
Abstract
Metal-catalyzed enantioselective construction of the loosening axial allene chirality spreading over three carbon atoms using a chiral ligand is still a significant challenge. In the literature, steric effect of the substrates is the major strategy applied for such a purpose. Herein, we present a general palladium-catalyzed asymmetrization of readily available racemic 2,3-allenylic carbonates with different types of non-substituted and 2-substituted malonates using (R)-(−)-DTBM-SEGPHOS as the preferred ligand to afford 1,3-disubstituted chiral allenes with 90~96% ee. This protocol has been applied to the first enantioselective synthesis of natural product, (R)-traumatic lactone. Control experiments showed that in addition to the chiral ligand, conducting this transformation via Procedure C, which excludes the extensive prior coordination of the allene unit in the starting allene with Pd forming a species without the influence of the chiral ligand, is crucial for the observed high enantioselectivity.
Similar content being viewed by others

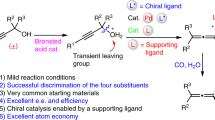
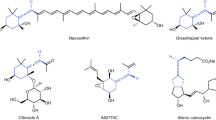
Introduction
Due to the rapid development in the last two decades1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17, allene chemistry has become an important branch in organic chemistry. Unlike other unsaturated hydrocarbons such as alkenes and alkynes, allenes are unique due to the intrinsic axial chirality. 1,3-Disubstituted allenes with synthetically useful functionalities are of particular high interest due to their common existence in natural products/drug molecules (Fig. 1)18 and their great potentials in the synthesis of natural products19,20,21,22,23,24. Thus, there is an urgent need for the development of catalytic highly enantioselective synthesis of such 1,3-disubstituted allenes.

Representative 1,3-disubstituted allenes. Natural products and a drug molecule
In view of the loosening nature of 1,3-substituted allene axial chirality spreading over three carbon atoms, the steric effect of the substrates is the major strategy used in such catalytic reactions of high ee (Fig. 2a). The highly enantioselective syntheses of such allenes with much less sterically bulky substituents are still a significant challenge and of high interest (Fig. 2b). Recently, CuBr2-catalyzed enantioselective allenylation of terminal alkynes (EATA) with aldehydes has been developed for such a purpose with very high ees and a wide scope, however, a stoichiometric amount of a sterically very bulky chiral amino alcohol, i.e., (R)- or (S)-diphenyl or dimethylprolinol, has to be applied (Fig. 2c)25,26. On the other hand, malonate is an extremely useful “sewing unit” in organic synthesis. Thus, a 1,3-disubstituted allene bearing a malonate unit with an unclosed reacting site is of high interest. Hayashi27, Imada28, and Trost29 have pioneered the Pd-catalyzed allene-asymmerization reaction of racemic non-terminal 2,3-allenol derivatives (or 2-(1,3-butadienyl) bromide) bearing a very bulky R group with rather bulky nucleophiles by applying a catalytic amount of certain chiral ligands27,28,29,30,31,32,33,34,35,36,37. However, the enantioselective reaction of substrates with R being the less sterically demanding yet very important 1°-alkyl group (Fig. 1) and unclosed parent malonate is still in its early infancy with ee of 60–70% (Fig. 2d)28,31,32. Furthermore, steric effect is also the strategy used in the catalyzed enantioselective synthesis of 4-mono-substituted 2,3-allenoates from stereo-defined enol triflates or 3-alkynoates by Frantz20, Tan38, Takemoto39, and their coworkers. This has also been observed in other development in this area40,41,42,43,44,45,46,47,48. In this paper, we wish to report a Pd-catalyzed protocol, in which the extensive prior coordination of the allene unit with palladium has to be excluded and DTBM-SEGPHOS has been identified as the best ligand, affording allene (Ra)-3 bearing the much less sterically bulky substituents in >90% ee. Such a strategy has been successfully applied to the enantioselective total synthesis of (R)-traumatic lactone (Fig. 2e).

Typical approaches to chiral 1,3-disubstituted allenes. a, b The current situation with such allene synthesis. c, d Reported strategies. e The approach developed in this study
Results
Optimization of reaction conditions
After some systematic screening on the ligands, solvents, Pd sources, and bases, we observed that the reaction of allenyl acetate 1a-OAc49,50,51 with diethyl malonate catalyzed by [Pd(π-cinnamyl)Cl)]2 and (R)-(−)-DTBM-SEGPHOS with K2CO3 as base in NMP at 30 °C afforded the 1,3-disubstituted allene (Ra)-3a-Et with 69% ee and 64% yield (Entry 1, Table 1)32 (defined as Procedure A). The reaction of allenyl carbonate 1a49,50,51 with diethyl malonate afforded (Ra)-3a-Et with a same level of ee although the yield was much higher (80%) (Entry 2, Table 1). Interestingly, the reaction following the Procedure B leading to a lower ee (60%) was observed (Entry 3, Table 1). Thus, we further adjusted the mixing sequence of the reactants and catalyst(s) and found that by using Procedure C, in which racemic allene substrate 1a was added at the end of the procedure, quite interestingly and unexpectedly, a much higher enantioselectivity of 87% was observed (Entry 4, Table 1). Running the reaction at 5 °C led to an ee of 90% and a yield of 85% for (Ra)-3aa (Entry 5, Table 1). We also evaluated the effect of the R group in malonate on the enantioselectivity (Entries 7–9, Table 1), confirming the steric effect of the parent malonate has no obvious effect.
Substrate scope
After establishing the optimal reaction conditions, the scope of allene substrates with the much less sterically hindered yet synthetically versatile 2-non-substituted dimethyl malonate was investigated elaborately. Allenylic carbonates 1 bearing a linear 1°-alkyl (R1) group reacted with dimethyl malonate 2a to afford (Ra)-3 with excellent ee values in good to excellent yields: Entries 1–9 showed that this protocol is very general for the different length of the carbon chain of R1 ranging from Me to n-C11H23. Common and practical protecting groups for the hydroxyl group, such as Bn, α-naphthylmethyl, and TBS, are also viable in this transformation affording allenes (Ra)-3ja, 3ka, and 3la in the ee value of 91%, 91%, and 92%, respectively (Entries 10, 11 and 12, Table 2). Expectedly, 1m with a secondary R1 group (Cy) and 1p with t-Bu afforded (Ra)-3ma and (Ra)-3pa with a slightly higher enantioselectivity, indicating again that the steric effect is not the major determining factor for ee (Entries 13–14, Table 2). Due to the unclosed nature of the malonate unit, these products 3aa-3pa could be used for further sewing of useful and versatile organosegment(s) to make new chiral allenes.
Next we turned to investigate the reaction of 2,3-allenylic carbonates with more sterically hindered 2-substituted malonates (Table 3). Generally, 2-substituted malonates gave the desired (Ra)-3 with higher ee values as expected, and synthetically attractive allyl, allenyl, propargyl, 3-phenylpropargyl are all compatible. The installation of such unsaturated C–C units in chiral (Ra)-3 elaborated the practical utilities of this approach via subsequent cycloaddition reaction, radical reaction, or cascade reaction, forming other chiral chemicals52,53,54.
Synthesis of racemic traumatic lactone
Such a method is applicable to the enantioselective synthesis of some naturally occurring allenes21,22,23. In order to show the potential in the syntheses of non-allene natural products, traumatic lactone was chosen as the target. In 1974, Masamune et al. isolated traumatic lactone from the roots of kidney bean (phaseolus vulgaris, beni-kintoki)55,56. The reported synthetic approaches to racemic traumatic lactone suffered from complicated starting materials, multi-steps, and harsh reaction conditions57,58,59. In addition, no enantioselective synthesis has been reported so far. Thus, we commenced to synthesize the racemic traumatic lactone (Fig. 3). Readily available racemic allenyl carbonate 1l49,50,51 reacted with malonate 2a under a set of similar reaction conditions at 25 °C with (±)-BINAP as the ligand to afford allene (±)-3la in 83% yield, which underwent alkaline hydrolysis, HOAc-mediated decarboxylation, and further alkaline hydrolysis to afford 4-allenoic acid (±)-4la in 78% yield. Then Au-catalyzed cycloisomerization of (±)-4la was applied to generate γ-butyrolactone bearing a trans C=C bond (±)-5la in 92% yield with 96:4 of E/Z selectivity24. After sequential hydrogenation of (±)-5la and Fe-TEMPO-catalyzed aerobic oxidation60,61 of the resulting hydrogenated product without purification, (±)-traumatic lactone was afforded in 79% yield.

The synthesis of (±)-traumatic lactone. Details of reagents, catalysts, solvent, and temperature are given in the reaction scheme
Enantioselective synthesis of (R)-traumatic lactone
Next, the current enantioselective protocol has been applied to the enantioselective synthesis of (R)-traumatic lactone (Fig. 4): Starting from readily available racemic allenyl carbonate 1j and malonate 2a under the optimal reaction conditions, allene (Ra)-3ja was produced with 91% ee in 84% yield, which was followed by alkaline hydrolysis and HOAc-mediated decarboxylation to afford 4,5-allenoic acid (Ra)-4ja in 91% yield. Then Au-catalyzed cycloisomerization of (Ra)-4ja generated γ-butyrolactone bearing a trans C=C bond (S,E)-5ja in 95% yield with 91% ee and 98:2 of E/Z selectivity24. After sequential hydrogenation of (S,E)-5ja and Fe-TEMPO-catalyzed aerobic oxidation60,61 of the resulting hydrogenated product without purification, (R)-traumatic lactone was afforded in 67% yield with 98% ee by one time recrystallization.

The first enantioselective synthesis of (R)-traumatic lactone. Details of reagents, catalysts, solvent, and temperature are given in the reaction scheme
Investigation of reaction mechanism
In order to rationalize the mechanism of this transformation, we conducted some control experiments (Fig. 5). A linear correlation between the ee of ligand and ee of 3aa was observed (Fig. 5a), which indicated that the active catalytic species was a monomeric palladium complex bearing a single chiral ligand62,63,64,65,66. When [Pd(π-cinnamyl)Cl)]2 with (R)-(−)-DTBM-SEGPHOS was stirred in THF for 2 h at 25 °C, after removal of THF via evaporation and recrystalization from CH2Cl2/n-hexane, (R)-(−)-DTBM-SEGPHOS·PdCl2·2CH2Cl2 was obtained as confirmed by X-ray diffraction study (Fig. 5b). However, no desired product was formed for the reaction of 1f and 2a with (R)-(−)-DTBM-SEGPHOS·PdCl2·2CH2Cl2 as the catalyst: 92% yield of 1f was recovered, indicating that (R)-(−)-DTBM-SEGPHOS·PdCl2·2CH2Cl2 is NOT the catalyst or pre-catalyst (Fig. 5c). On the other hand, the unique effect of the procedure for mixing these chemicals was re-confirmed (Fig. 5d). It has been reported that an allene unit may also function as ligand67,68. Thus, we reasoned that it is most likely that the allene unit in the stoichiometric amount of racemic 1f may compete with (R)-(−)-DTBM-SEGPHOS as ligand for extensive coordination with the palladium atom, leading to a lower ee (Procedure B). In fact, it was confirmed by the fact that a lower loading of the chiral ligand led to a lower ee (Fig. 5e). In addition, when Pd2(dba)3·CHCl3 was used instead of [Pd(π-cinnamyl)Cl)]2, the reaction gave a similar enantioselectivity albeit with a much lower yield, indicating that Pd(0) is indeed the catalytically active species and the in-situ generated Pd(0) is much more reactive69.

The studies on reaction mechanism. a The linear effect; b the complex formed from the Pd source and ligand; c the catalytic activity of this complex; d the effect of premixing; e confirming the role of the allene unit in the starting carbonate
Finally, a working model for the prediction of the axial chirality was proposed (Fig. 6). The monomeric palladium complex bearing a single chiral ligand, PdL*, would react with both (Ra)-1 and (Sa)-1 via SN2′-type oxidative addition from the back side of the C-OCO2Me bond to generate the same most favored E-η1-dienyl Pd, which would immediately yield the most favored delocalized intermediate α-alkynylidene-π-allyl palladium syn-η3-int A70 via the σ–π rearrangement involving the coordination of the terminal olefin with Pd. The front side of the terminal carbon atom in syn-η3-int A is blocked by L* ((R)-(−)-DTBM-SEGPHOS), thus, the malonate anion of 2 depronated by a base would attack from the back side the C–Pd bond in syn-η3-int A to genetate the chiral allene 3 in R configuration.

Asymmetric induction. Proposed mechanism and the model predicting the absolution configuration
Discussion
In conclusion, we have developed a non-substrate steric effect-based working strategy for the construction of allenes with non-sterically bulky substituents: due to the strong coordination ability of the allene unit with metals the exclusion of its extensive prior coordination with the in-situ generated Pd(0) should be avoid for the observed high enantioselectivity. We are actively extending this strategy to other challenging reactions constructing the allenes with less sterically bulky substituents.
Methods
The asymmetric synthesis of (R a)-3aa via Procedure C
To a dry Schlenk tube were added (R)-DTBM-SEGPHOS (70.5 mg, 0.06 mmol) and K2CO3 (276.2 mg, 2 mmol) in the glove box. Then [Pd(π-cinnamyl)Cl]2 (13.2 mg, 0.025 mmol) and 2a (263.8 mg, 2 mmol)/THF (3.5 mL) were added under nitrogen atmosphere sequentially. After being stirred at 25 °C for 30 min, the resulting mixture was stirred at 5 °C for another 10 min followed by the addition of 1a (226.2 mg, 1 mmol) and THF (1.5 mL) with stirring. After being stirred for 26 h at 5 °C, the reaction was complete as monitored by TLC. The resulting mixture was filtered through a short column of silica gel eluted with ethyl acetate (10 mL × 3). After evaporation, the residue was purified by flash column chromatography (eluent: petroleum ether (30–60 °C)/ethyl acetate = 30/1) on silica gel to afford (Ra)-3aa (217.6 mg, 77%) (eluent: petroleum ether/ethyl acetate = 30/1) as an oil: 90% ee (HPLC conditions: Chiralcel OD-H column, n-hexane/i-PrOH = 200/1, 0.5 mL/min, λ = 214 nm, tR(min) = 16.4 min, tR(major) = 17.9 min); [α]D20 = −54.2 (c = 1.08, CHCl3)); 1H NMR (300 MHz, CDCl3) δ 5.20–5.06 (m, 2H, CH=C=CH), 3.74 (s, 6H, 2 × OCH3), 3.51 (t, J = 7.4 Hz, 1H, CH), 2.61–2.54 (m, 2H, CH2), 2.00–1.90 (m, 2H, CH2), 1.43–1.19 (m, 10H, 5 × CH2), 0.88 (t, J = 6.5 Hz, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 204.0, 169.4, 169.3, 93.0, 87.3, 52.5, 51.3, 31.8, 29.12, 29.10, 29.08, 28.8, 28.0, 22.6, 14.1; IR (neat, cm−1) 2955, 2927, 2855, 1964, 1761, 1740, 1436, 1341, 1266, 1232, 1154, 1042; MS (EI, 70 eV) m/z (%) 282 (M+, 3.9), 138 (100); HRMS calcd. for C16H26O4 [M+]: 282.1831, found: 282.1830.
Data availability
All data that support the findings of this study are available in the online version of this paper in the accompanying Supplementary Information (including experimental procedures, compound characterization data).
References
Krause, N. & Hoffmann-Röder, A. Synthesis of allenes with organometallic reagents. Tetrahedron 60, 11671–11694 (2004).
Brummond, K. M. & DeForrest, J. E. Synthesizing allenes today (1982–2006). Synthesis 6, 795–818 (2007).
Ogasawara, M. Catalytic enantioselective synthesis of axially chiral allenes. Tetrahedron 20, 259–271 (2009).
Yu, S. & Ma, S. How easy are the syntheses of allenes? Chem. Commun. 47, 5384–5418 (2011).
Neff, R. K. & Frants, D. E. Recent advances in the catalytic syntheses of allenes: a critical assessment. ACS Catal. 4, 519–518 (2014).
Ye, J. & Ma, S. Conquering three-carbon axial chirality of allenes. Org. Chem. Front. 1, 1210–1224 (2014).
Chu, W., Zhang, Y. & Wang, J. Recent advances in catalytic asymmetric synthesis of allenes. Catal. Sci. Technol. 7, 4570–4579 (2017).
Lu, X., Zhang, C. & Xu, Z. Reactions of electron-deficient alkynes and allenes under phosphine catalysis. Acc. Chem. Res. 34, 535–544 (2001).
Bates, R. W. & Satcharoen, V. Nucleophilic transition metal based cyclization of allenes. Chem. Soc. Rev. 31, 12–21 (2002).
Ma, S. Some Typical advances in the synthetic applications of allenes. Chem. Rev. 105, 2829–2871 (2005).
Ma, S. Electrophilic addition and cyclization reactions of allenes. Acc. Chem. Res. 10, 1679–1688 (2009).
Alcaide, B., Almendros, P. & Aragoncillo, C. Exploiting [2+2] cycloaddition chemistry: achievements with allenes. Chem. Soc. Rev. 39, 783–816 (2010).
Krause, N. & Winter, C. Gold-catalyzed nucleophilic cyclization of functionalized allenes: a powerful access to carbo- and heterocycles. Chem. Rev. 111, 1994–2009 (2011).
Yu., S. & Ma, S. Allenes in catalytic asymmetric synthesis and natural product syntheses. Angew. Chem. Int. Ed. 51, 3074–3112 (2012).
Adams, C. S., Weatherly, C. D., Burke, E. G. & Schomaker, J. M. The conversion of allenes to strained three-membered heterocycles. Chem. Soc. Rev. 43, 3136–3163 (2014).
Tius, M. A. Allene ether Nazarov cyclization. Chem. Soc. Rev. 43, 2979–3002 (2014).
Zimmer, R. & Reissig, H. Alkoxyallenes as building blocks for organic synthesis. Chem. Soc. Rev. 43, 2888–2903 (2014).
Hoffmann-Röder, A. & Krause, N. Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. Int. Ed. 43, 1196–1216 (2004).
Ogasawara, M., Nagano, T. & Hayashi, T. A new route to methyl (R,E)-(-)-tetradeca-2,4,5-trienoate (Pheromone of Acanthoscelides obtectus) utilizing a palladium-catalyzed asymmetric allene formation reaction. J. Org. Chem. 70, 5764–5767 (2005).
Crouch, I. T., Neff, R. K. & Frantz, D. E. Pd-catalyzed asymmetric β-hydride elimination en route to chiral allenes. J. Am. Chem. Soc. 135, 4970–4973 (2013).
Tang, X. et al. CuBr2-catalyzed enantioselective routes to highly functionalized and naturally occurring allenes. Org. Chem. Front. 2, 688–691 (2015).
Jiang, X., Xue, Y. & Ma, S. Aerobic oxidation and EATA-based highly enantioselective synthesis of lamenallenic acid. Org. Chem. Front. 4, 951–957 (2017).
Yu, Q. & Ma, S. An enantioselective synthesis of (R)-5,6-octadecadienoic acid. Eur. J. Org. Chem. 2015, 1596–1601 (2015).
Zhou, J., Fu, C. & Ma, S. Gold-catalyzed stereoselective cycloisomerization of allenoic acids for two types of common natural γ-butyrolactones. Nat. Commun. 9, 1654–1664 (2018).
Huang, X. et al. General CuBr2-catalyzed highly enantioselective approach for optically active allenols from terminal alkynols. Chem. Commun. 51, 6956–6959 (2015).
Ma, D., Duan, X., Fu, C., Huang, X. & Ma, S. Dimethylprolinol versus diphenylprolinol in CuBr2-catalyzed enantioselective allenylation of terminal alkynols. Synthesis 50, 2533–2545 (2018).
Ogasawara, M., Ikeda, H., Nagano, T. & Hayashi, T. Palladium-catalyzed asymmetric synthesis ofaxially chiral allenes: a synergistic effect of dibenzalacetone on high enantioselectivity. J. Am. Chem. Soc. 123, 2089–2090 (2001).
Imada, Y., Ueno, K., Kutsuwa, K., & Murahashi, S. I. Palladium-catalyzed asymmetric alkylation of 2,3-alkadienyl phosphates. synthesis of optically active 2-(2,3-alkadienyl)malonates. Chem. Lett. 31, 140–141 (2002).
Trost, B. M., Fandrick, D. R. & Dinh, D. C. Dynamic kinetic asymmetric allylic alkylations of allenes. J. Am. Chem. Soc. 127, 14186–14187 (2005).
Ogasawara, M., Ueyama, K., Nagano, T., Mizuhata, Y. & Hayashi, T. Palladium-catalyzed asymmetric synthesis of axially chiral (allenylmethyl)silanes and chirality transfer to stereogenic carbon centers in SE′ reactions. Org. Lett. 5, 217–219 (2003).
Ogasawara, M., Ngo, H. L., Sakamoto, T., Takahashi, T. & Lin, W. Applications of 4,4′-(Me3Si)2-BINAP in transition-metal-catalyzed asymmetric carbon-carbon bond-forming reactions. Org. Lett. 7, 2881–2884 (2005).
Dai, J., Duan, X., Zhou, J., Fu, C. & Ma, S. Catalytic enantioselective simultaneous control of axial chirality and central chirality in allenes. Chin. J. Chem. 36, 387–391 (2018).
Ogasawara, M., Ge, Y., Uetake, K. & Takahashi, T. Vinyl ketones to allenes: preparation of 1,3-Dien-2-yl triflates and their application in Pd-catalyzed reactions with soft nucleophiles. Org. Lett. 7, 5697–5700 (2005).
Imada, Y., Nishida, M., Kutsuwa, K., Murahashi, S. & Naota, T. Palladium-catalyzed asymmetric amination and imidation of 2,3-allenyl phosphates. Org. Lett. 7, 5837–5839 (2005).
Imada, Y., Nishida, M. & Naota, T. Sequential asymmetric homoallenylation of primary amines with a palladium catalyst. Tetrahedron Lett. 49, 4915–4917 (2008).
Nemoto, T., Kanematsu, M., Tamura, S. & Hamada, Y. Palladium-catalyzed asymmetric allylic alkylation of 2,3-allenyl acetates using a chiral diaminophosphine oxide. Adv. Synth. Catal. 351, 1773–1778 (2009).
Wu, Z. et al. Palladium-catalyzed efficient enantioselective synthesis of chiral allenes: steric and electronic effects of ligands. ACS Catal. 4, 44–48 (2014).
Liu, H., Leow, D., Huang, K. W. & Tan, C. H. Enantioselective synthesis of chiral allenoates by guanidine-catalyzed isomerization of 3-alkynoates. J. Am. Chem. Soc. 131, 7212–7213 (2009).
Inokuma, T. et al. Bifunctional hydrogen-bond donors that bear a quinazoline or benzothiadiazine skeleton for asymmetric organocatalysis. Chem. Eur. J. 17, 10470–10477 (2011).
Nishimura, T., Makino, H., Nagaosa, M. & Hayashi, T. Rhodium-catalyzed enantioselective 1,6-addition of arylboronic acids to enynamides: asymmetric synthesis of axially chiral allenylsilanes. J. Am. Chem. Soc. 132, 12865–12867 (2010).
Li, C. et al. Iron porphyrin-catalyzed olefination of ketenes with diazoacetate for the enantioselective synthesis of allenes. J. Am. Chem. Soc. 129, 1494–1495 (2007).
Wang, Y., Zhang, W. & Ma, S. A room-temperature catalytic asymmetric synthesis of allenes with ECNU-Phos. J. Am. Chem. Soc. 135, 11517–11520 (2013).
Hashimoto, T., Sakata, K., Tamakuni, F., Dutton, M. J. & Maruoka, K. Phase-transfer-catalysed asymmetric synthesis of tetrasubstituted allenes. Nat. Chem. 5, 240–244 (2013).
Yao, Q. et al. Efficient synthesis of chiral trisubstituted 1,2-allenyl efficient synthesis of chiral trisubstituted 1,2-allenyl ketones by catalytic asymmetric conjugate addition of malonic esters to enynes. Angew. Chem. Int. Ed. 55, 1859–1863 (2016).
Liu, Y. et al. Synergistic kinetic resolution and asymmetric propargyl claisen rearrangement for the synthesis of chiral allenes. Angew. Chem. Int. Ed. 55, 4054–4058 (2016).
Tap, A., Blond, A., Wakchaure, V. N. & List, B. Chiral allenes via alkynylogous mukaiyama aldol reaction. Angew. Chem. Int. Ed. 55, 8962–8965 (2016).
Chu, W. D. et al. Enantioselective synthesis of trisubstituted allenes via Cu(I)-catalyzed coupling of diazoalkanes with terminal alkynes. J. Am. Chem. Soc. 138, 14558–14561 (2016).
Poh, J. S. et al. Rapid asymmetric synthesis of disubstituted allenes by coupling of flow-generated diazo compounds and propargylated amines. Angew. Chem. Int. Ed. 56, 1864–1868 (2017).
Kuang, J., Luo, H. & Ma, S. Copper (I) iodide-catalyzed one-step preparation of functionalized allenes from terminal alkynes: amine effect. Adv. Synth. Catal. 354, 933–944 (2012).
Li, Q., Fu, C. & Ma, S. Catalytic asymmetric allenylation of malonates with the generation of central chirality. Angew. Chem. Int. Ed. 51, 11783–11786 (2012).
Zhu, T. & Ma, S. 3,4-Alkadienyl ketones via the palladium-catalyzed decarboxylative allenylation of 3-oxocarboxylic acids. Chem. Commun. 53, 6037–6040 (2017).
Grissom, J. W., Klingberg, D., Huang, Da & Slattery, B. J. Tandem enyne allene-radical cyclization: low-temperature approaches to benz[e]indene and indene compounds. J. Org. Chem. 62, 603–626 (1997).
Aubert, C., Fensterbank, L., Garcia, P., Malacria, M. & Simonneau, A. Transition metal catalyzed cycloisomerizations of 1,n-allenynes and -allenenes. Chem. Rev. 111, 1954–1993 (2011).
Neff, R. K. & Frantz, D. E. Recent applications of chiral allenes in axial-to-central chirality transfer reactions. Tetrahedron 71, 7–18 (2015).
Gunstone, F. D. Fatty acids. Part I. 9-hydroxyoctadec-12-enoic acid,* a new hydroxy-acid occurring in strophanthus sarmentosus seed oil. J. Chem. Soc. 1952, 1274–1278 (1952).
Takasugi, M., Anetai, M., & Masamune, T. 9,10,13-Trihydroxy-11,15-octadecadienoic acid and related fatty acids in the roots of kidney bean. Chem. Lett. 3, 947–950 (1974).
Thakur, S. B., Jadhav, K. S. & Bhattacharyya, S. C. Simple synthesis of 2-(ω-carboxyhexyl)cyclopentenone, a prostaglandin synthon. Indian. J. Chem. 12, 893–894 (1974).
Deodhar, V. B., Dalavoy, V. S. & Nayak, U. R. A simple preparation of traumatic lactone. A prostanoid synthon. Org. Prep. & Proced. 9, 155–157 (1977).
Zhu, X. & Sayre, L. M. Long-lived 4-oxo-2-enal-derived apparent lysine michael adducts are actually the isomeric 4-ketoamides. Chem. Res. Toxicol. 20, 165–170 (2007).
Jiang, X., Zhang, J. & Ma, S. Iron catalysis for room-temperature aerobic oxidation of alcohols to carboxylic acids. J. Am. Chem. Soc. 138, 8344–8347 (2016).
Jiang, X. et al. Iron-catalyzed catalysis aerobic oxidation of aldehydes: single component catalyst and mechanism studies. Chin. J. Chem. 36, 15–19 (2018).
Wynberg, H. & Feringa, B. Enantiomeric recognition and interactions. Tetrahedron 32, 2831–2834 (1976).
Puchot, C. et al. Nonlinear effects in asymmetric synthesis. examples in asymmetric oxidations and aldolization reactions. J. Am. Chem. Soc. 108, 2353–2357 (1986).
Oguni, N., Matsuda, Y. & Kaneko, T. Asymmetric amplifying phenomena in enantioselective addition of diethylzinc to benzaldehyde. J. Am. Chem. Soc. 110, 7877–7878 (1988).
Girard, C. & Kagan, H. B. Nonlinear effects in asymmetric synthesis and stereoselective reactions: ten years of investigation. Angew. Chem. Int. Ed. 37, 2922–2959 (1998).
Shi, M. & Zhang, W. Asymmetric 1,4-addition of diethylzinc to cyclic enones catalyzed by Cu(I)-chiral sulfonamide-thiophosphoramide ligands and lithium salts. Adv. Synth. Catal. 347, 535–540 (2005).
Sato, I. et al. Highly enantioselective asymmetric autocatalysis of pyrimidin-5-yl alkanol induced by chiral 1,3-disubstituted hydrocarbon allenes. Helv. Chim. Acta 85, 3383–3387 (2002).
Cai, F. et al. Chiral allene-containing phosphines in asymmetric catalysis. J. Am. Chem. Soc. 133, 18066–18069 (2011).
Amatore, C., Jutand, A., M’Barki, M. A., Meyer, G., & Mottier, L. Importance of the presence of chloride ions in the first steps of palladium catalyzed nucleophilic allylic substitutions. Eur. J. Inorg. Chem. 2001, 873–880 (2001).
Ogasawara, M. et al. Synthesis, structure, and reactivity of (1,2,3-η3-butadien-3-yl)palladium complexes. Organometallics 26, 5025–5029 (2007).
Acknowledgements
Financial support from National Basic Research Program (21572202, 21690063 and 21772172) is greatly appreciated. Shengming Ma is a Qiu Shi Adjunct Professor at Zhejiang University. We thank Mr. Huanan Wang in our group for reproducing the results: (Ra)-3ha, (Ra)-3ka, and (Ra)-3nd.
Author information
Authors and Affiliations
Contributions
S.M. directed the research and developed the concept of the reaction with S.S., who also performed the experiments and prepared the Supplementary Information. S.S., Z.J., C.F. and S.M. checked the experimental data. S.S. and S.M. wrote the manuscript with contributions from the other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, S., Zhou, J., Fu, C. et al. Catalytic enantioselective construction of axial chirality in 1,3-disubstituted allenes. Nat Commun 10, 507 (2019). https://doi.org/10.1038/s41467-018-07908-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-07908-1
This article is cited by
-
Nickel-electrocatalysed C(sp3)–C(sp3) cross-coupling of unactivated alkyl halides
Nature Catalysis (2024)
-
Asymmetric α-allylic allenylation of β-ketocarbonyls and aldehydes by synergistic Pd/chiral primary amine catalysis
Nature Communications (2023)
-
Photo and copper dual catalysis for allene syntheses from propargylic derivatives via one-electron process
Nature Communications (2022)
-
Palladium-catalyzed allene synthesis enabled by β-hydrogen elimination from sp2-carbon
Nature Communications (2021)
-
Tetrasubstituted allenes via the palladium-catalysed kinetic resolution of propargylic alcohols using a supporting ligand
Nature Catalysis (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
