
Abstract
Selective creation of quaternary carbon centers has been a long-standing challenge in synthetic chemistry. We report here the chromium-catalyzed, para-selective formation of arylated quaternary carbon centers by alkylative reactions of benzamide derivatives with tertiary alkylmagnesium bromides at room temperature. The reaction, which was enabled by a low-cost chromium(III) salt combined with trimethylsilyl bromide, introduces a sterically bulky tertiary alkyl scaffold on the para-position of benzamide derivatives in a highly selective fashion without either isomerization of the tertiary alkyl group or formation of ortho-alkylated byproducts. Forming low-valent Cr species in situ by reaction of CrCl3 with t-BuMgBr accompanied by evolution of hydrogen can be considered, which serves as reactive species to promote the reaction. The para-alkylation likely occurs via a radical-type nucleophilic substitution of imino-coordination benzimidate intermediate.
Similar content being viewed by others
Introduction
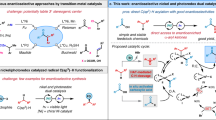
Transition-metal-catalyzed alkylative reactions are fundamental transformations in synthetic chemistry, and they represent a powerful tool with which to incorporate aliphatic scaffolds into molecules; such reactions have been used for the construction of pharmaceuticals and materials1,2,3. However, the introduction of bulky tertiary alkyl groups into motifs for the catalytic formation of quaternary centers has long been a prominent challenge because of the effect of steric hindrance, competing β-hydride elimination and the ease with which such moieties undergo isomerization4,5,6,7,8,9,10,11,12,13. To create arylated quaternary carbon centers, aryl halides, triflates, and organoborons are usually used as aromatic sources to react with tertiary alkyl nucleophiles or electrophiles. These approaches were pioneered by Biscoe14, Fu15, Gong16, and others17,18,19 and typically employed nickel catalysis (Fig. 1a). In contrast, the use of aromatic hydrocarbons for the catalytic formation of arylated quaternary carbon centers has rarely been studied.

Transition-metal-catalyzed formation of arylated quaternary carbon centers by alkylation. a Known examples of the formation of arylated quaternary carbon centers with nickel catalysis. b para-Selective alkylation of benzamide with nickel catalysis. c Cr-catalyzed para-alkylation of benzamides for the formation of arylated quaternary carbon centers
Given that aromatic hydrocarbons usually contain site-differentiated C–H bonds, regioselectivity in the incorporation of bulky tertiary alkyl groups is a formidable obstacle. The application of common methods involving ortho-alkylation to introduce arylated quaternary carbon centers may be challenging because of the effect of steric repulsion20. A landmark study by Nakao showed that arylated 2° carbon centers can be created in a para-selective manner with benzamides through nickel/aluminum co-catalyzed hydroarylation (Fig. 1b)21,22,23. We questioned whether it was possible to construct quaternary carbon centers at the para-position of benzamides by using a transition-metal-catalyzed alkylative reaction with sterically bulky tertiary alkyl nucleophiles.
Recently, transition-metal catalysis using abundant, low-cost base metals such as nickel, iron, and cobalt has appeared as a cost-effective tool for organic synthesis24,25,26,27,28,29. In contrast to the great achievements made with these first-row metals, synthetic chemistry of Group 6 metal chromium has still been underdeveloped30,31,32,33,34,35,36,37,38,39,40. Herein, we report that the para-selective formation of arylated quaternary carbon centers was enabled by using low-cost chromium(III) salt combined with trimethylsilyl bromide to achieve alkylative reaction of benzamides with tertiary alkylmagnesium bromides at room temperature (Fig. 1c). This reaction proceeded with high selectivity, with only the para-carbons of benzamides being alkylated without isomerization of bulky tertiary alkyl groups.
Results
Reaction optimization
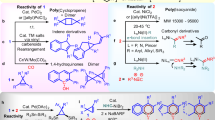
Based on our previous results, the treatment of chromium salt with PhMgBr allowed the formation of low-valent species, which show high catalytic activity in the selective cleavage of inert C–O and C–N bonds34,35. We initially probed the reactivity of chromium in promoting the alkylation of N-methylbenzamide (1) with t-butylmagnesium bromide (Table 1). With 10 mol% CrCl3, the alkylation of benzamide with t-BuMgBr did not occur (entry 1). Conducting the reaction with 2,3-dichlorobutane (2,3-DCB) as the additive did not give the alkylated compound (entry 2). Gratifyingly, chlorodimethyl(phenyl)silane could be used to promote the Cr-catalyzed alkylation, in which the bulky t-butyl group was selectively incorporated at the para-position of benzamide to afford the compound 3 in 44% yield (entry 3). In contrast, the alkylation did not take place using dichlorodiphenylsilane (entry 4). The inclusion of bromotrimethylsilane (TMSBr) greatly improved the transformation, giving 3 in preparatively useful yield (entry 6). The use of other chromium salts such as CrCl2 and Cr(acac)3 led to inferior result (entries 7 and 8). Other first-row transition-metal complexes such as Ni(cod)2 and CoCl2 were completely inactive in promoting the para-selective alkylation using t-BuMgBr (entries 9 and 10). Interestingly, the para-alkylation with 10 mol% of FeCl3, AlCl3, or AlMe3 also occurred, albeit with low conversions (entries 11–13).
Substrate scope
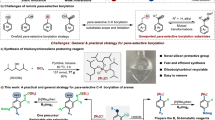
The substrate scope of the para-alkylation was then examined for the construction of structurally diverse substituted quaternary carbon centers with benzamides. As shown in Fig. 2, the benzamide derivative containing an ortho-methyl, benzyl, phenoxymethyl, or tert-butoxy(phenyl)methyl substituent reacted with t-BuMgBr smoothly to form the para-alkylated compound (4–6). Alkylation using benzamide bearing an electron-withdrawing fluoride substituent on the ortho-position gave an inferior result compared with those bearing electron-donating groups (7 and 8). Meanwhile, the incorporation of alkoxy and phenoxy groups into the ortho position of benzamides did not affect the para-selective alkylation of C–H bonds (8–11). Interestingly, the Cr-catalyzed reaction of 2-hydroxy-N-methylbenzamide occurred smoothly to give 2-hydoxyl and 4-tert-butyl-substituted benzamide derivative, albeit with a low conversion (12). The para-C–H bonds in the scaffolds of N-methylbenzamides bearing an ortho-methylthio or trimethylsilyl group can be effectively alkylated under present conditions, providing access to the desired products 13 and 14 in 61% and 52% yields, respectively. We were pleased to find that [1,1’-biphenyl]-2-carboxamide motifs containing functional substituents of methyl, phenyl, chloride, trifluoromethyl, methylthio, amino, and alkoxycarbonyl groups could couple with tert-butyl Grignard at the amide-bearing aromatics, the formation of diverse-substituted derivatives 15–23 in preparatively useful yields. In addition, ortho-naphthyl and thienyl-bearing benzamides were also amenable to the para-alkylative cross-coupling reaction (24 and 25). It was noteworthy that steric hindrance arising from the meta-substituents of benzamides did not affect the site-selectivity of alkylation, allowing for incorporating the bulky tert-butyl group at the para-position of benzamides (26–28). A broad range of functionalities, such as chloride, methylthio, trifluoromethyl, trifluoromethoxy, trimethylsilyl, hydroxyl, amino, alkoxycarbonyl, naphthyl, and thienyl were well retained under the reaction conditions. Importantly, this Cr-catalyzed para-selective alkylation can be applied to prepare tri-substituted N-methylbenzamide derivative that contains 2-fluoro-3-methoxy, 2,3-dimethoxy or dihydrobenzo[b][1,4]dioxine scaffold (29–31). Interestingly, the alkylation using N-methylthiophene-2-carboxamide led to the formation of a quaternary carbon center at the C5 position of thiophene, leading to 5-tert-butyl-substituted thiophene derivative 32. The chromium-catalyzed protocol is scalable, and can be applied to the synthesis of para-alkylated benzamide 3 on a gram scale.

Cr-catalyzed para-selective alkylation of benzamides with tert-butyl Grignard reagent. Conditions: N-methylbenzamide derivative (0.2 mmol), tert-butylmagnesium bromide (0.8 mmol), CrCl3 (0.02 mmol), TMSBr (0.6 mmol), THF (0.5 mL), rt, 24 h. Isolated yields are given. aYield of gram-scale reaction with 1a (10 mmol, 1.36 g). bTert-butylmagnesium bromide (1 mmol) was employed
In addition to t-butylmagnesium bromide, tertiary alkyl nucleophiles, such as 2-methyl-4-phenylbutan-2-yl, 2-methylhexan-2-yl, 2-methylnonan-2-yl, t-pentyl, 3-ethylpentan-3-yl, and methylcyclohexyl-substituted Grignard reagents also reacted with N-methylbenzamide smoothly under chromium catalysis, permitting the incorporation of sterically bulky tertiary alkyl scaffolds into the para-position of benzamide in the synthesis of related compounds 33–38 (Fig. 3). However, the para-alkylative reaction with adamantyl-substituted Grignard reagent furnished the coupling product 39 in low yield. Variation of N-methyl group to ethyl and substituted phenyl in the benzamide motifs did not hamper the para-selective transformation (40–43). Notably, these Cr-catalyzed alkylation reactions all proceeded with high selectivity without the formation of tertiary alkyl isomerized side products, and only the para-carbons of benzamides were alkylated at room temperature. However, the reaction between benzophenone and tert-butyl Grignard reagent did not afford the alkylated product. It was observed that primary and secondary alkylmagnesium bromide cannot react with N-methylbenzamide to give the alkylated compounds. Interestingly, the formation of para-TMS-substituted benzamide in low yield was obversed when using isopropyl Grignard reagent in the alkylation.

Cr-catalyzed para-alkylation of benzamides with tertiary alkylmagnesium bromides. Conditions: benzamide (0.2 mmol), tertiary alkylmagnesium bromide (0.8 mmol), CrCl3 (0.02 mmol), TMSBr (0.6 mmol), THF (0.5 mL), rt, 24 h. Isolated yields are given
Discussion
To probe the possibility of forming low-valent chromium species, the reaction of CrCl3 with t-BuMgBr was performed at room temperature (Fig. 4a). The evolution of hydrogen gas was observed and quantified by GC/MS analyses. The amount of hydrogen evolution was nearly equal to the amount of chromium(III) salt, which indicates that the formation of a low-valent chromium species through transmetalation of CrCl3 followed by β-Hydride elimination and hydride reduction could be considered. After the evolution of hydrogen, benzamide and TMSBr were added and the para-alkylation also occurred effectively to form 3 in 65% yield, suggesting that the reactive Cr species generated in situ could promote the transformation (Fig. 4b).

The formation of low-valent chromium species in situ for the para-selective alkylation. a Hydrogen evolution by the reaction of CrCl3 with t-BuMgBr. b para-Selective alkylation of benzamide that was promoted by the in situ generated low-valent Cr species
The reaction of N,N’-dimethylbenzamide (44) did not form the product, confirming that deprotonation of the NH group in benzamide by Grignard reagent to give benzimidate species is required to achieve the para-alkylation (Fig. 5a). The reaction of trimethylsilyl (Z)-N-phenylbenzimidate (46) with t-BuMgBr allowed the formation of the para-alkylated product 41 with Cr catalysis (Fig. 5b); whereas, the alkylation did not occur in the absence of either CrCl3 or TMSBr (Fig. 5c, d). These result shows that Cr and TMSBr play important roles in the latter transformation of para-C–H bond. Like Nakao’s reaction, the imino group on the benzimidate intermediate could ligate with the metal, and the coordination may enhance the reactivity of the electron-poor aromatic at the para position toward functionalization21,41.

Preliminary mechanistic studies. a Alkylation with N,N-dimethyl-substituted benzamide. b Alkylation with N-phenylbenzimidate. c Alkylation without CrCl3 salt. d Alkylation without TMSBr. e Quenching the alkylation using D2O. f Scavenger experiment with TEMPO. g Alkylation with deuterated N-methylbenzamide. h Alkylation with 1-methylcyclohex-1-ene. i Plausible reaction pathways
Quenching the alkylation by D2O showed that almost no deuterium was incorporated into the ortho- or para-positions of the product 3 and starting material 1. The related C–H bonds may not be metalated under present conditions (Fig. 5e). It was noteworthy that the addition of free radical inhibitor such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) into the reaction shut down the alkylation (Fig. 5f)42,43,44,45,46,47. The analysis of the alkylation of 1a after 4 h using EPR spectroscopy suggests the formation of radical species during this reaction (see Supplementary Fig. 5 for details)48,49,50. Meanwhile, radical species in the reaction of t-BuMgBr with CrCl3 and TMSBr was detected by EPR study51. Based on previous reports and these experimental results, the alkylation may proceed by a radical-type para-nucleophilic substitution of imino-coordination benzimidate to afford aryl radical, which undergoes a single electron transfer (SET)/proton transfer process to form para-alkylated compound (Fig. 5i)52,53. As to the role of trimethylsilyl bromide, in addition to the formation of benzimidate intermediate 46, we hypothesized that it may help to give tert-butyl radical and TMS-Cr intermediate by reaction with t-BuMgBr and low-valent Cr species in the catalytic cycle. The observation of para-TMS-substituted benzamide compound when using i-PrMgBr may indicate that the formation of TMS-Cr intermediate could be considered. It was found that no deuterium was incorporated into the tert-butyl group in 3-d4, and adding 1-methylcyclohex-1-ene into the reaction did not give hydroarylation products such as compound 38 (Fig. 5g, h). The addition of para-C–H bonds across olefin would not be involved in the alkylation. On the other hand, a small kinetic isotope effect (KIE ≈ 1.4) was observed in the Cr-catalyzed para-alkylation.
When kinetic studies on the reaction between 1a with t-BuMgBr were carried out, the data revealed a positive first-order dependence of the alkylation on the concentration of CrCl3 (Fig. 6a). This result suggests that the concentration of chromium likely determines the reaction rate and that a unimolecular event can be considered the turnover-liming step. The linear plot of log(kobs) versus the logarithm of [1] showed the reaction to have a slight first-order dependence on the concentration of benzamide (Fig. 6b).

Kinetic profile for chromium-catalyzed para-alkylative reaction. a Plot of initial rate vs concentration of CrCl3 reveal first-order kinetics for chromium salt. b Plot of initial rate vs concentration of 1 indicates a slightly first-order kinetics for benzamide
In conclusion, we have developed the para-selective alkylation of benzamide derivatives with chromium catalysis for the formation of arylated quaternary carbon centers. The use of low-cost chromium(III) salt as precatalyst combined with trimethylsilyl bromide allowed the alkylative reaction to occur smoothly at room temperature. The methodology provides a selective way to incorporate bulky tertiary alkyl groups into the para-position of benzamide derivatives without either isomerization or ortho-alkylation. The presented catalytic activity of low-valent Cr function as a redox shuttle in the para-selective formation of quaternary carbon centers should spur the development of synthetic strategies with chromium.
Methods
Cr-catalyzed para-selective formation of quaternary centers
A dried Schlenk tube was charged with N-methylbenzamide 1 (0.2 mmol), CrCl3 (3 mg, 0.02 mmol) and freshly distilled THF (0.5 mL). Tertiary alkylmagnesium bromide 2 (0.8–1 mmol) was dropwise added by syringe at room temperature. After stirring the mixture for 30 min, trimethylbromosilane (92 mg, 0.6 mmol) was added by syringe and the mixture was stirred at room temperature for 24 h. The resulting mixture was then quenched by an aqueous solution of NH4Cl and extraction with ethyl acetate (3 × 10 mL). The combined organic phase was dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by silica gel chromatography to give the para-alkylated product 3.
Spectroscopic methods
1H and 13C NMR spectra were recorded on a Bruker DRX-400 (operating at 400 MHz for 1H and 100 MHz for 13C). UV–vis spectra were recorded on a Thermo Fisher Nicolet 6700 FT-IR spectrometer using ATR (Attenuated Total Reflectance) method.
Single-crystal X-ray structure determinations
The crystal data of 3a were collected on a Bruker SMART CCD diffractometer with MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined on F2 using SHELXTL. All non-hydrogen atoms were refined anisotropically.
Data availability
The X-ray crystallographic coordinates for structures that support the findings of this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession code CCDC 1821836 (3). The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request.
References
Trost, B. M. Pd- and Mo-catalyzed asymmetric allylic alkylation. Org. Process Res. Dev. 16, 185–194 (2012).
Harutyunyan, S. R., den Hartog, T., Geurts, K., Minnaard, A. J. & Feringa, B. L. Catalytic asymmetric conjugate addition and allylic alkylation with Grignard reagents. Chem. Rev. 108, 2824–2852 (2008).
Dong, Z., Ren, Z., Thompson, S. J., Xu, Y. & Dong, G. Transition-metal-catalyzed C–H alkylation using alkenes. Chem. Rev. 117, 9333–9403 (2017).
Hayashi, T., Konishi, M., Yokota, K.-i. & Kumada, M. Cross-coupling of tertiary alkyl Grignard reagents with β-bromostyrene catalyzed by dichloro[1,1’-bis(diphenylphosphino)ferrocene]nickel(II). Chem. Lett. 9, 767–768 (1980).
Iwasaki, T., Takagawa, H., Singh, S. P., Kuniyasu, H. & Kambe, N. Co-catalyzed cross-coupling of alkyl halides with tertiary alkyl Grignard reagents using a 1,3-butadiene additive. J. Am. Chem. Soc. 135, 9604–9607 (2013).
Ren, P., Stern, L. A. & Hu, X. Copper-catalyzed cross-coupling of functionalized alkyl halides and tosylates with secondary and tertiary alkyl Grignard reagents. Angew. Chem. Int. Ed. 51, 9110–9113 (2012).
Hintermann, L., Xiao, L. & Labonne, A. A general and selective copper-catalyzed cross-coupling of tertiary Grignard reagents with azacyclic electrophiles. Angew. Chem. Int. Ed. 47, 8246–8250 (2008).
Iwasaki, T., Yamashita, K., Kuniyasu, H. & Kambe, N. Co-catalyzed cross-coupling reaction of alkyl fluorides with alkyl Grignard reagents. Org. Lett. 19, 3691–3694 (2017).
Rezazadeh, S., Devannah, V. & Watson, D. A. Nickel-catalyzed C-alkylation of nitroalkanes with unactivated alkyl iodides. J. Am. Chem. Soc. 139, 8110–8113 (2017).
Thapa, S. et al. Ligand-free copper-catalyzed Negishi coupling of alkyl-, aryl-, and alkynylzinc reagents with heteroaryl iodides. Angew. Chem. Int. Ed. 54, 8236–8240 (2015).
Martín, R. & Buchwald, S. L. An improved protocol for the Pd-catalyzed α-arylation of aldehydes with aryl halides. Org. Lett. 10, 4561–4564 (2008).
Peacock, D. M., Roos, C. B. & Hartwig, J. F. Palladium-catalyzed cross coupling of secondary and tertiary alkyl bromides with a nitrogen nucleophile. ACS Cent. Sci. 2, 647–652 (2016).
Zhao, C., Jia, X., Wang, X. & Gong, H. Ni-catalyzed reductive coupling of alkyl acids with unactivated tertiary alkyl and glycosyl halides. J. Am. Chem. Soc. 136, 17645–17651 (2014).
Joshi-Pangu, A., Wang, C.-Y. & Biscoe, M. R. Nickel-catalyzed Kumada cross-coupling reactions of tertiary alkylmagnesium halides and aryl bromides/triflates. J. Am. Chem. Soc. 133, 8478–8481 (2011).
Zultanski, S. L. & Fu, G. C. Nickel-catalyzed carbon–carbon bond-forming reactions of unactivated tertiary alkyl halides: Suzuki arylations. J. Am. Chem. Soc. 135, 624–627 (2014).
Wang, X., Wang, S., Xue, W. & Gong, H. Nickel-catalyzed reductive coupling of aryl bromides with tertiary alkyl halides. J. Am. Chem. Soc. 137, 11562–11565 (2015).
Zhou, Q., Cobb, K. M., Tan, T. & Watson, M. P. Stereospecific cross couplings to set benzylic, all-Carbon quaternary stereocenters in high enantiopurity. J. Am. Chem. Soc. 138, 12057–12060 (2016).
Primer, D. N. & Molander, G. A. Enabling the cross-coupling of tertiary organoboron nucleophiles through radical-mediated alkyl transfer. J. Am. Chem. Soc. 139, 9847–9850 (2017).
Lohre, C., Dröge, T., Wang, C. & Glorius, F. Nickel-catalyzed cross-coupling of aryl bromides with tertiary Grignard reagents utilizing donor-functionalized N-heterocyclic carbenes (NHCs). Chem. Eur. J. 17, 6052–6055 (2011).
Li, J. et al. N-Acyl amino acid ligands for ruthenium(II)-catalyzed meta-C–H tert-alkylation with removable auxiliaries. J. Am. Chem. Soc. 137, 13894–13901 (2015).
Okumura, S. et al. para-Selective alkylation of benzamides and aromatic ketones by cooperative nickel/aluminum catalysis. J. Am. Chem. Soc. 138, 14699–14704 (2016).
Okumura, S. & Nakao, Y. para-Selective alkylation of sulfonylarenes by cooperative nickel/aluminum catalysis. Org. Lett. 19, 584–587 (2017).
Yang, L., Semba, K. & Nakao, Y. para-Selective C−H borylation of (hetero)arenes by cooperative iridium/aluminum catalysis. Angew. Chem. Int. Ed. 56, 4853–4857 (2017).
Tobisu, M. & Chatani, N. Cross-couplings using aryl ethers via C–O bond activation enabled by nickel catalysts. Acc. Chem. Res. 48, 1717–1726 (2015).
Tasker, S. Z., Standley, E. A. & Jamison, T. F. Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014).
Gao, K. & Yoshikai, N. Low-valent cobalt catalysis: new opportunities for C–H functionalization. Acc. Chem. Res. 47, 1208–1219 (2014).
Kommagalla, Y. & Chatani, N. Cobalt(II)-catalyzed C–H functionalization using an N,N-bidentate directing group. Coord. Chem. Rev. 350, 117–135 (2017).
Nakamura, E. & Yoshikai, N. Low-valent iron-catalyzed C−C bond formation−addition, substitution, and C−H bond activation. J. Org. Chem. 75, 6061–6067 (2010).
Sun, C.-L., Li, B.-J. & Shi, Z.-J. Direct C–H transformation via iron catalysis. Chem. Rev. 111, 1293–1314 (2011).
Fürstner, A. Carbon−carbon bond formations involving organochromium(III) reagents. Chem. Rev. 99, 991–1046 (1999).
Zeng, X. & Cong, X. Chromium-catalyzed transformations with Grignard reagents–new opportunities for cross-coupling reactions. Org. Chem. Front 2, 69–72 (2015).
Yan, J. & Yoshikai, N. Phenanthrene synthesis via chromium-catalyzed annulation of 2-biaryl Grignard reagents and alkynes. Org. Lett. 19, 6630–6633 (2017).
Yan, J. & Yoshikai, N. Chromium-catalyzed migratory arylmagnesiation of unactivated alkynes. Org. Chem. Front 4, 1972–1975 (2017).
Cong, X. et al. Low-valent, high-spin chromium-catalyzed cleavage of aromatic carbon–nitrogen bonds at room temperature: a combined experimental and theoretical study. J. Am. Chem. Soc. 139, 15182–15190 (2017).
Cong, X., Tang, H. & Zeng, X. Regio- and chemoselective Kumada–Tamao–Corriu reaction of aryl alkyl ethers catalyzed by chromium under mild conditions. J. Am. Chem. Soc. 137, 14367–14372 (2015).
Steib, A. K., Kuzmina, O. M., Fernandez, S., Malhotra, S. & Knochel, P. Chemoselective chromium(II)-catalyzed cross-coupling reactions of dichlorinated heteroaromatics with functionalized aryl Grignard reagents. Chem. Eur. J. 21, 1961–1965 (2015).
Steib, A. K., Kuzmina, O. M., Fernandez, S., Flubacher, D. & Knochel, P. Efficient chromium(II)-catalyzed cross-coupling reactions between Csp2 centers. J. Am. Chem. Soc. 135, 15346–15349 (2013).
Murakami, K., Ohmiya, H., Yorimitsu, H. & Oshima, K. Chromium-catalyzed arylmagnesiation of alkynes. Org. Lett. 9, 1569–1571 (2007).
Kuzmina, O. M. & Knochel, P. Room-temperature chromium(II)-catalyzed direct arylation of pyridines, aryl oxazolines, and imines using arylmagnesium reagents. Org. Lett. 16, 5208–5211 (2014).
Li, Y., Deng, G. & Zeng, X. Chromium-catalyzed regioselective hydropyridination of styrenes. Organometallics 35, 747–750 (2016).
Fuson, R. C. & Tull, R. Para alkylation of hindered ketones by the Grignard reagent. J. Am. Chem. Soc. 71, 2543–2546 (1949).
Xu, W., Paira, R. & Yoshikai, N. Ortho-C–H benzylation of aryl imines with benzyl phosphates under cobalt–pyphos catalysis. Org. Lett. 17, 4192–4195 (2015).
Li, B.-J. et al. Cross-coupling of alkenyl/aryl carboxylates with Grignard reagent via Fe-catalyzed C-O bond activation. J. Am. Chem. Soc. 131, 14656–14657 (2009).
Perez Garcia, P. M., Di Franco, T., Orsino, A., Ren, P. & Hu, X. Nickel-catalyzed diastereoselective alkyl–alkyl Kumada coupling reactions. Org. Lett. 14, 4286–4289 (2012).
Ilies, L., Matsubara, T. & Nakamura, E. Nickel-catalyzed synthesis of diarylamines via oxidatively induced C–N bond formation at room temperature. Org. Lett. 14, 5570–5573 (2012).
Maji, M. S., & Studer, A. Transition-metal-free oxidative homocoupling of aryl, alkenyl, and alkynyl Grignard reagents with TEMPO. Synthesis 2009, 2467–2470 (2009).
Maji, M. S., Murarka, S. & Studer, A. Transition-metal-free Sonogashira-type coupling of ortho-substituted aryl and alkynyl Grignard reagents by using 2,2,6,6-tetramethylpiperidine-N-oxyl radical as an oxidant. Org. Lett. 12, 3878–3881 (2010).
Gough, R. G. & Dixon, J. A. Radical mechanisms in reactions of Grignard reagents. J. Org. Chem. 33, 2148–2149 (1968).
Ashby, E. C. & Bowers, J. R. Jr. Organometallic reaction mechanisms. 17. Nature of alkyl transfer in reactions of Grignard reagents with ketones. Evidence for radical intermediates in the formation of 1,2-addition product involving tertiary and primary Grignard reagents. J. Am. Chem. Soc. 103, 2242–2250 (1981).
Zhang, L. & Jiao, L. Pyridine-catalyzed radical borylation of aryl halides. J. Am. Chem. Soc. 139, 607–610 (2017).
Watabe, H., Terao, J. & Kambe, N. Formation of 1,4-disilyl-2-butenes from vinyl Grignard reagent and chlorosilanes catalyzed by a titanocene complex. Org. Lett. 3, 1733–1735 (2001).
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692–700 (2017).
Ortiz, F. L., Iglesias, M. J., Fernández, I., Sánchez, C. M. A. & Gómez, G. R. Nucleophilic dearomatizing (DNAr) reactions of aromatic C,H-systems. A mature paradigm in organic synthesis. Chem. Rev. 107, 1580–1691 (2007).
Acknowledgements
The support for this work by the National Natural Science Foundation of China (Nos. 21202128 and 21572175), SCU from a start-up fund and Beijing National Laboratory for Molecular Sciences is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
P.L., C.C. and X.C. performed the experiments and analysed the data. J.T. and X.Z. wrote the manuscript. P.L., C.C. and X.C. contributed equally. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, P., Chen, C., Cong, X. et al. Chromium-catalyzed para-selective formation of quaternary carbon centers by alkylation of benzamide derivatives. Nat Commun 9, 4637 (2018). https://doi.org/10.1038/s41467-018-07069-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-07069-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
