
Abstract
Shizukaols possess a common heptacyclic framework containing more than ten contiguous stereocenters and potential biological activities. Here we report that the total syntheses of shizukaols A (1) and E (2), two lindenane-type dimers from the Chloranthaceae family, are achieved via a modified biomimetic Diels–Alder reaction. The common crucial biomimetic diene 23 and ethylene species (6, 17) are obtained through either a highly Z-selective olefination of α-siloxy ketone with ynolate anions or an intramolecular Horner–Wadsworth–Emmons olefination from commercially available Wieland–Miescher ketone (7). This synthetic approach here opens up practical avenues for the total syntheses of the intriguing Chloranthaceae family members, as well as the understanding of their relevant biological action in nature.
Similar content being viewed by others
Introduction
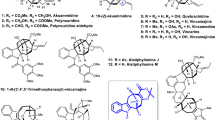
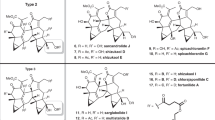
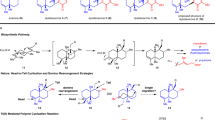
In 1990, shizukaol A (1) (Fig. 1) was isolated from Chloranthus japonicus1,2 as the first dimeric lindenane-type sesquiterpenoid. Subsequently, shizukaol E (2) (Fig. 1) was successively isolated from Chloranthus japonicus in 19953,4. To date, >110 congeners with a similar polycyclic core have been identified from the Chloranthaceae family5,6,7. These molecules possess a common heptacyclic framework containing >10 contiguous stereocenters. Noteworthy is that shizukaol E (2) showed potential anti-HIV-1 replication activities and significant inhibition effects against the currently most popular NNRTI-resistant HIV-1, together with the inhibitory activities on HCV replication, making it attractive as a potential therapy against the co-infection of HIV-1 and HCV8. In addition, most of these dimeric lindenane-type molecules show impressive bioactivities, such as remarkable cytotoxicity9, inhibition of the delayed rectifier K+ current10,11, and antimalarial activities12. Therefore, the promising bioactivities and synthetic challenges arising from these unprecedented molecules have initiated a significant interest to synthetic chemists13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30. However, only one total synthesis of two dimeric members30 and a number of relevant synthetic studies13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29 have been achieved. In 2017, Liu and coworkers reported the first total syntheses of the dimeric shizukaol D (3) and sarcandrolide J (4)30 through a cascade protocol featuring furan formation/alkene isomerization/Diels–Alder reaction. As shown in Fig. 2, the pyrolysis of shizukaol A (1) in a sealed tube resulted in a retro Diels–Alder fragmentation of the linking six-membered ring to furnish two products: the relatively unstable diene 5 and the previously known chloranthalactone A (6)1,2. This led to a biogenetic hypothesis for the lindenane dimeric family in which enzymatic endo-Diels–Alder reaction of two lindenane-type components (such as 5 and 6) forms a common linking six-membered ring. Based on this hypothesis, we have illustrated a practical strategy for constructing the heptacyclic framework of the Chloranthaceae family via an endo-Diels–Alder reaction as the pivotal step, as shown in Fig. 228,29. Encouraged by our preliminary results of endo-Diels–Alder reaction28,29, herein we accomplished the total syntheses of two members of this dimeric family, shizukaols A (1) and E (2), through mimicking a biosynthetic approach.

Structures of dimeric lindenane-type representives. Shizukaol family possesses a common heptacyclic framework with >10 contiguous stereocenters

Pyrolysis of shizukaol A. Shizukaol A was pyrolysized to afford relatively unstable diene 5 and known chloranthalactone A (6) via a retro Diels–Alder fragmentation of the linking six-membered ring in a sealed tube
Results
Retrosynthetic analysis
Inspired by our preliminary investigations28,29 on endo-Diels–Alder reaction to generate heptacyclic framework of the Chloranthaceae family, we considered to design more suitable precusors, being at least biogenetically much closer to components 5 and 6 for further transformation. Inspection of the structures of shizukaols A (1) and E (2) reveals a linking six-membered ring and a hydroxyl ketoester moiety to be the common structural features of many dimeric lindenane-type molecules. In consideration of the likely instability of 5, as well as the long synthetic steps in the conversion of our preliminary [4+2] cycloaddition products to shizukaol A (1), we reason that the synthesis of a crucial heptacyclic core with suitable functional groups for subsequent conversions to naturally occurring shizukaols A (1) and E (2) should be a more practical approach. Moreover, our preliminary investigation proved that the late-stage conversion of the epoxy unit in 9 (Fig. 3) to the hydroxyl ketoester unit of shizukaols A (1) and E (2), a common framework of the dimeric family members, is a more step-economic strategy for total syntheses. In addition, the epoxide moiety in triene 9 is expected to prevent a further [4+2] cycloaddition of relevant alkenyl unit. Therefore, on basis of these considerations and the aforementioned biogenetic hypothesis, we think that the critical step for the construction of skeleton relevant to that of shizukaols A (1) and E (2) would then be the expedient and pragmatic conversion of readily accessible precursor 10 and biomimetic epoxide 9 into the endo-Diels–Alder reaction product 11 via the relevant transition state. Upon endo-Diels–Alder reaction of 9 and 10, epoxy compound 11 could readily be obtained which then undergoes a biomimetic transformation of epoxy unit to the common hydroxyl ketoester species of the shizukaol family, eventually achieving shizukaols A (1) and E (2).

Retrosynthetic analysis toward shizukaols A and E. This proposed protocol is featuring with a modified biomimetic approach involving an endo-Diels–Alder reaction and biomimetic transformation
Total syntheses of shizukaols A and E
Based on the aforementioned synthetic strategy, we then commenced the synthesis of ethylene 17 from known compound 12 (Fig. 4)28,29, which was obtained through the sequence of classical Mitsunobu reaction31,32, nickel-mediated reduction33,34, and modified Julia–Kocienski olefination (see Supporting Information for details)35. Compound 12 was then treated with 9-BBN to form an inseparable mixture of diastereoisomers in a ratio of 8:1 (α/β). Deprotection with FeCl3•SiO2 led to alcohol 13 with an α-hydroxymethyl group as the major product. It is noteworthy that Zhao and coworkers reported the synthesis of the single stereoisomer of 13 with β-hydroxymethyl group under a similar hydroboration condition19. Therefore, in order to further identify the stereochemistry, protection of hydroxyl group in 13 with p-nitrobenzoyl group provided compound 13a in a quantitative yield. After many trials, compound 13a gave finally satisfactory single crystals from CH2Cl2, whose X-ray diffraction study convincingly showed an α-hydroxymethyl group in 13a to be consistent with its relevant orientation in shizukaol E (2). The stereoselectivity of this hydroboration is presumably caused by the presence of 1,3-syn-diaxial repulsion in alkene 12. Subsequent protection of the primary alcohol with a TBS group gave ketone 14, which reacted with Bredereck’s reagent and singlet oxygen successfully to form dione 15 in 77% yield36,37. The resulting dione 15 underwent Yamaguchi esterification38 with commercially available 2-(diethoxyphosphoryl)propanoic acid (15a) and 2,4,6-trichlorobenzoyl chloride (15b) to furnish an easily separable mixture of 16 and its regioisomer 16a in 59% and 25% yields, respectively. The undesired regioisomer 16a could be recycled into dione 15 in 81% yield in the presence of Na2CO3. Then, treatment of 16 with NaH in THF led to the desired ethylene 17 in 77% yield.

Synthesis of ethylene 17. 9-BBN 9-borabicyclo[3.3.1]nonane, TBS tert-butyldimethylsilyl, DMAP 4-dimethylaminopyridine, DMP Dess–Martin periodinane
With ethylene 17 in hand, we turned our attention to the synthesis of diene 23 from known enone 1828,29. As shown in Fig. 5, regio- and stereoselective dihydroxylation of enone 18 in the presence of a catalytic amount of OsO4 and one equivalent of NMO afforded exclusively the corresponding diol from the less-hindered α-face. Upon regioselective protection of the less sterically hindered secondary alcohol with a TBS group, α-siloxy ketone 19 was furnished in 70% yield. The regioselectivity of silylation could be ascribed to the steric hindrance of the angular methyl and the bulky silyl group. Next, treatment of α-siloxy ketone 19 with freshly prepared ynolate 19a led to a highly Z-selective olefination of 19 under a mild condition39,40,41. As a result, butenolide 20 was smoothly generated via a spontaneous silyl migration and lactonization. Moreover, the stereochemistry of 20 was confirmed through an X-ray crystallographic analysis of a single crystal of butenolide 20 grown from CH2Cl2. Lactone 20 was then allowed to undergo elimination of OTBS with DBU to generate 21 in 80% yield, which increased to 93% yield after recovery of 20. As shown in Fig. 3, we predicted that β-epoxide would facilitate subsequent transformation. Lamentably, we failed to form directly the β-epoxide from alkene 21 after considerable testing of epoxidation protocols. Similarly, we failed to convert the α-epoxide of compound 22 into β-epoxide after extensive efforts. We therefore planned to reverse its relevant stereochemistry at a later stage. Epoxidation of 21 with m-CPBA led exclusively to α-epoxide 22. Then, dehydration of 22 with Martin sulfurane smoothly gave rise to triene 2342,43, which was very unstable and easy to dimerize in its neat form or undiluted solution. Fortunately, diene 23 was relatively stable when diluted in toluene or xylene and stored at −20 °C for immediate use without obvious dimerization or polymerization.

Synthesis of diene 23. NMO N-methylmorpholine N-oxide, TBS tert-butyldimethylsilyl, DMAP 4-dimethylaminopyridine, DBU 1,8-diazabicyclo[5.4.0]undec-7-ene, and m-CPBA meta-chloroperbenzoic acid
Next, we investigated the possibility of an endo-Diels–Alder reaction between electron-deficient electrophilic diene 23 and electron-rich C8′–C9′ double bond of nucleophilic ethylene 1744,45,46. As shown in Fig. 6, treatment of compounds 17 and 23 with butylated hydroxytoluene (BHT) at 160 °C in xylene successfully led to the endo-Diels–Alder reaction cycloadduct 24 as a single detectable isomer. Noteworthy, the homo-dimerization product of diene 23 was observed as the major by-product of this Diels–Alder reaction. Therefore, in order to reduce the homo-dimerization of diene 23, it is essential for endo-Diels–Alder reaction that a high dilute solution of 23 was added into a refluxing dilute solution of 17 very slowly. Then, removal of the silyl group in 24 led to the crucial alcohol 25 in 87% yield as a solid. After many trials, alcohol 25 gave eventually satisfactory single crystals from CH2Cl2. Its X-ray diffraction study confirmed that alcohol 25 was the desired endo-Diels–Alder reaction product. However, efforts to improve this Diels–Alder reaction using Lewis acid/base catalysts caused decomposition of the sensitive substrates. Therefore, this thermal Diels–Alder reaction appears to be an acceptable approach for generating 24 as a single detectable dimer. With alcohol 25 in hand, it was then allowed to undergo hydrolysis with K2CO3 to generate the corresponding diol, thereby installing the crucial hydroxyl ketoester moiety of the natural product family in a biomimetic transformation. Selective acetylation of the primary alcohol then provided monoacetate 26 in 72% yield in two steps. Next, efforts to invert the secondary α-hydroxyl group in 26 were unsuccessful, probably because the hydroxy group at C9 of 26 was sterically hindered. Even the corresponding mesylate and triflate, which were easily epimerized, did not give positive results when treated with alkaline reagents such as DBU and KNO247. Therefore, oxidation of alcohol 26 with Dess–Martin periodinane led to the dione 27. Treatment of 27 with Zn(BH4)2 eventually gave the synthetic shizukaol E (2) with a C9 β-hydroxyl group, together with recyclable compound 26 in 92% yield with an α/β ratio of 2.8:1 (see Supplementary Table 3). The synthetic shizukaol E (2) was fully characterized, and its NMR spectroscopic data are identical to those reported in the literature3,4. However with the exception of Zn(BH4)2, other reductive reagents such as NaBH4, L-selectride, etc. resulted in much lower stereoselectivity than that of Zn(BH4)2. Some reducing reagents such as LiAl(tBuO)3H even led only to compound 26 with α-selectivity. Presumably, the stereoselectivity of this reduction results from coordination between Zn(BH4)2 and the two carbonyl groups at C8 and C9 in dione 27. The relevant DFT calculations of the reduction of dione 27 with Zn(BH4)2 was performed using Gaussian 09 to understand the observed stereoselectivity (see Supplementary Table 12 and Supplementary Figure 4).

Total synthesis of shizukaol E (2). BHT butylated hydroxytoluene, DMP Dess–Martin periodinane, DMAP 4-dimethylaminopyridine
With the completion of the total synthesis of shizukaol E (2), we then concerned on the total synthesis of shizukaol A (1). As shown in Fig. 7, when ethylene glycol in compound 12 was removed with FeCl3•SiO2 in acetone, the resulting ketone underwent a conversion similar to the transformation of 14 to 15. Subsequent sun-lamp irradiation afforded exclusively dione 28 in 75% overall yield for two steps. Upon a similar procedure as described above, regioselective Yamaguchi esterification of dione 28 followed by intramolecular Horner–Wadsworth–Emmons olefination48 finally gave rise to chloranthalactone A (6) as nucleophilic ethylene for Diels–Alder reaction. During workup and purification process, 6 was partially oxidized to the respective epoxide in air. Then, exposure of 6 and 23 under a thermal condition provided exclusively the desired endo-Diels–Alder cycloadduct 29. To better understand the observed stereochemical outcome of the endo-Diels–Alder reaction between diene 23 and ethylene species 17 or 6, we carried out computational studies of diene 23 and ethylene 6 using Gaussian 0949, and the relevant results were consistent with our experimental observations (see Supporting Information for details). Subsequently, a biomimetic hydrolysis with KOH in MeOH gave compound 30 in 72% yield. Oxidation of 30 with Dess–Martin periodinane followed by reduction of dione 31 with Zn(BH4)2, eventually achieved shizukaol A (1) and alcohol 30 in 95% yield with an α/β ratio of 4.6:1. The spectroscopic data of synthetic shizukaol A (1), including MS, IR, and NMR, are in full agreement with those reported1,2.

Total synthesis of shizukaol A (1). DMAP 4-dimethylaminopyridine, BHT butylated hydroxytoluene, DMP Dess–Martin periodinane
Discussion
In summary, we achieved the first total syntheses of shizukaols A (1) and E (2) from commercially available Wieland–Miescher ketone (7) through mimicking the modified biogenetic pathway in 0.1% overall yield over 24 steps and 0.15% overall yield over 28 steps in the longest linear sequence, respectively. According to the structural features of the shizukaol family, an efficient butenolide formation was developed for the synthesis of the common biomimetic diene 23 and ethylene species (6 and 17) using either a highly Z-selective olefination of α-siloxy ketone with ynolate anions or an intramolecular Horner–Wadsworth–Emmons olefination. Inspired by the hypothetical biogenetic pathway for the shizukaol family and the pyrolysis results, we proposed and validated a modified biomimetic approach involving an endo-Diels–Alder reaction and biomimetic transformation. This methodology may be practical for the syntheses of other members in the intriguing Chloranthaceae family, as well as other potential lead molecules for understanding of their relevant biological action and biogenetically synthetic protocol in nature.
Methods
General
All reagents and solvents were of reagent grade. Further purification and drying following the guidelines of Perrin and Armarego50 were used when necessary. Organic solvents were concentrated under reduced pressure on a rotary evaporator in a water bath no more than 40 ℃ unless otherwise specified. Thin-layer chromatography (TLC) was performed on E. Merck silica gel 60 F254 (0.25 mm thickness) coated on aluminum plates. Chromatographic purification of products was performed on Macherey–Nagel–Kieselgel 60 M (230–400 mesh). Visualization of the developed chromatogram was performed by acidic ceric ammonium molybdate and subsequent heating. Melting points were measured with a Stuart Melting Point Apparatus (SMP40) in Celsius degrees and were uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker ADVANCE-III NMR spectrometer at 400.13 MHz (1H) or at 100.6 MHz (13C). All NMR measurements were carried out in CDCl3 and internally referenced to residual solvent signals (referenced at δ 7.26 ppm in 1H, and δ 77.16 ppm for central line of the triplet in 13C). Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets, ddd = doublet of doublets of doublets, m = multiplet), integration, coupling constant (Hz), and assignment. Data for 13C NMR are reported in terms of chemical shift. Mass spectrometry (MS) and high-resolution mass spectrometry (HRMS) were measured on a ThermoFinnigan MAT 95XL. Elemental analyses were carried out by Shanghai Institute of Organic Chemistry, the Chinese Academy of Science, PRC. Selected crystals for X-ray analyses were used for intensity data collection on either a Bruker AXS Kappa Apex II Duo diffractometer or Bruker D8 Venture X-Ray Diffractometer at 173 K using frames of oscillation range 0.3°, with 2° < θ < 28°. Infrared spectra (IR) were recorded on a Nicolet 420 FT-IR spectrometer as thin film on potassium bromide discs.
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 1827569 for compound 13a, CCDC 1833455 for compound 20, and CCDC 1833456 for compound 25. These data are provided free of charge by The Cambridge Crystallographic Data Centre. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
References
Kawabata, J., Fukushi, Y., Tahara, S. & Mizutani, J. Shizukaol A, a sesquiterpene dimer from Chloranthus japonicus. Phytochemistry 29, 2332–2334 (1990).
Guo, Y. Q. et al. Natural nitric oxide (NO) inhibitors from Chloranthus japonicus. Bioorg. Med. Chem. Lett. 26, 3163–3166 (2016).
Kawabata, J., Fukushi, E. & Mizutani, J. Sesquiterpene dimers from Chloranthus japonicus. Phytochemistry 39, 121–125 (1995).
Xu, Y.-J. et al. Mono- and Di-sesquiterpenoids from Chloranthus spicatus. J. Nat. Prod. 70, 1987–1990 (2007).
Zhou, B., Liu, Q.-F., Dalal, S., Cassera, M. B. & Yue, J.-M. Fortunoids A−C, three sesquiterpenoid dimers with different carbon skeletons from Chloranthus fortunei. Org. Lett. 19, 734–737 (2017).
Shen, C.-P., LuoJ.-G., Yang M.-H. & KongL.-Y. Sesquiterpene dimers from the roots of Chloranthus holostegius with moderate anti-inflammatory activity. Phytochemistry 137, 117–122 (2017).
Zhuo, Z.-G. et al. Two new sesquiterpenoids and a new lindenane sesquiterpenoid dimer from Chloranthus japonicus. Phytochem. Lett. 20, 133–138 (2017).
Yan, H. et al. Lindenane sesquiterpenoid dimers from Chloranthus japonicus inhibit HIV-1 and HCV replication. Fitoterapia 115, 64–68 (2016).
Li, C.-J. et al. Bis-sesquiterpenes and diterpenes from Chloranthus henryi. Phytochemistry 69, 2867–2874 (2008).
Yang, S.-P. et al. Chlorahololides A and B, two potent and selective blockers of the potassium channel isolated from Chloranthus holostegius. Org. Lett. 9, 903–906 (2007).
Yang, S.-P., Gao, Z.-B., Wu, Y., Hu, G.-Y. & Yue, J.-M. Chlorahololides C.-F: a new class of potent and selective potassium channel blockers from Chloranthus holostegius. Tetrahedron 64, 2027–2034 (2008).
Zhou, B. et al. Nanomolar antimalarial agents against chloroquine-resistant plasmodium falciparum from medicinal plants and their structure-activity relationships. J. Nat. Prod. 80, 96–107 (2017).
enlon, T. W. et al. Studies towards the synthesis of Lindenene: unexpected stereochemical outcome of an intramolecular cyclopropanation reaction and synthesis of (±)-epi-Lindenene and (±)-iso-Lindenene. Synlett 2679–2682 (2007).
Fenlon, T. W., Jones, M. W., Adlington, R. M. & Lee, V. Synthesis of rac-Lindenene via a thermally induced cyclopropanation reaction. Org. Biomol. Chem. 11, 8026–8029 (2013).
Liu, Y. & Nan, F.-J. Synthetic studies towards Chlorahololides A: practical synthesis of a lindenane-type sesquiterpenoid core framework with a 5,6-double bond. Tetrahedron Lett. 51, 1374–1376 (2010).
Zhang, H.-Z. & Nan, F.-J. Synthesis toward the lindenane-type sesquiterpenoid monomer of Chlorahololide A. Chin. J. Chem. 31, 84–92 (2013).
Qian, S. & Zhao, G. Synthetic studies toward the total synthesis of Chlorahololide A. Synlett 722–724 (2011).
Qian, S. & Zhao, G. Total synthesis of (+)-chloranthalactone F. Chem. Commun. 48, 3530–3532 (2012).
Qian, S. & Zhao, G. Enantioselective total synthesis of (+)-sarcandralactone A. Tetrahedron 69, 11169–11173 (2013).
Yue, G., Yang, L., Yuan, C., Jiang, X. & Liu, B. Total synthesis of (±)-chloranthalactone A. Org. Lett. 13, 5406–5408 (2011).
Yue, G., Yang, L., Yuan, C., Du, B. & Liu, B. Total syntheses of lindenane-type sesquiterpenoids: (±)-chloranthalactones A, B, F, (±)-9-hydroxy heterogorgiolide, and (±)-shizukanolide E. Tetrahedron 68, 9624–9637 (2012).
Du, B. et al. Asymmetric total synthesis of onoseriolide, bolivianine, and isobolivianine. Chem. Eur. J. 20, 2613–2622 (2014).
Yuan, C., Du, B., Yang, L. & Liu, B. Bioinspired total synthesis of Bolivianine: a Diels−Alder/intramolecular hetero-Diels−Alder cascade approach. J. Am. Chem. Soc. 135, 9291–9294 (2013).
Yang, L. et al. Synthetic studies toward lindenane-type sesquiterpenoid dimers. Synlett 25, 2471–2474 (2014).
Ramesh, S. & Mehta, G. A total synthesis of sarcandralactone A: a general, concise, RCM enabled approach to lindenanolide sesquiterpenoids. Tetrahedron Lett. 56, 3941–3944 (2015).
Eagan, J. M., Hori, M., Wu, J., Kanyiva, K. S. & Snyder, S. A. Synthesis and applications of Hajos–Parrish ketone isomers. Angew. Chem. Int. Ed. 54, 7842–7846 (2015).
Demertzidou, V. P. & Zografos, A. L Platinum-catalyzed cycloisomerizations of a common enyne: a divergent entry to cyclopropane sesquiterpenoids. Formal. Synth. sarcandralactone A. Org. Biomol. Chem. 14, 6942–6946 (2016).
Lu, Y.-S. & Peng, X.-S. A concise construction of the chlorahololide heptacyclic core. Org. Lett. 13, 2940–2943 (2011).
Lu, Y.-S. & Peng, X.-S. Corrections to “A concise construction of the chlorahololide heptacyclic core”. Org. Lett. 18, 5447–5448 (2016).
Yuan, C., Du, B., Deng, H., Man, Y. & Liu, B. Total syntheses of sarcandrolide J and shizukaol D: lindenane sesquiterpenoid [4+2] dimers. Angew. Chem. Int. Ed. 56, 637–640 (2017).
Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1, 1–28 (1981).
Mitsunobu, O. & Yamada, M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 40, 2380–2382 (1967).
Yamashita, S., Iso, K. & Hirama, M. A concise synthesis of the pentacyclic framework of cortistatins. Org. Lett. 10, 3413–3415 (2008).
Shigehisa, H., Mizutani, T., Tosaki, S.-Y., Ohshima, T. & Shibasaki, M. Formal total synthesis of (+)-wortmannin using catalytic asymmetric intramolecular aldol condensation reaction. Tetrahedron 61, 5057–5065 (2005).
Kocienski, P. J., Lythgoe, B. & Waterhouse, I. The influence of chain-branching on the steric outcome of some olefin-forming reactions. J. Chem. Soc. Perkin Trans. 1, 1045–1050 (1980).
Wasserman, H. H. & Ives, J. L. A novel method for converting ketones to α-diketones. The reaction of enamino ketones with singlet oxygen. J. Am. Chem. Soc. 98, 7868–7869 (1976).
Wasserman, H. H. & Ives, J. L. Reaction of singlet oxygen with enamino carbonyl systems. A general method for the synthesis of α-keto derivatives of lactones, esters, amides, lactams, and ketones. J. Org. Chem. 50, 3573–3580 (1985).
Inanaga, J., Hirata, K., Saeki, H., Katsuki, T. & Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 52, 1989–1993 (1979).
Shindo, M., Sato, Y., Yoshikawa, T., Koretsune, R. & Shishido, K. Stereoselective olefination of unfunctionalized ketones via ynolates. J. Org. Chem. 69, 3912–3916 (2004).
Shindo, M. et al. Heteroatom-guided torquoselective olefination of α-oxy and α-amino ketones via ynolates. Chem. Eur. J. 12, 524–536 (2006).
Shindo, M., Matsumoto, K. & Shishido, K. Generation of ynolate and Z-selective olefination of acylsilanes: (Z)-2-methyl-3-thrmethylsilyl-2-butenoic acid. Org. Synth. 84, 11–21 (2007).
Evans, D. A. & Black, W. C. Total synthesis of (+)-A83543A [(+)-lepicidin A]. J. Am. Chem. Soc. 115, 4497–4513 (1993).
Nicolaou, K. C., Toh, Q.-Y. & Chen, D. Y.-K. An expedient asymmetric synthesis of platencin. J. Am. Chem. Soc. 130, 11292–11293 (2008).
Domingo, L. R. & Saez, J. A. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 7, 3576–3583 (2009).
Tian, Z. et al. An enzymatic [4+2] cyclization cascade creates the pentacyclic core of pyrroindomycins. Nat. Chem. Biol. 11, 259–265 (2015).
Domingo, L. R., Saéz, J. A., Zaragozá, R. J. & Arnó, M. Understanding the participation of quadricyclane as nucleophile in polar [2σ+2σ+2π] cycloadditions toward electrophilic π molecules. J. Org. Chem. 73, 8791–8799 (2008).
Moriarty, R. M. et al. Inversion of configuration of α-trisubstituted(neopentyl) type secondary alcohols. Tetrahedron Lett. 34, 8029–8032 (1993).
Wadsworth, W. S. & Emmons, W. D. The utility of phosphonate carbanions in olefin synthesis. J. Am. Chem. Soc. 83, 1733–1738 (1961).
Domingo, L. R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 4, 32415–32428 (2014).
Perrin, D. D., Armarego, W. L. F. & Perrin, D. R. Purification of Laboratory Chemicals (Butterworth-Heinemann, Oxford, 1997).
Acknowledgements
We thank warmly Professor Henry N. C. Wong (The Chinese University of Hong Kong) for his great contribution in the development of Organic Chemistry in China on the occasion of his retirement from the Chinese University of Hong Kong after a career over a span of 35 years, Grants from the National Natural Science Foundation of China (Project No. 21672181), Shenzhen Science and Technology Innovation Committee (JCYJ20160608151520697), NSFC/RGC Joint Research Scheme (N_CUHK451/13), Research Grants Council (GRF Projects 403012/CUHK14309216/CUHK14303815 and CRF PolyU C5023-14G), and Innovation and Technology Commission (a grant to the State Key Laboratory of Synthetic Chemistry and GHP/004/16GD) are gratefully acknowledged. BT also acknowledges the financial support from the NSF of Ningbo (2017A610070), the International Doctoral Innovation Centre, Ningbo Education Bureau, Ningbo Science and Technology Bureau, and the University of Nottingham. We also thank Professor Thomas C. W. Mak (The Chinese University of Hong Kong) and Dr. Chun-Kit Hau (The Hong Kong Baptist University) for X-ray crystallographic analysis. We thank Professor Li Dang (Shantou University) for the preliminary computation on Diels–Alder reaction. We also thank Professors Jian-Min Yue (Shanghai Institute of Materia Medica) and Jun Kawabata (Hokkaido University) for providing us with the authentic 1H and 13C NMR spectra of 1 and 2, respectively.
Author information
Authors and Affiliations
Contributions
J.-L.W. and Y.-S.L. contributed equally to this work. J.-L.W. and Y.-S.L. performed the synthesis and characterization, and analyzed the experimental results. J.-L.W. wrote the draft of the manuscript. B.T. performed the theoretical calculations and is responsible for the computational part. X.-S.P. provided overall supervision and composed the manuscript with inputs from all authors. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, JL., Lu, YS., Tang, B. et al. Total syntheses of shizukaols A and E. Nat Commun 9, 4040 (2018). https://doi.org/10.1038/s41467-018-06245-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-06245-7
This article is cited by
-
Picomolar antimalarial agent from a Chinese medicinal plant
Science China Chemistry (2022)
-
Natural disesquiterpenoids: an overview of their chemical structures, pharmacological activities, and biosynthetic pathways
Phytochemistry Reviews (2020)
-
A unified strategy toward total syntheses of lindenane sesquiterpenoid [4 + 2] dimers
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
