lett. 7, 4171–4177 (2016).
Chong, S.-H. & Ham, S. Anomalous dynamics of water confined in protein-protein and protein-DNA interfaces. J. Phys. Chem. Lett. 7, 3967–3972 (2016).
Shen, Y. R. The Principles of Nonlinear Optics (Wiley, New York, 1984).
Lambert, A. G., Davies, P. B. & Neivandt, D. J. Implementing the theory of sum frequency generation vibrational spectroscopy: a tutorial review. Appl. Spectrosc. Rev. 40, 103–145 (2005).
Yan, E. C. Y., Fu, L., Wang, Z. G. & Liu, W. Biological macromolecules at interfaces probed by chiral vibrational sum frequency generation spectroscopy. Chem. Rev. 114, 8471–8498 (2014).
Ding, B., Jasensky, J., Li, Y. X. & Chen, Z. Engineering and characterization of peptides and proteins at surfaces and interfaces: a case study in surface-sensitive vibrational spectroscopy. Acc. Chem. Res. 49, 1149–1157 (2016).
Schach, D. K. et al. Reversible activation of a cell-penetrating peptide in a membrane environment. J. Am. Chem. Soc. 137, 12199–12202 (2015).
Ye, S. J., Li, H. C., Yang, W. L. & Luo, Y. Accurate determination of interfacial protein secondary structure by combining interfacial-sensitive amide I and amide III spectral signals. J. Am. Chem. Soc. 136, 1206–1209 (2014).
McGuire, J. A. & Shen, Y. R. Ultrafast vibrational dynamics at water interfaces. Science 313, 1945–1948 (2006).
Bonn, M. et al. Structural inhomogeneity of interfacial water at lipid monolayers revealed by surface-specific vibrational pump-probe spectroscopy. J. Am. Chem. Soc. 132, 14971–14978 (2010).
Eftekhari-Bafrooei, A. & Borguet, E. Effect of hydrogen-bond strength on the vibrational relaxation of interfacial water. J. Am. Chem. Soc. 132, 3756–3761 (2010).
Singh, P. C., Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Interfacial water in the vicinity of a positively charged interface studied by steady-state and time-resolved heterodyne-detected vibrational sum frequency generation. J. Chem. Phys. 141, 18C527 (2014).
Tan, J. J., Zhang, B. X., Luo, Y. & Ye, S. J. Ultrafast vibrational dynamics of membrane-bound peptides at the lipid bilayer/water interface. Angew. Chem. Int. Ed. 56, 12977–12981 (2017).
Perry, A., Ahlborn, H., Space, B. & Moore, P. B. A combined time correlation function and instantaneous normal mode study of the sum frequency generation spectroscopy of the water/vapor interface. J. Chem. Phys. 118, 8411–8419 (2003).
Engelhardt, K., Peukert, W. & Braunschweig, B. Vibrational sum-frequency generation at protein modified air-water interfaces: effects of molecular structure and surface charging. Curr. Opin. Colloid Interface Sci. 19, 207–215 (2014).
Wang, J. et al. Detection of amide I signals of interfacial proteins in situ using SFG. J. Am. Chem. Soc. 125, 9914–9915 (2003).
Meister, K. et al. Observation of pH-induced protein reorientation at the water surface. J. Phys. Chem. Lett. 8, 1772–1776 (2017).
Ding, B. et al. Probing site-specific structural information of peptides at model membrane interface in situ. J. Am. Chem. Soc. 137, 10190–10198 (2015).
Sengupta, D., Leontiadou, H., Mark, A. E. & Marrink, S. J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 1778, 2308–2317 (2008).
Hill, E. H., Whitten, D. G. & Evans, D. G. Computational study of bacterial membrane disruption by cationic biocides: structural basis for water pore formation. J. Phys. Chem. B 118, 9722–9732 (2014).
Wildman, K. A. H., Lee, D. K. & Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 42, 6545–6558 (2003).
Ghosh, A., Qiu, J., DeGrado, W. F. & Hochstrasser, R. M. Tidal surge in the M2 proton channel, sensed by 2D IR spectroscopy. Proc. Natl Acad. Sci. USA 108, 6115–6120 (2011).
Dunkelberger, E. B., Woys, A. M. & Zanni, M. T. 2D IR cross peaks reveal hydrogen-deuterium exchange with single residue specificity. J. Phys. Chem. B 117, 15297–15305 (2013).
Demmers, J. A., Haverkamp, J., Heck, A. J., Koeppe, R. E. & Killian, J. A. Electrospray ionization mass spectrometry as a tool to analyze hydrogen/deuterium exchange kinetics of transmembrane peptides in lipid bilayers. Proc. Natl. Acad. Sci. USA 97, 3189–3194 (2000).
Tan, J. J., Li, C. Z., Zhang, J. H. & Ye, S. J. Real-time observation of protein transport across membranes by femtosecond sum frequency generation vibrational spectroscopy. Chin. J. Chem. Phys. 31, 523–528 (2018).
Lórenz-Fonfria, V. A. et al. Temporal evolution of helix hydration in a light-gated ion channel correlates with ion conductance. Proc. Natl Acad. Sci. USA 112, E5796–E5804 (2015).
Torii, H. Amide I vibrational properties affected by hydrogen bonding out-of-plane of the peptide group. J. Phys. Chem. Lett. 6, 727–733 (2015).
Manas, E. S., Getahun, Z., Wright, W. W., DeGrado, W. F. & Vanderkooi, J. M. Infrared spectra of amide groups in α-helical proteins: evidence for hydrogen bonding between helices and water. J. Am. Chem. Soc. 122, 9883–9890 (2000).
Ghosh, A., Smits, M., Bredenbeck, J. & Bonn, M. Membrane-bound water is energetically decoupled from nearby bulk water: an ultrafast surface-specific investigation. J. Am. Chem. Soc. 129, 9608–9609 (2007).
Nienhuys, H. K., Woutersen, S., van Santen, R. A. & Bakker, H. J. Mechanism for vibrational relaxation in water investigated by femtosecond infrared spectroscopy. J. Chem. Phys. 111, 1494–1500 (1999).
Lock, A. J., Woutersen, S. & Bakker, H. J. Ultrafast energy equilibration in hydrogen-bonded liquids. J. Phys. Chem. A 105, 1238–1243 (2001).
Rubtsov, I. V. & Hochstrasser, R. M. Vibrational dynamics, mode coupling, and structural constraints for acetylproline-NH2. J. Phys. Chem. B 106, 9165–9171 (2002).
DeCamp, M. F. et al. Amide I vibrational dynamics of N-methylacetamide in polar solvents: the role of electrostatic interactions. J. Phys. Chem. B 109, 11016–11026 (2005).
Rubtsov, I. V., Wang, J. P. & Hochstrasser, R. M. Dual-frequency 2D-IR spectroscopy heterodyned photon echo of the peptide bond. Proc. Natl Acad. Sci. USA 100, 5601–5606 (2003).
Rubtsov, I. V., Wang, J. P. & Hochstrasser, R. M. Vibrational coupling between amide-I and amide-A modes revealed by femtosecond two color infrared spectroscopy. J. Phys. Chem. A 107, 3384–3396 (2003).
Wynne, K. & Hochstrasser, R. M. The theory of ultrafast vibrational spectroscopy. Chem. Phys. 193, 211–236 (1995).
Smits, M. et al. Ultrafast energy flow in model biological membranes. N. J. Phys. 9, 390 (2007).
Liu, Y. et al. Influenza A M2 transmembrane domain tunes its conformational heterogeneity and structural plasticity in the lipid bilayer by forming loop structures. Chem. Commun. 54, 5903–5906 (2018).
Scheiner, S. & Čuma, M. Relative stability of hydrogen and deuterium bonds. J. Am. Chem. Soc. 118, 1511–1521 (1996).
Mirkin, N. G. & Krimm, S. Ab initio vibrational analysis of isotopic derivatives of aqueous hydrogen-bonded trans-N-methylacetamide. J. Mol. Struct. 377, 219–234 (1996).
Han, W. G., Jalkanen, K. J., Elstner, M. & Suhai, S. Theoretical study of aqueous N-acetyl-L-alanine N′-methylamide: structures and Raman, VCD, and ROA spectra. J. Phys. Chem. B 102, 2587–2602 (1998).
Cazade, P. A., Hédin, F., Xu, Z. H. & Meuwly, M. Vibrational relaxation and energy migration of N‑methylacetamide in water: the role of nonbonded interactions. J. Phys. Chem. B 119, 3112–3122 (2015).
Sieler, G. & Schweitzer-Stenner, R. The amide I mode of peptides in aqueous solution involves vibrational coupling between the peptide group and water molecules of the hydration shell. J. Am. Chem. Soc. 119, 1720–1726 (1997).
Kubelka, J. & Keiderling, T. A. Ab initio calculation of amide carbonyl stretch vibrational frequencies in solution with modified basis sets. 1. N-methyl acetamide. J. Phys. Chem. A 105, 10922–10928 (2001).
Chen, X. G., Schweitzer-Stenner, R., Krimm, S., Mirkin, N. G. & Asher, S. A. N-methylacetamide and its hydrogen-bonded water molecules are vibrationally coupled. J. Am. Chem. Soc. 116, 11141–11142 (1994).
Chen, X. G., Schweitzer-Stenner, R., Asher, S. A., Mirkin, N. G. & Krimm, S. Vibrational assignments of trans-V-methylacetamide and some of its deuterated isotopomers from band decomposition of IR, Visible, and resonance Raman spectra. J. Phys. Chem. 99, 3074–3083 (1995).
Hamm, P., Lim, M. & Hochstrasser, R. M. Vibrational energy relaxation of the cyanide ion in water. J. Chem. Phys. 107, 10523–10531 (1998).
Yu, P. Y., Yang, F., Zhao, J. & Wang, J. P. Hydration dynamics of cyanoferrate anions examined by ultrafast infrared spectroscopy. J. Phys. Chem. B 118, 3104–3114 (2014).
King, J. T., Ross, M. R. & Kubarych, K. J. Water-assisted vibrational relaxation of a metal carbonyl complex studied with ultrafast 2D-IR. J. Phys. Chem. B 116, 3754–3759 (2012).
Piatkowski, L. & Bakker, H. J. Vibrational dynamics of the bending mode of water interacting with ions. J. Chem. Phys. 135, 214509 (2011).
Larsen, O. F. A. & Woutersen, S. Vibrational relaxation of the H2O bending mode in liquid water. J. Chem. Phys. 121, 12143–12145 (2004).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0303500, 2018YFA0208700), National Natural Science Foundation of China (21873090, 21633007, 21790350), Fundamental Research Funds for the Central Universities (WK2340000064), CAS (2016HSC-IU003), and Anhui Initiative in Quantum Information Technologies (AHY090000).
Author information
Authors and Affiliations
Contributions
J.T., J.Z. and C.L. collected the SFG spectra. S.Y. processed and analyzed the data. S.Y. and Y.L. designed the study. S.Y. and Y.L. wrote the paper. All authors discussed the results of the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Journal peer review information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, J., Zhang, J., Li, C. et al. Ultrafast energy relaxation dynamics of amide I vibrations coupled with protein-bound water molecules. Nat Commun 10, 1010 (2019). https://doi.org/10.1038/s41467-019-08899-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-08899-3
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
