
Abstract
The type VI secretion system (T6SS) is a multi-protein complex that injects bacterial effector proteins into target cells. It is composed of a cell membrane complex anchored to a contractile bacteriophage tail-like apparatus consisting of a sharpened tube that is ejected by the contraction of a sheath against a baseplate. We present structural and biochemical studies on TssA subunits from two different T6SSs that reveal radically different quaternary structures in comparison to the dodecameric E. coli TssA that arise from differences in their C-terminal sequences. Despite this, the different TssAs retain equivalent interactions with other components of the complex and position their highly conserved N-terminal ImpA_N domain at the same radius from the centre of the sheath as a result of their distinct domain architectures, which includes additional spacer domains and highly mobile interdomain linkers. Together, these variations allow these distinct TssAs to perform a similar function in the complex.
Similar content being viewed by others


Introduction
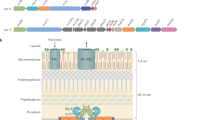
Contractile bacteriophages of the family Myoviridae (i.e. T4), R-type pyocins and the type VI secretion system (T6SS) of Gram-negative bacteria are evolutionarily related nano-scale injection machines that puncture target cell membranes using a shared contraction mechanism1,2,3. These injection devices are comprised of an inner tube, surrounded by a contractile sheath, that are both assembled on a platform known as the baseplate. The inner tube is sharpened with spike proteins at the baseplate proximal end, which facilitates its penetration of target cells upon contraction of the sheath against the baseplate2,3,4,5.
The T6SS secretion machinery is formed from multiple copies of 12 core subunits (TssA-TssG, TssI-TssM) and a single PAAR tip protein6,7,8,9 and can be subdivided into two main components. One of these, the membrane complex, consists of 10 subunits each of TssJ, TssL, and TssM that assemble into a chamber-like structure with five-fold symmetry which serves to anchor the injection machinery at the cell envelope as well as providing an exit channel for translocated subunits and effectors10,11,12,13,14,15. The other component, the injection machinery, consists of two sub-complexes. One sub-complex consists of the inner tube, which is comprised of stacked hexameric rings of TssD (Hcp), capped by the trimeric hub protein, TssI (VgrG), and sharpened by the PAAR subunit, surrounded by repeating TssBC heterodimers that form the contractile sheath1,3,5,16,17. The latter consists of a six-start helix that possesses six-fold symmetry, giving a cogwheel-like appearance when viewed end-on1,18,19,20,21. Both the inner tube and sheath exhibit the same degree of helical twist thereby ensuring a six-fold symmetry match along the entire length of the tube-sheath complex21. The other sub-complex is the baseplate, which consists of TssE, TssF, TssG and TssK, and contains a central channel through which the sharpened inner tube passes upon contraction of the sheath3,17,22,23,24. The sheath is subsequently recycled by the AAA+ ATPase, TssH (ClpV)1,18,25.
Until recently, relatively little was known about the location and role of the TssA subunit within the T6SS complex. TssA subunits are enigmatic as they possess a conserved N-terminal region of unknown function, previously identified as ImpA_N (PFAM: PF0681226), whereas sequences located C-terminal to this region are highly divergent6,27,28. Consistent with this, phylogenetic analysis has suggested that the TssA family can be subdivided into three clades (TssA1, TssA2 and TssA3)28. The C-terminal regions of TssA1 and TssA2 have been shown to be required for assembly of these TssA subunits into higher order oligomers and both subunits are required for T6SS function27,28. However, the TssA3 subunit has not been previously investigated.
Recent studies on the TssA2 subunit of enteroaggregative Escherichia coli (EAEC), Ec042_4540, have provided structures for two of its putative three domains (the middle (Nt2) and the C-terminal domain (CTD)), leaving the structure of the highly conserved N-terminal domain (Nt1), yet to be determined. These structural studies showed that the CTD assembles into a dodecamer that resembles a six-pointed star. Further analysis showed that TssA2 interacts with components of the baseplate, inner tube, sheath and the T6SS membrane complex27. This led to the proposal of a capping model whereby TssA2 initially interacts with the core TssJLM membrane complex, thereby triggering baseplate recruitment. According to the model, TssA2 subsequently serves to coordinate the assembly of the inner tube and contractile sheath, during which it migrates away from the baseplate complex, remaining in contact with the distal end of the polymerising tube27,29. In a separate study, the TssA1 subunit of Pseudomonas aeruginosa, PA0082, was shown to form a ring-like complex of indeterminate stoichiometry and symmetry, and in contrast to TssA2, was proposed to serve as the T6SS counterpart of the phage T4 baseplate protein gp6, in a baseplate-associated model of TssA function28. However, in the absence of a high-resolution structure for TssA1 or TssA3, any relationship between their CTDs and the Nt2 and CTDs of TssA2 subunits are, as yet, unclear.
In this paper, we present evidence that there are only two main TssA clades (TssA1 and TssA2) and that each clade can be sub-divided into two distinct sub-clades, referred to here as A and B. We provide the first structure for any TssA1 subunit (TssA1B) to show that it is composed of two domains instead of three: the conserved N-terminal domain (Nt1), containing the ImpA_N region, that is tethered by a long linker to a distinct CTD that assembles into a ring containing 32 subunits with 16-fold symmetry. Furthermore, we present the first T6SS subunit interaction analysis of a TssA1B member. We also provide the structure of an Nt2-CTD TssA2A construct, demonstrating that 10 subunits assemble into a complex with distinct five-fold symmetry rather than the six-fold symmetry seen in EAEC TssA2B. The structure further reveals a striking range of conformational mobility of the TssA2A Nt2 domain with respect to the CTD oligomer. Despite the differences in symmetry of TssAs belonging to the different clades, the similar subunit interaction network and disposition of their conserved domains suggests that they function by a related mechanism to coordinate inner tube/sheath assembly.
Results
Bioinformatic and proteolytic analysis of TssA family proteins
Bioinformatic analysis suggests that all TssA subunits contain an N-terminal conserved region of ~120 amino acids (ImpA_N) composed of three shorter motifs of significant sequence similarity (ImpA_N1–3) (Fig. 1a and Supplementary Fig. 1). However, these proteins possess one of two distinct C-terminal regions based on amino acid sequence conservation, and are referred to here as TssA1 and TssA2 (Fig. 1a and Supplementary Fig. 2). In members of the TssA1 clade, ImpA_N forms part of a large N-terminal domain (Nt1) of ~250 amino acids which is connected by a central region of variable length (40–60 amino acids) and sequence to a conserved C-terminal region of 60–75 amino acids containing a signature EPxxP motif (Supplementary Fig. 2a). Phylogenetic analysis shows that TssA1 subunits fall into one or other of two sub-clades, referred to here as A and B (Fig. 1b). In TssA1A members, such as P. aeruginosa PA0082 (Pa TssA1A), the EPxxP motif is EPSxP, whereas in TssA1B members, such as B. cenocepacia I35_RS01755 (Bc TssA1B), an alternative EP(H/Q)SP motif is present (Supplementary Fig. 2a). TssA1A corresponds to TssA1 in the previously proposed nomenclature28, whereas TssA1B corresponds to TssA3. In comparison, TssA2 orthologues are longer (480–540 amino acids) and are predicted to contain three domains: Nt1 (including the ImpA_N region), a middle domain (Nt2) and CTD, each separated by regions of variable length and sequence (Fig. 1a and Supplementary Fig. 1 and 2b). However, our phylogenetic analysis reveals that TssA2 proteins are also subdivided into two sub-clades (Fig. 1b) with members of sub-clade B, represented by the EAEC TssA2 subunit (EAEC TssA2B) having an extension of 20–40 amino acids at the C-terminus compared to sub-clade A members, such as A. hydrophila AHA1844 (Ah TssA2A).

Phylogeny and domain organisation of TssA family proteins. a Proposed domain organisation of TssA1A, TssA1B, TssA2A and TssA2B subunits based on structural studies, secondary structure prediction and amino acid sequence alignment. The interdomain linkers comprise regions of variable length and amino acid sequence. The N-terminal conserved region, ImpA_N (comprised of three major regions of conservation, ImpA_N1-3), Nt1, Nt2, CTD and EX (C-terminal extension present in TssA2B proteins) are indicated. Scale bar corresponds to 50 amino acids. b Maximum-likelihood phylogenetic tree from amino acid sequences of 47 TssA family members, showing the subdivision into three major clades as observed previously 28but with a revised nomenclature. The TssA2 clade is further divided into sub-clades A and B, based on patterns of sequence identity. TssA members discussed in detail in this study are highlighted in dark yellow. Scale bar represents 0.2 substitutions per site
To provide support for the bioinformatics, analysis of protein fragments derived by either limited or adventitious proteolysis was used to identify putative domain boundaries. Analysis of Bc His6.TssA1B degradation products suggested that TssA1B possesses a large folded Nt1 domain (~31 kDa) connected to a small CTD (~8.5 kDa) by an interdomain linker of ~40 amino acids (Supplementary Fig. 3a, c, and d, Supplementary Table 1).
Products of limited proteolysis carried out on purified Ah His6.TssA2A showed that residues in regions corresponding to positions R178-G190 and K378-E399, two regions of high sequence variability, were particularly susceptible to protease cleavage. This is consistent with the predicted three-domain architecture with approximate boundaries of M1-R178 for Nt1, D231-L374 for Nt2, and I402-K478 for the CTD (Supplementary Fig. 3b and e).
Interactions of TssA1B and its domains with other T6SS components
The interactions of a TssA1B subunit with other components of the T6SS have not been previously investigated. To address this, a two-hybrid analysis was performed. Complementation of CyaA function was assayed on maltose MacConkey indicator plates and by performing β-galactosidase assays, both of which indicated that TssA1B may interact with TssC, Hcp, TssE, TssF, the conserved core region of VgrG (VgrGC), TssK, TssL and the N-terminal cytoplasmic region of TssM (TssMN) (Fig. 2a, b). To validate these results, co-IP experiments were performed using FLAG-tagged TssA1B in pairwise combinations with other epitope-tagged subunits. The results supported the in vivo observations. Thus, epitope-tagged TssC, Hcp, TssF, VgrGC, TssK, TssL and TssMN were all co-immunoprecipitated with FLAG.TssA1B (Fig. 2c). Demonstration of an interaction between TssA1B and TssE required solubilisation of TssE by fusion to the MBP solubility tag. In summary, the protein-protein interaction analysis suggests that TssA1B makes contact with components of the baseplate, the membrane-anchoring complex, the inner tube and its cap, and the surrounding sheath (Fig. 2d). Two-hybrid analysis was then performed to investigate the role of the N- and C-terminal regions of TssA1B in interactions with other T6SS subunits. This showed that TssA1B CTD interacts with Hcp, TssF and VgrG (and also with VgrGC) but interactions with other subunits were not observed for the Nt1 domain (Supplementary Fig. 6).

Interaction of TssA1B with other T6SS subunits. a Two-hybrid analysis (maltose phenotypes). Hybrid proteins are represented by a green or yellow coloured motif representing the CyaA fragment (T25 and T18, respectively) linked to a white rectangle labelled according to the fused T6SS subunit, as shown in the key at the bottom of (b). T6SS subunits are indicated by a single letter corresponding to the suffix used in the Tss nomenclature (i.e. A1 corresponds to TssA1B, B corresponds to TssB etc) except for H (Hcp), V (VgrG) and VC (VgrG core region). MN and MC represent the N-terminal cytoplasmic and C-terminal periplasmic regions of TssM, respectively. The efficiency of complementation of representative combinations (phenotypes shown in red font) were determined by β-galactosidase assay. b Two-hybrid analysis (β-galactosidase activity). Data is representative of three independent experiments (n = 3) performed in duplicate and values correspond to the mean ± standard deviation. Nomenclature as in (a). Z represents the Zip control. Values are presented in Supplementary Data 1. c Co-immunoprecipitation analysis. FLAG-tagged Bc TssA1B and potential interacting T6SS subunits (TssX) tagged with the VSV-g or HA epitope tags, or with MBP, were co-expressed in E. coli. FLAG.TssA1B was immunoprecipitated from cell lysates and recovered prey proteins (IP) were detected with the appropriate epitope antibody by western blotting. MBP was included as a control and was detected with MBP pAb. Proteins present in the cell lysate (Tot) and the unbound material (UB) were also analysed. Blots were also probed with FLAG mAb (an example of such a blot following co-expression of FLAG.TssA1B and VSVg.TssMN is shown in the bottom panel). Uncropped images of the blots are shown in Supplementary Fig. 4. d Bc TssA1B interaction network. Interactions between Bc TssA1B and other T6SS subunits are indicated by red arrows. Previously reported interactions are indicated by grey connectors. Note that VgrG and TssK have been shown to interact with the TssF-TssG complex22 but for simplicity are linked to TssF in this figure
TssA1B is an essential component of the T6SS
P.a TssA1A, V. cholerae TssA2A (VCA0119) and EAEC TssA2B have been shown to be essential for T6SS activity27,28,30. Therefore, the requirement for TssA1B for a fully functional T6SS was explored by assaying the ability of a B. cenocepacia tssA1B mutant to secrete Hcp—an indicator of T6SS activity. The results showed that, unlike the wild-type strain, Hcp was absent from the culture supernatant of the tssA1B mutant, but it was secreted when tssA1B mutant bacteria contained a plasmid expressing tssA1B (Fig. 3), thereby confirming the requirement for this subunit for a functional T6SS31.

Effect of TssA1B inactivation on T6SS secretion activity. Cell associated protein (CA) and spent supernatants (SN) from cultures of B. cenocepacia H111 (WT) and the isogenic tssA1B mutant (tssA::Tp), each with or without pBBR1MCS (VC) or pBBR1MCS-tssA1B (ptssA), were fractionated by SDS-PAGE, blotted onto PVDF membrane and probed with a Hcp pAb or a mAb specific for the RNAP β subunit (lysis control). Detection of bound antibodies was with HRP-conjugated secondary antibodies. The tssM mutant (ΔM) was included as a control as TssM has been shown to be an essential component of the B. cenocepacia T6SS31. Uncropped images of the blots are shown in Supplementary Fig. 5
Structure determination of the TssA1B Nt1 domain
SEC and two-hybrid analysis of the overexpressed Bc TssA1B Nt1 domain indicated that it is monomeric (Supplementary Fig. 7a, b). Analysis of the crystal structure of the TssA1B Nt1 domain, determined to 1.80 Å (Supplementary Table 2), revealed a monomer composed of 11 α-helices that are organised into two subdomains, an N-terminal subdomain 1 (Sd1) connected to a larger subdomain 2 (Sd2) (Fig. 4a, b). Sd1 (M1-P113) is comprised of five antiparallel α-helices (α1–α5) and contains the ImpA_N1 (L8-I31) and the ImpA_N2 (D55–P113) motifs (Fig. 4a–c).

High-resolution structure of Bc TssA1B Nt1 and its similarity to Ah TssA2 Nt2. a Bc TssA1B Nt1 sequence annotated to show the elements of secondary structure. Location of α-helices are shown by coloured rectangles above the amino acid sequence with the region corresponding to Sd1 highlighted in yellow and Sd2 highlighted in red. The three ImpA_N motifs are indicated in orange font and conserved ImpA_N domain residues (see Supplementary Fig. 1) are shown in black boxes. b X-ray structure of Bc TssA1B Nt1 (PDB: 6HS5 https://www.ebi.ac.uk/pdbe/entry/pdb/6hs5). The two subdomains, Sd1 and Sd2, are shown in yellow and red, respectively. c X-ray structure of Bc TssA1B Nt1 represented as in (b) showing location of ImpA_N1-ImpA_N3 regions in orange. For clarity, the helices of Sd2 (α6-α11) located C-terminal to ImpA_N3 are made more transparent than those in Sd1. d Bc TssA1B Sd1 (orange) superimposed on the Ah TssA2A Nt2 domain (PDB: 6G7B https://www.ebi.ac.uk/pdbe/entry/pdb/6g7b; cyan) showing the close structural similarity of helices α1-α5 of Bc TssA1B Nt1 (majority of ImpA_N) to helices α2-α6 of Ah TssA2A Nt234. α-helices are shown as cylinders
Sd2 of Bc TssA1B (L114-Q251) contains six α-helices (α6–α11), of which the three longest (α6, α10 and α11) form a five-helix bundle with α4–α5 of Sd1. Residues from ImpA_N3 (R123–R133) (Fig. 4a–c) form the start of α6, a helix which includes a sharp kink between L124 and G125. The C-terminal helix of Sd2 (α11) terminates at Q251 and is thereafter followed by the connecting interdomain linker to the CTD. The sequence conservation in the ImpA_N region across the TssA clades (Supplementary Fig. 1) implies that all Nt1 domains are closely related and adopt a similar fold, which includes the entirety of Sd1, and the N-terminal part of the first helix of Sd2 of Bc TssA1B. Mapping of the 34 conserved residues in the ImpA_N region onto the structure of Nt1, shows that approximately two-thirds are involved in packing interactions in the core, with the remainder located predominantly at one end of the molecule in the loops between helices α1–α2, α3–α4, and α5–α6 (Fig. 4a, c).
TssA1B CTD forms a double-layered ring with D16 symmetry
SEC-MALLS analysis of native Bc TssA1B indicated an apparent molecular mass of 1.24 MDa (Supplementary Fig. 7c), suggesting that the TssA1B oligomer contains ~30 subunits, and negative stain EM revealed rosette-like structures in which globular domains were irregularly distributed around a central ring (Fig. 5a). Two-hybrid analysis demonstrated that oligomerisation of TssA1B required the CTD (Supplementary Fig. 7b), suggesting that the inner ring is composed of polymerised CTDs. This was confirmed by negative stain EM of the isolated CTD which was observed to form rings without peripherally located globular domains (Supplementary Fig. 7e).

Structure of the Bc TssA1B CTD ring. a Negative stain EM of TssA1B particles. The magnified view indicates the location of the Nt1 domains (arrows) presented around the CTD oligomeric ring (circle). Scale bar corresponds to 50 nm. b Bc TssA1B CTD monomers assemble to form a double-layered ring containing 32 subunits exhibiting D16 symmetry (PDB: 6HS6 [https://www.ebi.ac.uk/pdbe/entry/pdb/6hs6]). The views shown correspond to a top view down the 16-fold axis and a side view perpendicular to the 16-fold axis, with the dimensions of the ring shown. c, d Dimerisation of Bc TssA1B CTD monomers. c Side view from inside the Bc TssA1B CTD ring showing interaction of residues from helices α12, α13 and α14 within a dimer (chain A, purple; chain B, cyan). Interacting surfaces are highlighted (chain A, light pink; chain B, blue). d Formation of the salt bridge between R306 and E324 that caps both ends of the dimer interface. e, f Interactions between dimers to form the Bc TssA1B CTD ring. e Expanded view of the two chains shown in purple and orange in (b), to show interactions leading to ring assembly. f Side view from inside the Bc TssA1B CTD ring showing the interlocking interaction between helices α13, α14 and α15 from two dimers which are critical to ring formation around the 16-fold axis: dimer 1, purple/cyan; dimer 2, blue/orange. The interfacing regions of the purple and orange monomers are shown in light pink and yellow, respectively. g Structure-based sequence alignment of the Pa TssA1A and Bc TssA1B CTD domains. The secondary structure of TssA1B CTD is highlighted above the sequence75
The structure of a Bc TssA1B CTD domain construct (I303-S373) was determined to 3.08 Å (Fig. 5b and Supplementary Table 3) and the asymmetric unit was observed to contain eight CTD monomers (chains A–H) (Supplementary Fig. 8a–c). Each chain folds into four α-helices (α12-α15) that lie in approximately the same plane, and form two antiparallel pairs (Fig. 5e). The eight monomers in the asymmetric unit further assemble into a higher order oligomer of 32 subunits, consistent with the SEC-MALLS analysis of TssA1B and TssA1B CTD oligomers (Supplementary Fig. 7c, d), with D16 symmetry using the crystallographic two-fold axes to make up a double-layered ring with 16 subunits in each layer (Fig. 5b and Supplementary Figure 8a). In the assembled Bc TssA1B CTD ring, analysis by PISA32 revealed an extensive dimer interface (~950 Å2) between monomers in the different layers, which principally involves the interaction of residues located in helices α12–α13 with their two-fold related subunits forming a region of hydrophobic packing with hydrophilic interactions at the periphery (Fig. 5c, d). Further less extensive interactions (~570 Å2) were identified between dimers that facilitate formation of the ring, with each pair of helices α14-α15 in one dimer, together with a small number of residues from α13, interacting with their equivalents in adjacent 16-fold related dimers on either side (Fig. 5e, f). This type of interlocking series of interactions is repeated in a cyclic arrangement to form a double-layered ring (Supplementary Fig. 8d). Nested deletion of consecutive helical segments of TssA1B CTD revealed that loss of α15 resulted in the failure of the protein to assemble into a high molecular weight ring, and structure determination of the construct H12–H14 (helices α12–α14) and of that consisting of α12 and α13 alone (residues 303–347), revealed them both to be dimers (Supplementary Figure 8e–h and Supplementary Tables 4 and 5).
In the crystal structure of the TssA1B CTD oligomer, the double ring is ~25 Å thick with a lumenal diameter of ~110 Å and an outer diameter of ~200 Å (Fig. 5b), in agreement with the dimensions of Bc TssA1B and purified CTD oligomers determined by negative stain EM analysis (Supplementary Fig. 7e, f). The C-termini of the TssA1B CTD monomers are located at the inner face of the ring, while their N-termini are orientated on the outside, consistent with EM analysis, which places the Nt1 domain at the periphery of the ring in the intact complex (Fig. 5a). The irregular arrangement of the globular domains and the poor resolution of the region linking them to the inner ring are consistent with the presence of a flexible linker connecting the Nt1 domain to the CTD. Analysis of the CTD sequences of Bc TssA1B and Pa TssA1A identified 17 conserved residues, of which 11 are involved in interactions around the subunit interfaces, including the highly conserved EPxxP motif (Fig. 5g and Supplementary Fig. 2a). The Bc TssA1B CTD structure shows that this motif defines a loop that connects α12 and α13, and includes a salt bridge between E324 from this motif and a conserved arginine, R306, from a symmetry-related subunit (Fig. 5d). The conservation of these residues suggests that the quaternary structures of TssA1A and TssA1B are related. Moreover, although it has been previously proposed that the C-terminal region of Pa TssA1A is structurally similar to the C-terminal part of the gp6 subunit of the T4 phage baseplate on the basis of low level sequence similarity28, the similarity in sequence between TssA1A and TssA1B and the difference in structure of the TssA1B CTD to gp6 indicates that this is not the case.
TssA2A Nt2 is structurally similar to the TssA1B Nt1 domain
The structure of an Ah TssA2A Nt2 domain construct (D231-L374) was solved to 1.76 Å with four subunits (chains A–D) in the asymmetric unit (Supplementary Table 6) arranged as two independent dimers, consistent with previous SEC analysis33, that showed that Ah TssA2A Nt2 exists as dimers in solution. Each monomer folds into a cluster of seven α-helices (α1–α7), the arrangement of which is very similar to that of the EAEC TssA2B Nt2 domain (RMSD = 2.0 Å) as determined by structural alignment27,34. Subsequent comparison using the Dali structural alignment server35 identified a match between the Sd1 domain of Bc TssA1B Nt1 and the Nt2 domains of both Ah TssA2A and EAEC TssA2B (RMSD = 2.7 and 2.6 Å, respectively) despite there being essentially no amino acid sequence similarity between them. Specifically, the related regions include helices α1-α5 of Bc TssA1B Sd1 (i.e. most of the ImpA_N region) and α2–α6 of Ah TssA2A Nt2 (Fig. 4d). This similarity suggests that an ancient gene duplication event may have played a role in the evolution of the Nt1 and Nt2 domains of TssA2 subunits.
TssA2A CTD assembles into an oligomer with D5 symmetry
Two-hybrid and SEC MALLS analysis indicated that Ah TssA2A CTD self-associates and dictates the overall stoichiometry of the TssA2A complex (Supplementary Fig. 9a, b, c, d). This is also a feature of EAEC TssA2B 27. However, unlike EAEC TssA2B, the molecular weight estimations of Ah TssA2A, Ah TssA2A CTD and an Ah TssA2A Nt2-CTD fusion construct corresponded to a subunit stoichiometry of 10 rather than 12. Consistent with this, negative stain EM showed that Ah TssA2A CTD and Ah TssA2A Nt2-CTD assembled into complexes that resembled a five-pointed star (Fig. 6a and Supplementary Fig. 9e, f). The structure of crystals of an Ah His6.TssA2A Nt2-CTD construct (R232-K478) was successfully determined to 3.13 Å through molecular replacement using the coordinates of the Ah TssA2A Nt2 dimer as a search model (Supplementary Table 7). This identified the position of five dimers of the TssA2A Nt2 domain (R232 to L374, helices α1–α7) in the asymmetric unit, which did not appear to be arranged with any obvious symmetry with respect to each other. Following refinement, these dimers could be seen to be essentially identical in structure to that of the dimer formed by the isolated TssA2A Nt2 domain (RMSD = 0.4 Å). The remaining C-terminal residues of each of the 10 subunits (chains A-J) were organised into a separate domain (the CTD) consisting of five α-helices (G388-L472, α8–α12). The 10 CTDs were, in turn, assembled into a flat ring resembling a star with D5 symmetry (Fig. 6b), consistent with the negative stain EM and the SEC-MALLS analysis.

Structure of the Ah TssA2A CTD ring. a Negative stain EM particle averaging of Ah TssA2A CTD indicating five-fold symmetry. b Structure of 10 Ah TssA2A CTD monomers assembled to form a decameric oligomer exhibiting D5 symmetry (PDB: 6G7C https://www.ebi.ac.uk/pdbe/entry/pdb/6g7c). Boxed region 1 (Interface 1), two-fold axis formed by dimerisation via the WEP motif (red and blue chains). Boxed region 2 (Interface 2), two-fold axis generated through packing of helices α9, α10 and α11 (blue and orange chains). The N-terminus of chain A is indicated and the dimensions of the ring are shown. c Interface 1 with key interacting residues indicated (chain A, pink: chain B, light blue). d Side view of interface 2 with interacting surfaces highlighted in light blue and sand. e Superposition of WEP-mediated dimers from Ah TssA2A (cyan) and EAEC TssA2B (PDB: 4YO5, green) about the WEP interface. The monomers exhibit a different relative orientation of ~35° about the WEP interface. View corresponds to chains A and B shown in (b). f Structural similarity of the CTD monomers of Ah TssA2A (cyan) and EAEC TssA2B (PDB: 4YO5, green). Helices α13 and α14 are unique to EAEC TssA2B and form the C-terminal extension (magenta), which is important for the different quaternary structures assembled by TssA2A and TssA2B. g, h Side by side views of the Ah TssA2A (g) and EAEC TssA2B (h) CTD oligomers with their respective five-fold and six-fold axes vertical. Assembly of Ah TssA2A WEP-mediated dimers (cyan) around the five-fold axis involves a rotation relative to EAEC TssA2B (green) of ~50° around the dimer axis as illustrated by the dotted black line. The neighbouring subunits with which each monomer within a WEP-mediated dimer interacts are shown in grey
In the assembled TssA2A CTD decamer two distinct two-fold interfaces can be identified (Fig. 6b). In the first, a two-fold related interface (shown by PISA32 to be 530 Å2) is formed primarily through interactions between residues from the extended loop linking α11 and α12 (Fig. 6b, c) and the N-terminal end of α10 and their symmetry-related partners. This loop includes a WEP motif (W461, E462 and P463) that is conserved across both TssA2 sub-clades, where W461 is partially buried within a hydrophobic pocket formed by the conserved residues L456, L458 and L465, and an interaction with F423, a non-conserved residue, in the two-fold related monomer. The entrance to this pocket is capped by a salt bridge between E462 of the motif and R420, two residues that are again conserved (Fig. 6c and Supplementary Fig. 2b). In the second, more extensive interface (950 Å2), the WEP-mediated dimers assemble around the five-fold axis through interactions between the surface of helices α9, α10 and α11 (Fig. 6b, d) and their two-fold related symmetry mates to form a mixed interface of hydrophobic/hydrophilic contacts. The TssA2A CTD (S381-K478) ring has a thickness of ~30 Å, an outer diameter of ~110 Å, and a lumenal diameter of 25 Å (Fig. 6b, g). As in the structure of the Bc TssA1B CTD ring, the N-terminus of each TssA2A CTD monomer is orientated on the outside of the CTD oligomer resulting in the presentation of the Nt2-CTD interdomain linker at the periphery of the molecule (Fig. 6b).
Despite the differences in symmetry between TssA2 CTD oligomers assembled by members of sub-clades 2A and 2B, the fold of the Ah TssA2A CTD monomer is closely related to that of its counterpart in EAEC TssA2B (PDB = 4YO5, RMSD = 1.6 Å). Moreover, close inspection shows that the conserved WEP motif is maintained at the dimer interface in members of both TssA2 sub-clades, albeit that the two monomers of Ah TssA2A are arranged in a slightly different orientation with respect to each other, corresponding to a rotation of ~35° compared to their TssA2B counterparts (Fig. 6e).
The way in which the dimers assemble around their respective five-fold and six-fold symmetry axes in the Ah and EAEC TssA2 CTDs are strikingly different, in contrast to the similarity seen around their two-fold axes. In Ah TssA2A, two monomers, related by a WEP-mediated two-fold axis, pack adjacent to one another, parallel to the five-fold axis, resulting in the formation of an oligomer which resembles two interpenetrating rings (Fig. 6g). The face of Ah TssA2A, used in subunit assembly around the five-fold axis (residues from α9, α10, and α11), interacts with a symmetry related equivalent (Fig. 6g). In comparison, EAEC TssA2B CTD contains a 41 residue C-terminal extension which folds into two additional helices (α13 and α14) (Fig. 6f). These dominate the interactions around the six-fold axis, packing against the face of helices α9, α10 and α11 of an adjacent subunit (Fig. 6h). Consequently, this causes an incline of the WEP-mediated dimer by ~50°. Therefore, in EAEC TssA2B, two monomers, related by a WEP-mediated two-fold axis, stack on top of one another, parallel to the six-fold axis, with the dimers then assembling around the latter to form a double-layered ring (Fig. 6h). Interestingly, despite the difference in symmetry we note that the lumenal diameter and outer diameter of the TssA2A and TssA2B CTD oligomers are similar (Fig. 6g, h), thereby placing the Nt2-CTD interdomain linker at the same distance from the centre of the complex.
A flexible linker in TssA2A Nt2-CTD mediates domain mobility
Despite the electron density for the Nt2 and CTD domains being clear, density was observed for only part of the Nt2-CTD interdomain linker, and, depending on the chain, between 6 and 13 residues (Q375-A387) could not be identified within this region. However, restrictions imposed by the maximum length of the absent linker residues and the relative orientations of the Nt2 and CTD domains allowed the unambiguous assignment of the Nt2 domains to their corresponding CTD (i.e. in the same polypeptide chain). This revealed that in the decameric Ah TssA2A Nt2-CTD complex, the Nt2 domains are not related by the five-fold symmetry exhibited by their corresponding CTDs (Fig. 7a). Instead, dimers of Nt2 make no contacts with their own CTDs but rather contact either TssA2A Nt2 or CTD domains of other decamers in the crystal lattice. Moreover, as a result of the flexible linker, the Nt2 dimers have quite different orientations with respect to the five-fold axis, with angular rotations of up to ~60° about the vertical axis (Fig. 7b), and ~45° and ~60° about the horizontal and torsional axes, respectively. Although, the observed Nt2 domain orientations described above are believed to be an artefact of crystal packing, the range of motion seen is believed to reflect the inherent flexibility between CTD and Nt2 domains. This is consistent with negative stain EM analysis that revealed a lower calculated correlation coefficient for strict five-fold symmetry when comparing an Nt2-CTD construct to a CTD construct (Supplementary Fig. 9e–h).

Schematic representation of TssA ring dimensions and domain flexibility. a Arrangement of the Ah TssA2A Nt2 domains around the CTD oligomer as observed in the asymmetric unit. The Nt2 domains do not follow strict five-fold symmetry. The decameric CTD ring is shown in dark grey perpendicular to the five-fold (left) and two-fold (right) axes with each peripheral Nt2 dimer shown in a different colour. The diagram between the two structures shows the relationship between dimer-forming Nt2 domains (represented by coloured squares) and their corresponding C-terminal domains in the TssA2A Nt2-CTD decamer. b Displacement of the Ah TssA2A Nt2 dimers perpendicular to the five-fold axis of the CTD oligomer. The CTD ring is shown in light grey with one WEP-mediated CTD dimer indicated in cyan to which is tethered an Nt2 dimer. The Nt2 dimer (light green) is shown at its maximum displacement of ~60° either side of the plane of the CTD ring (orange line). Other observed positions of Nt2 are indicated by translucent shading. c Top and side view representations showing the domain architecture of TssA subunits from clades 1B, 2A and 2B modelled on the baseplate-distal end of the T6SS tube/sheath assembly during polymerisation. The V. cholerae TssBC extended sheath (green) surrounding a Hcp hexamer (yellow) (PDB: 5MXN) is shown on the left. Onto this, is modelled the TssA1B CTD oligomer (orange) with 16-fold symmetry, showing one stacked CTD dimer attached to a pair of Nt1 domains via flexible linkers, the TssA2A CTD oligomer (blue) with five-fold symmetry, showing a WEP-mediated CTD dimer connected to the two monomers of an Nt2 dimer via flexible linkers, each of which are in turn connected to an Nt1 domain, and the TssA2B CTD oligomer (red) displaying six-fold symmetry, showing a CTD dimer connected to the two monomers of an Nt2 dimer via flexible linkers, each of which are in turn connected to an Nt1 domain. Labels are: C, CTD dimers (top view); N1, Nt1; N2, Nt2; CTD, CTD oligomer (side view). Scale bar corresponds to 65 Å
Sequence alignments suggest that the Nt2-CTD interdomain linker is longer in TssA2B subunits than in TssA2A subunits (Supplementary Fig. 2b). For example, the linker in Ah TssA2A is composed of ~21 residues whereas it contains ~37 residues in EAEC TssA2B. The most likely explanation for the presence of a longer linker in TssA2B is to overcome a restricted range of motion of Nt2 dimers that would be imposed by a shorter linker due to the possible physical interference between the Nt2 dimer and the ‘taller’ double-layered ring arrangement formed by EAEC TssA2B CTD compared to the ‘squat’ interpenetrating ring assembled by Ah TssA2A CTD (Fig. 6g, h).
The quite different orientation of the CTD WEP-mediated dimers in TssA2A and TssA2B oligomers has one further consequence, which is that the orientation of the interdomain linker between Nt2 and CTD is necessarily rotated by ~50° (Fig. 6g, h). We assume that the flexibility of the linker allows this structural difference to be maintained without affecting the biological role of the protein. The finding that in the Ah TssA2A Nt2-CTD structure the Nt2 domains consistently occur as dimers further suggests that the Nt2 domains remain associated as part of the biological role of TssA, in contrast to the proposal by Zoued and co-workers27, who interpreted low resolution negative stain EM data to suggest that they separate to form monomers27. A rigorous test will require a high-resolution EM reconstruction from TssA particles embedded in vitreous ice, although this could be challenging given the apparent flexibility of domain linkers.
Discussion
The structural studies presented here show that TssA1B, TssA2A and TssA2B possess structurally diverse CTDs that lead to distinct overall architectures for their respective complexes. Nevertheless, all these subunits contain a conserved N-terminal ImpA_N region that is peripherally located in the assembled complexes. The question that arises from this is whether these TssAs play the same role in the function of the T6SS? The presence of the ImpA_N region in the Nt1 domain of subunits from all TssA clades strongly suggests that they have a common function that is of critical importance. This is further supported by the studies described here which reveal that TssA1B interacts with the baseplate components TssE, TssF and TssK, the inner tube subunit, Hcp, the tip protein, VgrG, the sheath subunit TssC and cytoplasmic components of the cell envelope-anchoring complex. This is strikingly similar to the TssA2B subunit that was shown to interact with the same components except for TssF27. Indeed the TssA1B and TssA2B CTD oligomers were both shown to interact with Hcp and VgrG despite their quite different structures. This would argue for a similarity of function despite the divergent architecture of the two proteins. Given the sequence similarity and predicted structural relationship between TssA1A and TssA1B, this suggests that all three clades of TssA perform related roles within the T6SS. If the role of these quite different TssA subunits is similar, then how is this achieved given the structural variations that are observed?
To date, two distinct models for the role of TssA have been proposed. In the first, baseplate-associated model, it was proposed that TssA is a component of the T6SS baseplate required for priming the inner tube and sheath for assembly, but not migrating during polymerisation28. This model was based partly on predicted similarities in structure for the CTDs of TssA1A and the phage T4 baseplate protein gp6, to which it was believed that this TssA subunit was related. The structure we have obtained for TssA1B, which is closely related to TssA1A, reveals that it has a quite different structure to gp6. This suggests that TssA1A is very unlikely to be a gp6 orthologue. Due to the absence of structural similarity to baseplate proteins and the conservation of the Nt1 domain among all TssA subunits, it is therefore likely that TssA1 subunits fulfil a similar role to TssA2 subunits.
In the alternative model proposed for a TssA subunit that corresponds to TssA2B, TssA acts as a coordinator of inner tube and sheath assembly, in which the CTD oligomer of TssA2B would lie close to the centre of the tail assembly, above the position occupied by Hcp where it would coordinate polymerisation of the inner tube and sheath27,29,36,37. In one version of this proposal (the cap model) eversion of CTD monomers, from a closed form of the CTD oligomer to an open form, is then proposed to permit the passage of Hcp through the lumen of the open ring. This would allow sequential stacking of Hcp hexamers, with the N-terminal domains of TssA2B interacting with the polymerising sheath. However, as yet, an open form of TssA2B has not been observed and the proposed reorientation of monomers in the CTD oligomer remains speculative.
The studies described here show that the fold of TssA2A retains clear structural similarity to TssA2B yet generates a decamer possessing five-fold symmetry in contrast to the six-fold symmetry of the TssA2B dodecamer27. Therefore, while it could be argued that it might lie in the same position in the complex as TssA2B, the difference in symmetry implies there must be changes to the complementary surfaces of surrounding TssB/-C/-D subunits to allow pseudo equivalent interactions. This situation becomes even more extreme given the quite different organization of the TssA1B ring oligomer with its sixteen-fold symmetry. One possibility is that the structure of TssA1B we have determined represents an open form of the TssA CTD oligomer, leaving a closed form of the TssA1B CTD oligomer, which might pack on top of a Hcp hexamer, still to be identified. Alternatively, if some features of the cap model are correct, and TssA1A/TssA1B and TssA2A also serve to coordinate tube and sheath assembly, then, given their quite different symmetries and architectures, rationalising how they function in an equivalent way to that proposed for TssA2B is problematic. One way for this dilemma to be resolved is if the different structures of these distinct TssAs represent different ways in which a common function can be maintained.
Comparing the TssAs of clades 1 and 2, these structures indicate that the Nt1 domain is linked either to a large diameter CTD ring (200 Å) of high symmetry, via a single flexible segment, as seen in sub-clade TssA1B, or to a smaller (100–110 Å) diameter CTD of lower symmetry, through a middle domain (Nt2) flanked by two flexible linkers, as seen in sub-clades 2A and 2B. Despite these two alternative architectures, the radial position of the Nt1 domain from the centre of the complex is similar in both TssA1 and TssA2 clades (Fig. 7c). This suggests that a role of the Nt2 domain is to act as a spacer connecting the Nt1 domain of TssA2A and TssA2B to their corresponding CTDs.
In light of the above, and given the conservation of the Nt1 domain, we suggest that the common feature shared by all these TssAs is that the Nt1 domain is positioned around the periphery of the ring-forming CTD oligomers, furthest away from the symmetry axis but at an equivalent distance. Given the apparent symmetry mismatch between the tube/sheath assembly and the TssA CTD oligomers of some TssA species, it is important to consider how equivalent interactions between these components can be maintained. The simplest explanation is to consider a model whereby the interactions between TssA subunits and the tube/sheath assembly are solely the property of the conserved Nt1 domains, which have been suggested to participate in assembly of the latter27. This would mean that the role of the CTD or Nt2-CTD oligomers of clades 1 or 2, respectively, are to provide a platform for the presentation of the Nt1 domains above the growing tube/sheath complex at a similar distance from the centre of each TssA complex (Fig. 7c). In such a model it would not be necessary for the CTDs to interact directly with the tube/sheath assembly except perhaps in a transient manner as indicated by interaction studies. This agrees with surface plasmon resonance data for EAEC TssA2B where the interaction of the Nt1-Nt2 region with the TssBC sheath is significantly greater than that of the corresponding CTD with Hcp27.
This proposal in which one domain or component of a protein subunit (CTD/Nt2-CTD) associates into a platform that facilitates the role of a second domain (Nt1) in subunit incorporation by a polymerising protein assembly, is a theme that occurs in the mechanism of flagellum assembly by the HAP2 cap protein27,29,38,39. An important aspect of the flagellin-capping model is the observation of domain mobility, a feature clearly shown here for both the relative motion of Nt1 with respect to the CTD in TssA1B and between the CTD and Nt2 in TssA2A and TssA2B, and the predicted flexibility of the linker between Nt2 and Nt1 in TssA2A/TssA2B. This would provide the necessary movement of the Nt1 domains for dynamic interactions with the TssBC subunits during sheath biogenesis. This might involve the displacement of Nt1 domains and their linkers, consequently allowing passage of Hcp, in addition to further TssB/C subunits to be incorporated into the growing sheath. This model does not necessarily require eversion of the CTD rings. However, we do not rule out the possibility that TssA2A/TssA2B CTD oligomers can re-orientate to allow access of Hcp hexamers to the growing tube. In this regard, we note that the TssA1B CTD ring oligomer can adequately accommodate Hcp hexamers within its lumen.
A question arising from the dihedral symmetry shown by all TssA structures that have been determined to date, is how this leads to a uni-directional assembly of the tube/sheath complex. While any initial interaction between TssA and other subunits of the complex can use either two-fold related face of the TssA oligomer, once contact has been made with T6SS components, the symmetry is broken. This would occur when TssA interacts with cytoplasmic components of the TssJLM membrane complex that occurs during the initial steps in T6SS assembly27, as one surface of the TssA ring faces the membrane while the other faces the cytosol. According to the current model, this asymmetry is reinforced once the baseplate is recruited to the membrane complex, whereupon one face of the TssA oligomer is located adjacent to the cytoplasmic face of the baseplate27. Here TssA is ready to coordinate inner tube and sheath assembly once the first Hcp ring is recruited to the base of the VgrG trimer located at the hub of the baseplate40. Although the precise mode of action of TssA subunits remains unknown, they are likely to lock the inner tube/sheath structure in the high energy form prior to firing29. Thus, TssA may possess chaperone-like activity.
A final question is to consider the mismatch in symmetry between the assemblies formed by members of different TssA clades, on the one hand, and other components of the T6SS, on the other, and how this impacts on the mechanism by which TssA executes its role. For example, the six-fold symmetry of the TssA2B oligomer is consistent with the symmetry of the tube-sheath structure but not with the five-fold symmetry proposed for the TssJLM membrane complex10,27. In contrast, the symmetry of the TssA2A oligomer matches that of the membrane complex but not the contractile machinery. In this regard it is interesting to note that, as seen with the different symmetries of TssA2A/TssA2B, both five-fold and six-fold symmetrical variants of the HAP2 capping protein can interact with the common 11-fold symmetrical flagellin filament41,42 suggesting that the apparent symmetry mismatch between oligomers formed by some TssA species and the tube/sheath assembly is not necessarily problematic. Based on these considerations we suggest that a flagellin-like capping model is a plausible mechanism for the role of all TssA subunits. Ultimately, these proposals need to be confirmed by the structural analysis of other complexes of secretion systems from each of the different TssA clades. Once this has been achieved, the manner in which the difference in structure and symmetries among TssA subunits resolves to provide equivalent function will be revealed.
Methods
Bacterial strains and growth conditions
Bacterial strain details are given in Supplementary Table 8. E. coli strains JM83 and DH5α were used for routine cloning steps. B. cenocepacia strain H111 and A. hydrophila ATCC 7966 were the source of the TssA1B and TssA2A subunit genes, respectively. LB was routinely used for growing bacteria except where indicated43. Lennox medium was used with glucose included at 0.2%44. D-BHI broth was made by dissolving 9.25 g brain-heart infusion (BBL) in 25 ml water and dialysed (12,000–14,000 MWCO) against 500 ml water overnight45. Antibiotics were used at the following concentrations for E. coli: ampicillin, 100 μg/ml; kanamycin, 50 μg/ml; chloramphenicol, 25 μg/ml; trimethoprim, 25 μg/ml (selection for trimethoprim resistance was applied in/on iso-sensitest (IST) broth/agar (Oxoid)). Bacto MacConkey agar base used for two-hybrid assays was obtained from Becton, Dickinson and Co.
Plasmids and primers
All oligonucleotides and plasmids used in this study are listed in Supplementary Tables 9,10. Construction of plasmids is also described in Supplementary Table 10. Plasmids were transferred to E. coli by transformation46 and to B. cenocepacia by conjugation47,48.
Construction of a B. cenocepacia tssA1 B mutant
To construct the B. cenocepacia H111 tssA::Tp mutant, a segment of the tssA1B ORF was amplified with primers iotAfor3 and iotArev and cloned between the HindIII and BamHI sites of pBBR1MCS (to give pBBR1MCS-‘tssA1B) whereupon it was disrupted by insertion of the dfrB2 (TpR) cassette from p34E-Tp (generating pBBR1MCS-‘tssA1B::Tp). The tssA1B::Tp fragment was then transferred to the suicide vector pSHAFT2 and the resultant plasmid (pSHAFT2-‘tssA1B::Tp) conjugally mobilised into H111 using E. coli S17–1(λpir). Ex-conjugants were selected on M9-glucose agar containing trimethoprim (25 μg/ml). Mutants were identified by PCR screening chloramphenicol-sensitive ex-conjugants using primers iotAfor and iotArev2 that anneal to sequences located outside the genomic region contained on the suicide vector. Construction of the tssM deletion mutant was performed by conjugally mobilising the allelic replacement plasmid, pSNUFF-ΔtssM, containing a tssM deletion allele (codons 506–1216 removed) into strain H111 using E. coli SM10(λpir). Ex-conjugants containing the integrated allelic replacement vector were selected on M9-glucose agar containing trimethoprim (25 μg/ml) and were then plated on M9-glucose agar containing p-chlorophenylalanine to identify recombinants in which a second crossover had resulted in excision of the integrated plasmid49. Mutants were identified by diagnostic PCR using primers tssM-OPfor and tssM-OPrev2.
Hcp secretion assay
D-BHI broth was inoculated with overnight cultures of B. cenocepacia strains to give a starting OD600 of 0.03, and grown to an OD600 of 1.0 at 37 °C. Cultures were centrifuged to collect the cells and the supernatant was filter-sterilised. To precipitate secreted proteins, sodium deoxycholate was added to the cleared supernatant to a final concentration of 0.2 mg/ml followed by incubation on ice for 30 min. Trichloroacetic acid was then added to a final concentration of 10% (v/v), and the suspension was incubated overnight at −20 °C. Precipitated proteins were recovered by centrifugation for 15 min at 14,000 × g at 4 °C and washed with ice cold acetone. Pellets were air-dried and resuspended in 0.001 of the original volume of SDS-PAGE sample buffer. Cell-associated proteins were isolated by resuspending the initial bacterial pellet in a volume of SDS-PAGE sample buffer that was 20-fold less than the starting culture volume and transferred to a microcentrifuge tube. All protein samples were boiled for 5 min, centrifuged for 10 min at 14,000 × g to remove cell debris and then fractionated by electrophoresis in a 15% discontinuous SDS-PA gel followed by electroblotting onto a PVDF membrane (Millipore). Membranes were blocked with 5% (w/v) non-fat skimmed milk in TBS containing Tween 20 (0.05% (v/v)) solution for 1 h. Hcp was detected by probing with a custom polyclonal rat antibody raised to purified His6.Hcp (1:1000) and goat anti-rat HRP secondary antibody (1:5000). Antibody to the cytoplasmically located β subunit of E. coli RNA polymerase (1:2000) that cross-reacted to the Burkholderia spp. subunit was also used as a lysis control, with a rabbit anti-mouse HRP secondary antibody (1:5000). Detection of chemiluminescence was performed using an EZ-ECL detection kit and a XRS+ imaging system (BioRad). Uncropped blots are shown in Supplementary Fig. 5.
Two-hybrid analysis
The bacterial adenylate cyclase two-hybrid (BACTH) system was employed50. B. cenocepacia T6SS subunit genes (tssA-tssC, hcp, tssE-tssG, vgrG, tssJ-tssM) or domain coding sequences were fused to the N- and C-terminal coding sequences of the B. pertussis CyaA T25 or T18 fragments present in plasmids pKT25, pKNT25, pUT18 and pUT18C. To avoid membrane or periplasmic targeting of CyaA fusion proteins, signal sequences and TMDs were omitted from the TssJ, TssL and TssM hybrid proteins. As a consequence, the cytoplasmic N-terminal region of TssM (TssMN) and the periplasmic C-terminal region (TssMC) were fused to the CyaA domains separately. Hybrid proteins were generated with full-length VgrG and also with the conserved core region corresponding to the phage T4 gp27 and gp5 proteins (VgrGC). DNA encoding full-length A. hydrophila TssA2A and the TssA2A Nt1 domain were also cloned into all four vectors while DNA encoding the Nt2 and CTDs were cloned into pKT25 and pUT18C only. Further details on plasmid constructions are given in Supplementary Table 10. The E. coli cya− strain BTH101 was transformed with the two compatible plasmids expressing fusion proteins and plated on MacConkey indicator agar containing 1% (w/v) maltose, ampicillin (100 μg/ml) and kanamycin (50 μg/ml). Colony phenotypes were scored after 3 and 5 nights incubation at 30 °C (Mal+ (Cya+) colonies are purple and Mal− (Cya−) colonies are white). The appearance of purple colonies after 3 days incubation indicated a strong Cya+ phenotype. The positive control combination (Zip control), where each plasmid encoded the leucine zipper segment of the yeast transcriptional activator protein, GCN4, fused to T25 and T1850 was always included as were all negative control combinations (two compatible empty vectors and one empty vector in combination with a plasmid specifying a hybrid protein).
The efficiency of CyaA complementation was quantitated by measurement of the β-galactosidase activity in broth cultures. Cells were grown in LB broth containing antibiotics and 0.5 mM IPTG at 30 °C with aeration for 14–16 h. The cultures were then chilled on ice before being diluted in LB medium to OD600 0.4–0.6. 50 μl of each diluted culture was added to test tubes containing 950 μl of Z buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 0.001 M MgSO4 and 0.27% (v/v) β-mercaptoethanol), chloroform (30 μl) and 0.1% SDS (30 μl). The mixtures were vortexed for 10 s and equilibrated at 30 °C for 15 min whereupon 200 μl of ONPG (4 mg/ml in Z buffer) was added to initiate the reaction. Reactions were stopped by addition of 500 μl 1 M Na2CO3 and the absorbance at 420 and 550 nm was measured. The β-galactosidase activity (in Miller units) was calculated using the equation 1000 (OD420–1.75 × OD550/t x v × OD600) where t corresponds to the duration of the reaction in minutes and v corresponds to the volume of culture used in the assay43. The background activity measured in the negative controls (a pair of empty vectors or one empty vector in combination with a plasmid encoding a hybrid protein) was 75–90 Miller units (Mu). Each assay reaction was performed in duplicate on three biological replicates (n = 3), and the mean, along with standard deviation, was calculated.
Co-immunoprecipitation assay
Co-IP analysis was performed on cell lysates following co-expression of N-terminal FLAG-tagged TssA1B as bait and an epitope-tagged T6SS subunit (or domain) as prey in E. coli. FLAG.TssA1B was expressed from the second T7 RNAP-dependent promoter contained on pACYCDuet-1 while the prey protein was expressed from the upstream promoter on the same plasmid (except where indicated below). Prey proteins were generally tagged at their N-terminus with the vesicular stomatitis virus glycoprotein (VSV-g) epitope tag (TssC was C-terminally tagged). TssF was tagged at the N- or C-terminus with the human influenza haemagglutinin (HA) epitope tag. In two cases, a separate plasmid was used to express the prey protein: N-terminal HA-tagged TssF was expressed from the downstream T7 promoter of pETDuetΔO whereas MBP-TssE was expressed from the tac promoter present on pMAL-c5X. Further details on plasmid constructions are given Supplementary Table 10.
Expression of all proteins was induced by addition of 0.5–1 mM IPTG (0.1 mM for the TssC co-IP) to E. coli BL21(DE3) cells grown to OD600 0.5–0.7 in 50 ml BHI broth at 37 °C (for the Hcp, TssK and TssL co-IPs), 30 °C (for the TssE, TssF and VgrGC co-IPs) or 22 °C (for the TssC and TssMN co-IPs), according to the solubility of the prey protein, and incubation was continued at the same temperature for 2–3 h. Bacterial cells were harvested and stored frozen at −20 °C overnight. Defrosted E. coli cell pellets containing co-expressed proteins were resuspended in TBS (50 mM Tris-Cl (pH 7.4), 150 mM NaCl (or 500 mM NaCl for VgrGC and TssMN)) with EDTA-free protease inhibitor tablets (Roche) at a ratio of 5 ml TBS to 1 g cell pellet. Following sonication, the soluble fraction was cleared by centrifugation at 20,000 g for 15 min at 4 °C, and MgCl2 was added to a final concentration of 2.5 mM. After gently mixing by inversion, the mixture was centrifuged at 13,000 g for 10 min. The supernatant was then incubated with Tween 20 (0.2%) for one hour with gentle mixing on a rotating wheel at room temperature, and followed by centrifugation at 15,000 g for 10 min. The supernatant was used for co-immunoprecipitation with the anti-FLAG affinity gel. Fifty microlitres of the anti-FLAG M2 affinity resin suspension equilibrated in TBS was added to 200–1000 μl cell lysate (depending on the abundance of FLAG-tagged TssA1B in the lysate) and the final volume was brought to 1 ml by adding TBS. After 2–3 h gentle mixing at 4 °C to allow for precipitation, the mixture was centrifuged at 7000 × g for 30 s, and the supernatant was removed (unbound material). The resin was then washed three times with 1 ml TBS with gentle mixing for 5–15 min at 4 °C between each wash followed by centrifugation at 7000 × g for 30 s. After discarding the supernatant of the final wash, the immunoprecipitated material was dissociated from the resin by boiling with 25 μl of SDS-PAGE sample buffer with or without 5% β-mercaptoethanol for 3 min. Finally, the mixture was centrifuged at 7000 × g for 30 s to clear the suspension. The supernatant (immunoprecipitated material) was subjected to SDS-PAGE alongside a pre-stained protein ladder, blotted onto PVDF membrane and probed with tag-specific antibody (anti-VSVg, anti-HA or anti-FLAG, each at 1:5000–1:10,000 dilution) and detected with HRP-conjugated secondary antibody (1:5000–1:6666 dilution). MBP fusion proteins were detected with HRP-conjugated anti-MBP (1:5000 dilution). The sizes of the tagged B. cenocepacia T6SS subunits in kDa (excluding the N-terminal formyl-methionine) are: TssA1B, 42.6; TssC, 56.1; Hcp, 19.8; TssE, 61.6; TssF, 69.8 (both versions); VgrGC, 60.7; TssK, 51.3; TssL, 24.4; TssMN, 44.5; MBP, 45.5 kDa. Uncropped blots are shown in Supplementary Fig. 4.
Antibodies
Polyclonal antibody to B. cenocepacia Hcp was raised in rats at the University of Sheffield Biological Services unit using N-terminal His-tagged Hcp which was purified following its overproduction in E. coli as described below. Rabbit anti-FLAG mAb and anti-VSVg pAb were obtained from Sigma-Aldrich (cat. no. F7425 and V4888, respectively), whereas rabbit anti-HA mAb was purchased from Cell Signaling Technology (cat. no. C29F4). HRP-conjugated mouse anti-MBP polyclonal antibodies were obtained from New England Biolabs (cat. no. E8038S). The mouse mAb to the β subunit of E. coli RNA polymerase was obtained from Neoclone (cat. no. W0023). Proteins harbouring a polyhistidine tag were detected using the HisProbe-HRP conjugate (Thermo Fisher, cat. no. 15165) or mouse anti-KLH-conjugated His-tag peptide (BioLegend, 652501). HRP-linked goat-derived (secondary) antibodies that cross-react to rat and rabbit IgG were obtained from SouthernBiotech (cat. no. 3050–05) and Vector Laboratories Ltd (cat. no. PI-1000), respectively. HRP-conjugated rabbit anti-mouse IgG secondary antibody were purchased from Thermo Fisher (cat. no. 31450).
Expression and purification of Hcp for antibody production
His6.Hcp was overproduced following a 4 h induction of the T7 promoter on pET14b-His6.TssD2 carried by E. coli BL21(DE3) cells growing in BHI at 37 °C. His6.Hcp, which lacks the first three native Hcp amino acids, was purified by sequential IMAC on nickel-sepharose (HisTrap HP, GE Healthcare Life Sciences) and SEC using a Superose-6 10/300 GL column equilibrated in PBS. Prior to the SEC step, the protein was concentrated using a 10,000 MWCO centrifugal concentrator.
Expression and purification of TssA subunits and domains
Expression and purification of His6.TssA2A (1–478), His6.TssA2A Nt2 (223–387), His6.TssA2A Nt2-CTD (223–478) and TssA2A CTD (381–478) was carried out as previously described33. Both MBP.TssA1B CTD (303–373) and His6.TssA1B Nt1 (1–255) were overexpressed and purified, with TssA1B CTD (303–373) being separated from the MBP solubility tag by Factor Xa cleavage51. MBP.TssA1B CTD (294–373) was produced and purified in a similar manner as MBP.TssA1B CTD (303–373). However, following Factor Xa cleavage of MBP.TssA1B CTD (294–373), TssA1B CTD oligomers were separated from the MBP solubility tag for EM analysis using a 100 kDa MWCO concentrator (Amicon) rather than SEC and the additional amylose column purification step was not included. Truncated TssA1B CTD derivatives were overproduced with an N-terminal MBP-His6 tag in a similar manner to that described for MBP.TssA1B CTD (303–373) but were purified by IMAC on a HisTrap HP column with imidazole gradient elution rather than by amylose affinity chromatography. For crystallisation, MBP.His6.TssA1B CTD H12–H14 (amino acids 303–358) was purified by amylose affinity chromatography, and eluted with 10 mM maltose. His6.TssA1B CTD H12–H14 was proteolytically removed from the MBP tag by treatment with Factor Xa, followed by purification on a nickel column with imidazole gradient elution and SEC on a Superdex 200 column prior to concentration and final buffer exchange51.
TssA2A Nt1-Nt2.His6 (1–374), His6.TssA2A CTD (381–478) and the TssA1B variants (Native TssA1B (1–373), His6.TssA1B (1–373) and linkerHis6.TssA1B (1–373)) were overproduced by inducing the T7 promoter present on the appropriate expression vector derivative in E. coli BL21(DE3) cells growing in BHI broth at 37 °C by addition of IPTG (final concentration 1 mM) to the culture at OD600 0.5–0.7 and incubation was continued for a further 2–3 h (30 °C was used for inducing the linker His-tagged derivative). Harvested cells were resuspended in 50 mM Tris-Cl (pH 8.0), 100 mM NaCl (5 ml/g cell paste) and lysed by sonication. If the construct contained a His-tag, the lysate was cleared by centrifugation at 35,000 × g for 30 min. The clarified lysate was applied to nickel-sepharose (HisTrap HP, GE Healthcare Life Sciences) and eluted with a linear gradient of imidazole (10–500 mM).
For purification of native TssA1B (1–373), nucleic acids were removed from the clarified supernatant by precipitation with polyethyleneimine (0.15% (v/v)) at pH 8.0. After centrifugation at 40,000 g for 30 min at 4 °C, ammonium sulphate was added to the supernatant to a final concentration of 30% to precipitate TssA. The precipitated protein was recovered by centrifugation at 40,000 × g for 30 min at 4 °C and resuspended in a small volume of 50 mM Tris-Cl (pH 8.0) containing 50 mM NaCl, which was then applied onto a Superose 6 column equilibrated with the same buffer. SEC was performed at 0.5 ml/min flow-rate. TssA1B eluted from the column as a single protein peak at ~13.75 min corresponding to a size of ~1.9 MDa.
SEC
Size estimation of His6.TssA1B Nt1 (1–255) was performed on a Superose 12 GL column (GE Healthcare) in 50 mM Tris-Cl (pH 8.0) containing 500 mM NaCl. The following standards were used (molecular weights in kDa): thyroglobulin, 669; apoferritin, 443; alcohol dehydrogenase, 149.5; conalbumin, 75; ovalbumin, 43; carbonic anhydrase, 29; ribonuclease, 13.7.
SEC-MALLS
For size exclusion chromatography-multiangle laser light scattering (SEC-MALLS), native TssA1B (1–373) and TssA1B CTD (294–373) were purified as described above and analysed on a Superose-6 10/300 column in 10 mM phosphate buffer (pH 7.4) containing 2.7 mM KCl and 137 mM NaCl, whilst purified His6.TssA2A (4.5 mg/ml), His6.TssA2A Nt2-CTD (6.8 mg/ml) and His6.TssA2A CTD (6.0 mg/ml), were filtered (0.2 µm pore size) and analysed on an S200 10/300 column (GE Healthcare) in 50 mM Tris.HCl (pH 8.0), 500 mM NaCl. These were carried out at ~20 °C using a system comprising a Dawn Heleos-II multi-angle light scattering detector, an Optilab rEX refractive index detector and a QELS Plus module (all items by Wyatt Technology) linked to a Shimadzu HPLC system (SPD-20A UV detector, LC20-AD isocratic pump system, DGU-20A3 degasser and SIL-20A autosampler) at the Molecular Interactions Lab, University of York, UK. Peak areas chosen for analysis were defined by Astra software (Wyatt Technology).
Limited proteolysis
Twenty microlitres of purified His6.TssA2A at 3.7 mg/ml in 16.7 mM Tris-HCl (pH 8.0), 175 mM NaCl was mixed with 1 μl of glutamyl endopeptidase (V8 protease or Glu-C, Sigma), glycyl endopeptidase (Gly-C, Protogen) and trypsin type I (Sigma) prepared at 1 mg/ml in water. The reaction was allowed to proceed for 90 min at room temperature and then stopped by addition of 20 μl 10% acetic acid. Digestion products were fractionated by 4–20% SDS-PAGE and blotted on to a PVDF membrane for N-terminal sequence analysis or analysed by mass spectrometry.
Mass spectrometry and N-terminal analysis
Mass spectrometry (MS) analysis of TssA1B and TssA2A cleavage products in ultrapure water was carried out on an electrospray Q-TOF mass spectrometer at the Astbury Centre for Structural Molecular Biology, University of Leeds, UK. Samples were diluted into 50% acetonitrile/0.1% formic acid, and analysed by Z-spray nanoelectrospray ionisation MS using a quadrupole-IMS-orthogonal time-of-flight MS (Synapt HDMS, Waters UK Ltd., Manchester, U.K.) and in-house fabricated gold/palladium coated nanospray capillaries. The MS was operated in positive TOF mode using a capillary voltage of 1.2 kV, cone voltage of 20 V, nanoelectrospray nitrogen gas pressure of 0.1 bar, backing pressure of 2.47 mbar and a trap bias of 4 V. The source and desolvation temperatures were set at 80 °C and 150 °C, respectively. During TOF-MS acquisition, argon was used as the buffer gas, at a pressure of 4.0 × 10–3 mbar in the trap and transfer regions. Mass calibration was performed by a separate injection of sodium iodide at a concentration of 2 µg/µl. Data processing was performed using MassLynx v4.1 software.
Sequencing of TssA1B and TssA2A cleavage products was performed using an ABI Procise Sequencer operating standard Edman degradation procedures. After electrophoresis, proteins were electroblotted from the gel on to PVDF membrane (ABI ProBlott) using CAPS buffer at pH 11.0. Blotting was performed for 1–2 h at 450 mA with water cooling. Proteins were located on the membrane by staining with 1% Coomassie blue for 1 min, followed by destaining in 50% methanol. Transfer efficiency was verified by staining the gel after the transfer. The bands of interest were excised from the blot using a scalpel blade and transferred to a blot cartridge for sequence determination.
Electron microscopy (EM)
0.75% (w/v) uranyl formate was used as a negative stain. Carbon-coated grids were glow discharged for 30 s in a Cressington 208 carbon coater system. Purified protein samples (~5 μl of 5–50 μg/ml in 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 10% glycerol (LinkerHis6.TssA1B (1–373), His6.TssA2A Nt2-CTD (223–478) and His6.TssA2A CTD (381–478)) or 20 mM Tris-HCl (pH 7.4), 200 mM NaCl, 1 mM EDTA (TssA1B CTD (294–373))) were incubated on the grids for 1 min. Excess protein sample was removed by blotting and the grid was then washed twice in distilled water and once in negative stain (1 s for each wash) before staining for 20 s. After final blotting to remove excess stain, the grids were vacuum dried and imaged on Philips CM100 transmission electron microscope equipped with a Gatan Mulitiscan 794 1k × 1k charge-coupled device camera, at nominal magnification 28500 and defocus between 500 and 900 nm or Philips CM200 FEG transmission electron microscope using a Gatan UltraScan 890 (US4000SP) 2k × 2k charge-coupled device camera, at a nominal magnification of 66,000× and defocus between −200 and −800 nm. Apart from the initial screening of His6.TssA2A CTD and His6.TssA2A Nt2-CTD, all other images were captured on the CM200 FEG. Alignments and averaging for EM analysis were carried out using the IMAGIC-5 software package52,53.
X-ray crystallography
Native data on crystals of purified His6.TssA1B Nt1 (1–255)51 were collected on beamline I02 at the Diamond Light Source, and 150° of data were processed in xia2–3d54,55,56,57 to 1.8 Å in spacegroup P21212 (Supplementary Table 2). Crystals from the same conditions were subjected to sublimation of elemental iodine crystals overnight. Crystals were cryoprotected in 0.16 M calcium acetate, 0.08 M sodium cacodylate buffer (pH 6.5), 16.4% (w/v) PEG 8000 and 30% glycerol. Iodine SAD data were collected at 1.7 Å wavelength on beamline I03 at the Diamond Light Source, and data were processed in FastDP58 to 2.04 Å in spacegroup P212121 (Supplementary Table 2). The structure was solved using SHELX59 alone and as part of FastEP based on the identification of seven potential iodine sites. The subsequent poly-alanine model was partially refined and used to solve the structure of the 1.8 Å native data. This model then underwent rounds of model building in Coot60 and refinement using Refmac561,62 to produce the final model (Supplementary Table 2). The asymmetric unit contains one molecule of His6.TssA1B Nt1 (1–255), with eight N-terminal tag residues, 119 water molecules, two calcium ions and an ethylene glycol molecule.
Native data on crystals of purified TssA1B CTD (303–373)51 and the selenomethionine derivative were collected on beamline I24 at the Diamond Light Source. Data were processed in xia2–3da54,55,56,63,64 (Supplementary Table 3) to 3.08 Å, in spacegroup I222. Selenomethionine peak SAD data were collected at 0.97922 Å wavelength on beamline I03 at the Diamond Light Source. Data were processed in xia2–3d54,55,56,57 (Supplementary Table 3) to 3.03 Å, in spacegroup I222. The SHELX program suite, SHELXC, D and E59, was used to identify 15 selenium sites and produce an initial electron density map. An initial model was built using Coot60, Buccaneer65 and employing a structure of a shorter Bc TssA1B CTD derivative comprising residues 303–358 which had been solved separately, as a guide. This model was used as input to Phenix66,67, and subsequent output was subjected to rounds of building in Coot60, NCS averaging and solvent flattening in DM68. Refinement to the native data at 3.08 Å was performed using Refmac561,62. Between 4 and 6 residues of the protein remain unmodelled at the C-terminus of each chain.
Purified His6.TssA1B CTD H12–H14 (303–358), together with 12 additional residues from the tag, was set down in sitting-drop crystallisation trials and produced crystals in conditions 0.18 M tri-ammonium citrate, 20% (w/v) PEG 3350, which were cryoprotected in 0.2 M di-ammonium citrate, 22% (w/v) PEG 3350, 25% (v/v) ethylene glycol. Data were collected on beamline I02 at the Diamond Light Source, and processed in xia2 3dii54,55,56,57,63,64 to 2.35 Å in spacegroup P6 (Supplementary Table 4). The structure was solved in spacegroup P62 by molecular replacement, in Phaser69, using the previously solved structure of TssA1B CTD containing helices ɑ12 and ɑ13. The model was built and refined to native data at 2.35 Å in Coot60 and Refmac61,62, with two molecules in the asymmetric unit.
The structure of a TssA1B CTD fragment corresponding to residues 303–347 was determined as follows. TssA1B CTD (303–373), together with 4 additional residues from the tag, also produced crystals in condition 0.1 M citric acid (pH 5.0), 20% (w/v) PEG 6000 which were cryoprotected in 0.1 M citric acid (pH 5.0), 22% (w/v) PEG 6000, 25% (v/v) ethylene glycol. Native data were collected on beamline I04–1 at the Diamond Light Source, and processed in xia2 3daii54,55,56,57,63,64 to 1.78 Å in spacegroup I222 (Supplementary Table 5). Selenomethionine-incorporated MBP.TssA1B CTD (303–373) was overexpressed and purified using a similar strategy to that described previously51. Crystals were produced in condition 0.1 M citric acid (pH 5.0), 20% (w/v) PEG 6000, and were cryoprotected in 0.1 M citric acid (pH 4.0), 22% (w/v) PEG 6000, 25% (v/v) glycerol. Selenomethionine MAD peak, inflection and high energy remote data were collected on beamline I03 at the Diamond Light Source and data were processed in xia2 3da or 3daii54,55,56,57,63,64 (Supplementary Table 5). The structure was solved by MAD using the SHELX program suite59, followed by building in Coot60 and refinement in Refmac561,62 to the native data at 1.78 Å. Analysis of the electron density map strongly suggested that the protein had been clipped after W347.
To generate a derivative for crystals produced from purified His6.TssA2A Nt2 (223–387)33, flakes of ethyl mercury phosphate powder were added to a drop containing crystals for 5 min, before subsequent cryoprotection in 0.04 M potassium phosphate monobasic, 16% (w/v) PEG 8000, 30% (v/v) glycerol. Hg SAD data were collected at 1.0088 Å wavelength on beamline I03 at the Diamond Light Source, and data were processed in xia2-DIALS54,55,56,57,58 to 2.16 Å in spacegroup P21 (Supplementary Table 6). The structure of mercury bound Nt2 was solved using SHELX59 based on the identification of one mercury site for each of the two monomers in the asymmetric unit and built using Buccaneer and Coot60,65. Refinement was performed using Refmac561,62
Purified TssA2A Nt1-Nt2.His6 (1–374) containing a C-terminal LEHHHHHH tag crystallised in 0.2 M sodium chloride, 0.1 M HEPES buffer (pH 7.0), and 20% (w/v) PEG 6000. Crystals were cryoprotected in 0.2 M sodium chloride, 0.1 M HEPES buffer (pH 7.0), and 20% (w/v) PEG 6000 and 25% ethylene glycol. Native data were collected on beamline I04 at the Diamond Light Source, and data were processed in xia2-DIALS54,55,56,57,58 to 1.76 Å in spacegroup P21. The structure of these crystals was solved by molecular replacement69 using the coordinates of the mercury bound TssA2A Nt2 dimer as a search model. The model was iteratively built using Coot and refined to 1.8 Å using Refmac561,62 to produce the final model (Supplementary Table 6). The asymmetric unit contains 4 molecules of TssA2A Nt1-Nt2.His6 (231–374), and 380 water molecules. Despite the fact that the construct covered both the Nt1 and Nt2 domains the purified protein had undergone proteolytic cleavage leading to the loss of the Nt1 domain. The model therefore contains the entire Nt2 domain (residues 231–374).
Native data on crystals of His6.TssA2A Nt2-CTD (223–478)33 were collected on beamline I03 at the Diamond Light Source. Data were processed in xia2-DIALS54,55,56,58 to 3.13 Å, in spacegroup P21 (Supplementary Table 7). Molecular replacement69 using a search model of the TssA2A Nt2 dimer identified the position of five copies of the TssA2A Nt2 dimer in the asymmetric unit with additional density being seen for 10 copies of the CTD. An initial model was built using Coot60 and subsequently was subjected to rounds of re-building60, and NCS averaging with solvent flattening in Parrot70. Refinement to the native data at 3.13 Å was performed using Refmac561,62 (Supplementary Table 7). Representative electron density for each deposited model is shown in Supplementary Fig. 10. All structures were validated using Molprobity71, with average B-factors calculated using Baverage72. Structural alignment was carried out with LSQKAB using secondary structure matching using Cα of the main chain34. PISA was used to calculate interface areas between molecules32. Structural homologues were discovered using the Dali structural recognition server35. Diagrams shown in this paper were generated using the PyMOL Molecular Graphics System, (Schrödinger, LLC).
Bioinformatics
Phylogenetic analysis was carried out using MEGA 673 and sequence alignment utilised Clustal Omega74. ESpript 3 was used for secondary structure mapping of conserved residues75.
Data availability
The X-ray crystal structures of TssA1B Nt1, TssA1B CTD, His6.TssA1B CTD H12–H14, TssA1B CTD fragment (303–347), TssA2A Nt2, and TssA2A Nt2-CTD and associated data have been deposited in the Protein Data Bank under the ID codes 6HS5 https://www.ebi.ac.uk/pdbe/entry/pdb/6hs5, 6HS6 https://www.ebi.ac.uk/pdbe/entry/pdb/6hs6, 6H8E https://www.ebi.ac.uk/pdbe/entry/pdb/6h8e, 6H8F https://www.ebi.ac.uk/pdbe/entry/pdb/6h8f, 6G7B https://www.ebi.ac.uk/pdbe/entry/pdb/6g7b and 6G7C https://www.ebi.ac.uk/pdbe/entry/pdb/6g7c, respectively. Other data that support the findings of the study are available from the corresponding author(s) upon request.
References
Basler, M., Pilhofer, M., Henderson, G. P., Jensen, G. J. & Mekalanos, J. J. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483, 182–186 (2012).
Ge, P. et al. Atomic structures of a bactericidal contractile nanotube in its pre- and postcontraction states. Nat. Struct. Mol. Biol. 22, 377–382 (2015).
Leiman, P. G. et al. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl Acad. Sci. USA 106, 4154–4159 (2009).
Kanamaru, S. et al. Structure of the cell-puncturing device of bacteriophage T4. Nature 415, 553–557 (2002).
Shneider, M. M. et al. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353 (2013).
Shalom, G., Shaw, J. G. & Thomas, M. S. In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153, 2689–2699 (2007).
Zheng, J. & Leung, K. Y. Dissection of a type VI secretion system in Edwardsiella tarda. Mol. Microbiol. 66, 1192–1206 (2007).
Boyer, F., Fichant, G., Berthod, J., Vandenbrouck, Y. & Attree, I. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genom. 10, 104 (2009).
Cianfanelli, F. R. et al. VgrG and PAAR Proteins Define Distinct Versions of a Functional Type VI Secretion System. PLoS Pathog. 12, 1–27 (2016).
Durand, E. et al. Biogenesis and structure of a type VI secretion membrane core complex. Nature 523, 555–560 (2015).
Aschtgen, M. S., Bernard, C. S., De Bentzmann, S., Lloubes, R. & Cascales, E. SciN is an outer membrane lipoprotein required for Type VI secretion in Enteroaggregative Escherichia coli. J. Bacteriol. 190, 7523–7531 (2008).
Ma, L. S., Lin, J. S. & Lai, E. M. An IcmF family protein, ImpLM, is an integral inner membrane protein interacting with ImpKL, and its Walker a motif is required for type VI secretion system-mediated Hcp secretion in Agrobacterium tumefaciens. J. Bacteriol. 191, 4316–4329 (2009).
Aschtgen, M. S., Gavioli, M., Dessen, A., Lloubès, R. & Cascales, E. The SciZ protein anchors the enteroaggregative Escherichia coli Type VI secretion system to the cell wall. Mol. Microbiol. 75, 886–899 (2010).
Aschtgen, M.-S., Zoued, A., Lloubès, R., Journet, L. & Cascales, E. The C-tail anchored TssL subunit, an essential protein of the enteroaggregative Escherichia coli Sci-1 Type VI secretion system, is inserted by YidC. Microbiologyopen 1, 71–82 (2012).
Durand, E. et al. Structural characterization and oligomerization of the TssL protein, a component shared by bacterial type VI and type IVb secretion systems. J. Biol. Chem. 287, 14157–14168 (2012).
Brunet, Y. R., Hénin, J., Celia, H. & Cascales, E. Type VI secretion and bacteriophage tail tubes share a common assembly pathway. EMBO Rep. 15, 315–321 (2014).
Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D. & Mekalanos, J. J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl Acad. Sci. 104, 15508–15513 (2007).
Bönemann, G., Pietrosiuk, A., Diemand, A., Zentgraf, H. & Mogk, A. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 28, 315–325 (2009).
Lossi, N. S. et al. The HsiB1C1 (TssB-TssC) complex of the Pseudomonas aeruginosa type VI secretion system forms a bacteriophage tail sheathlike structure. J. Biol. Chem. 288, 7536–7548 (2013).
Kube, S. et al. Structure of the VipA/B type VI secretion complex suggests a contraction-state-specific recycling mechanism. Cell Rep. 8, 20–30 (2014).
Wang, J. et al. Cryo-EM structure of the extended type VI secretion system sheath-tube complex. Nat. Microbiol. 2, 1507–1512 (2017).
Brunet, Y. R., Zoued, A., Boyer, F., Douzi, B. & Cascales, E. The Type VI secretion TssEFGK-VgrG phage-like baseplate is recruited to the TssJLM membrane complex via multiple contacts and serves as assembly platform for tail tube/sheath polymerization. PLoS Genet. 11, 1–21 (2015).
Taylor, N. M. I. et al. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 533, 346–352 (2016).
English, G., Byron, O., Cianfanelli, F. R., Prescott, A. R. & Coulthurst, S. J. Biochemical analysis of TssK, a core component of the bacterial Type VI secretion system, reveals distinct oligomeric states of TssK and identifies a TssK–TssFG subcomplex. Biochem. J. 461, 291–304 (2014).
Kapitein, N. et al. ClpV recycles VipA/VipB tubules and prevents non-productive tubule formation to ensure efficient type VI protein secretion. Mol. Microbiol. 87, 1013–1028 (2013).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Zoued, A. et al. Priming and polymerization of a bacterial contractile tail structure. Nature 531, 59–63 (2016).
Planamente, S. et al. TssA forms a gp6-like ring attached to the type VI secretion sheath. EMBO J. 35, 1613–1627 (2016).
Zoued, A. et al. TssA: The cap protein of the Type VI secretion system tail. Bioessays 39, 1600262 (2017).
Zheng, J., Ho, B. & Mekalanos, J. J. Genetic Analysis of Anti-Amoebae and Anti-Bacterial Activities of the Type VI Secretion System in Vibrio cholerae. PLoS ONE 6, e23876 (2011).
Aubert, D. F., Hu, S. & Valvano, M. A. Quantification of type VI secretion system activity in macrophages infected with Burkholderia cenocepacia. Microbiology 161, 2161–2173 (2015).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Dix, S. R. et al. TssA from Aeromonas hydrophila: expression, purification and crystallographic studies. Acta Crystallogr. Sect. F. Struct. Biol. Commun. 74, 578–582 (2018).
Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A 32, 922–923 (1976).
Holm, L. & Rosenström, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010).
Vettiger, A., Winter, J., Lin, L. & Basler, M. The type VI secretion system sheath assembles at the end distal from the membrane anchor. Nat. Commun. 8, 1–9 (2017).
Nazarov, S. et al. Cryo‐EM reconstruction of Type VI secretion system baseplate and sheath distal end. EMBO J. 37, e97103 (2018).
Yonekura, K. et al. The bacterial flagellar cap as the rotary promoter of flagellin self-assembly. Science 290, 2148–2152 (2000).
Yonekura, K., Maki-Yonekura, S. & Namba, K. Growth mechanism of the bacterial flagellar filament. Res. Microbiol. 153, 191–197 (2002).
Renault et al. The gp27-like Hub of VgrG Serves as Adaptor to Promote Hcp Tube Assembly. J. Mol. Biol. 430, 3143–3156 (2018).
Song, W. S., Cho, S. Y., Hong, H. J., Park, S. C. & Yoon, S. Self-oligomerizing structure of the Flagellar cap protein FliD and its implication in filament assembly. J. Mol. Biol. 429, 847–857 (2017).
Yonekura, K., Maki-Yonekura, S. & Namba, K. Complete atomic model of the bacterial flagellar filament by electron cryomicroscopy. Nature 424, 643–650 (2003).
Miller, J. Experiments in Molecular Genetics. (Cold Spring Harb. Lab. Press, Cold Spring Harb, NY, 1972).
Lennox, E. S. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1, 190–206 (1955).
Sokol, P. A., Ohman, D. E. & Iglewski, B. H. A more sensitive plate assay for detection of protease production by Pseudomonas aeruginosa. J. Clin. Microbiol. 9, 538–540 (1979).
Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 (1983).
Herrero, M., De Lorenzo, V. & Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 172, 6557–6567 (1990).
de Lorenzo, V. & Timmis, K. N. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235, 386–405 (1994).
Barrett et al. Genetic Tools for Allelic Replacement in Burkholderia Species. Appl. Environ. Microbiol. 74, 4498–4508 (2008).
Karimova, G., Pidoux, J., Ullmann, A. & Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl Acad. Sci. 95, 5752–5756 (1998).
Owen, H. J. et al. TssA from Burkholderia cenocepacia: expression, purification, crystallization and crystallographic analysis. Acta Crystallogr. Sect. F. Struct. Biol. Commun. 74, 536–542 (2018).
van Heel, M. & Keegstra, W. IMAGIC: a fast, flexible and friendly image analysis software system. Ultramicroscopy 7, 113–129 (1981).
Van Heel, M., Harauz, G., Orlova, E. V., Schmidt, R. & Schatz, M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 116, 17–24 (1996).
Nonaka, T. The CCP4 suite-computer programs for protein crystallography. Nihon Kessho Gakkaishi 36, 224–227 (1994).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D. Biol. Crystallogr. 62, 72–82 (2006).
Kabsch, W. Xds. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 125–132 (2010).
Winter, G. Xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 (2010).
Winter, G. & McAuley, K. E. Automated data collection for macromolecular crystallography. Methods 55, 81–93 (2011).
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 64, 112–122 (2008).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D. Biol. Crystallogr. 67, 355–367 (2011).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D. Biol. Crystallogr. 53, 240–255 (1997).
Sauter, N. K., Grosse-Kunstleve, R. W. & Adams, P. D. Robust indexing for automatic data collection. J. Appl. Crystallogr. 37, 399–409 (2004).
Zhang, Z., Sauter, N. K., Van Den Bedem, H., Snell, G. & Deacon, A. M. Automated diffraction image analysis and spot searching for high-throughput crystal screening. J. Appl. Crystallogr. 39, 112–119 (2006).
Cowtan, K. Fitting molecular fragments into electron density. Acta Crystallogr. Sect. D.-Biol. Crystallogr. 64, 83–89 (2008).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, (213–221 (2010).
Echols, N. et al. Graphical tools for macromolecular crystallography in PHENIX. J. Appl. Crystallogr. 45, 581–586 (2012).
Cowtan, K. ‘Dm’: an automated procedure for phase improvement by density modification. Jt. CCP4 ESF-EACBM Newsl. Protein Crystallogr. 31, 34–38 (1994).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Cowtan, K. Recent developments in classical density modification. Acta Crystallogr. D. Biol. Crystallogr. 66, 470–478 (2010).
Chen, V. B. et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 12–21 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D.-Biol. Crystallogr. 67, 235–242 (2011).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Sievers, F. & Higgins, D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116 (2014).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
We would like to acknowledge James Ault, Andrew Leech and Arthur Moir for carrying out the mass spectrometry, SEC-MALLS and protein sequencing analysis, and for helpful discussions. We would like to thank the Diamond Light Source for access to beamlines I02, I03, I04, I04–1 and I24 that contributed to these results, and the beamline scientists for their assistance with data collection. This work was supported by BBSRC Doctoral Training Grants BB/J014443/1 and BB/F016840/1 awarded to H.J.O. and H.L.S., respectively, a scholarship from the Libyan Ministry of Higher Education awarded to A.A. and a University of Sheffield scholarship awarded to S.R.D. H.J.O was also supported by Newton Fund grants R/151699 and R/152968.
Author information
Authors and Affiliations
Contributions
M.S.T. conceived the investigation. S.R.D., H.J.O., R.S., A.A., S.S., H.L.S., S.B.T. and M.S.T. designed the experiments; S.R.D., H.J.O., R.S., A.A., S.S., M.J.H., S.L.B., H.L.S., D.J.M., S.B.T., T.A.B. and S.E.S. performed the experiments; S.R.D., H.J.O., R.S., A.A., S.S., H.L.S., S.B.T., P.J.B., P.A.B., D.W.R and M.S.T. interpreted the data and S.R.D., H.J.O., R.S., H.L.S., D.W.R. and M.S.T. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dix, S.R., Owen, H.J., Sun, R. et al. Structural insights into the function of type VI secretion system TssA subunits. Nat Commun 9, 4765 (2018). https://doi.org/10.1038/s41467-018-07247-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-07247-1
This article is cited by
-
Structure of a type IV secretion system core complex encoded by multi-drug resistance F plasmids
Nature Communications (2022)
-
The Bradyrhizobium Sp. LmicA16 Type VI Secretion System Is Required for Efficient Nodulation of Lupinus Spp.
Microbial Ecology (2022)
-
TssA–TssM–TagA interaction modulates type VI secretion system sheath-tube assembly in Vibrio cholerae
Nature Communications (2020)
-
A comparative genomics methodology reveals a widespread family of membrane-disrupting T6SS effectors
Nature Communications (2020)
-
Bidirectional contraction of a type six secretion system
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
