
Abstract
Electrochemical reduction of N2 to NH3 provides an alternative to the Haber−Bosch process for sustainable, distributed production of NH3 when powered by renewable electricity. However, the development of such process has been impeded by the lack of efficient electrocatalysts for N2 reduction. Here we report efficient electroreduction of N2 to NH3 on palladium nanoparticles in phosphate buffer solution under ambient conditions, which exhibits high activity and selectivity with an NH3 yield rate of ~4.5 μg mg−1Pd h−1 and a Faradaic efficiency of 8.2% at 0.1 V vs. the reversible hydrogen electrode (corresponding to a low overpotential of 56 mV), outperforming other catalysts including gold and platinum. Density functional theory calculations suggest that the unique activity of palladium originates from its balanced hydrogen evolution activity and the Grotthuss-like hydride transfer mechanism on α-palladium hydride that lowers the free energy barrier of N2 hydrogenation to *N2H, the rate-limiting step for NH3 electrosynthesis.
Similar content being viewed by others

Introduction
Due to the limited supply of fossil fuels, there is a critical demand to use renewable energy to drive the chemical processes that have heavily relied on the consumption of fossil fuels1. One such energy-intensive chemical process is the Haber−Bosch process2, 3, which produces NH3 from N2 and H2 using iron-based catalysts under high temperature (350−550 °C) and high pressure (150−350 atm). NH3 is one of the most highly produced inorganic chemicals in the world, because of its vast need in fertilizer production, pharmaceutical production, and many other industrial processes4, 5. In 2015, around 146 million tons of NH3 was produced globally through the Haber−Bosch process6, which consumes 3−5% of the annual natural gas production worldwide, approximating to 1−2% of the global annual energy supply2, 5. This industrial process is also responsible for >1% of the global CO2 emissions2. Therefore, it is highly desirable to develop an alternative, efficient process for NH3 synthesis using renewable energy7,8,9,10,11, which can simultaneously reduce the CO2 emissions.
One alternative approach to the Haber−Bosch process is to use electrical energy to drive the NH3 synthesis reaction under ambient conditions12,13,14,15, which can reduce the need for high temperature and pressure, and, thereby lower the energy demand. When powered by electricity from renewable energy sources such as solar and wind, electrochemical synthesis of NH3 from N2 and H2O can facilitate sustainable, distributed production of NH3, as well as the storage of renewable energy in NH3 as a carbon-neutral liquid fuel16,17,18, owing to its high energy density (4.32 kWh L−1), high hydrogen content (17.6 wt%), and facile liquidation (boiling point: −33.3 °C at 1 atm). However, the development of the process has been impeded by the lack of efficient electrocatalysts for N2 reduction reaction (N2RR). Although electrochemical synthesis of NH3 has been demonstrated on various materials including Ru, Pt, Au, Fe, and Ni19,20,21,22,23,24,25,26,27,28,29,30,31, most of them showed low activity and selectivity (typically, Faradaic efficiency <1%) for NH3 production26,27,28,29,30,31. Therefore, major improvement in N2RR catalysts is essential for the development of low-temperature NH3 electrolyzers, which necessitates a better understanding and control of the catalytic materials and the reaction kinetics.
There are two major challenges associated with electrochemical NH3 synthesis in aqueous media32, 33. From the thermodynamic point of view, the splitting of the strong N≡N bond requires a reduction potential where the hydrogen evolution reaction (HER) readily occurs, leading to an extremely low Faradaic efficiency for N2RR under ambient conditions26,27,28,29,30,31. Therefore, it would be optimal to find a catalytic system that can promote N2RR at low overpotentials and suppress the competing HER. From the kinetic perspective, it is suggested that the rate-determining step for N2RR is the formation of *N2H through a proton-coupled electron transfer process (H+ + e− + * + N2 → *N2H)33, 34, where * signifies an active site on the catalyst surface. It involves a proton from the electrolyte, an electron transferred from the electrode, and a N2 molecule in the solution. The strong solvent reorganization required for the endergonic charge transfer steps has a low probability of occurrence, leading to the sluggish kinetics. Instead, if the atomic *H species can be formed on a catalyst surface and directly react with N2, it may largely accelerate the kinetics to form *N2H: *H + N2 → *N2H. Indeed, there have been several investigations of metal hydride complexes for N2 reduction in a homogenous medium35, 36. Similarly, NH3 synthesis has been achieved at a low temperature by LiH-mediated N2 transfer and hydrogenation on the transition metals37. Therefore, it is imperative to examine such a hydrogenation pathway for the electrochemical N2RR in aqueous electrolyte systems under ambient conditions.
Here we report an ambient electrochemical reduction of N2 to NH3 on carbon black-supported Pd nanoparticles (Pd/C), which can form Pd hydrides under certain potentials and promote surface hydrogenation reactions. Operating in a N2-saturated phosphate buffer solution (PBS) electrolyte, the Pd/C catalyst enables NH3 production with a yield rate of around 4.5 μg mg−1Pd h−1 and a high Faradaic efficiency of 8.2% at 0.1 V vs. the reversible hydrogen electrode (RHE), which corresponds to a low overpotential of 56 mV. This catalytic performance is enabled by an effective suppression of the HER activity in the neutral PBS electrolyte and the Grotthuss-like hydride transfer mechanism on α-PdH for N2 hydrogenation. All potentials reported in this study are with respect to the RHE scale.
Results
Synthesis and characterization of Pd/C catalyst
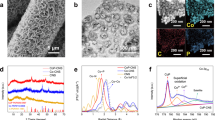
The Pd/C catalyst was prepared using polyol reduction method (see Methods section for experimental details). Figure 1a shows a representative transmission electron microscopy (TEM) image of the obtained Pd/C catalyst, which suggests that the Pd nanoparticles are homogeneously dispersed on the carbon black. The nanoparticle sizes have a narrow distribution between 4 and 9 nm, with an average size of around 6 nm (see the inset of Fig. 1a). A high-resolution TEM image in Fig. 1b shows the atomic lattice fringes of the particles with lattice plane spacings determined to be 0.225 nm, corresponding to the (111) lattice spacing of Pd. An X-ray diffraction (XRD) pattern of the Pd/C catalyst is shown in Fig. 1c, in which the peaks with 2θ values of 40.1o, 46.6o, 68.1o, 82.1o, and 86.6o can be indexed to the diffraction from the (111), (200), (220), (311), and (222) lattice planes of Pd, respectively (PDF#65-2867). X-ray photoelectron spectroscopy (XPS) was used to examine the elemental composition of the Pd/C catalyst. As shown in Fig. 1d, only Pd, C, and O were observed in the survey spectrum, where the binding energies of 335.4 and 340.9 eV correspond to the 3d5/2 and 3d3/2 levels of metallic Pd0. Of note, no N species were observed within the detection limit of the XPS (~0.1 atomic percent), as shown in Supplementary Fig. 1.

Structural and compositional characterizations of the Pd/C catalyst. a TEM and b high-resolution TEM images of the Pd/C catalyst. Inset a: particle size distribution. Scale bars, a 50 nm; b 2 nm. c XRD pattern and d XPS survey spectrum of the Pd/C catalyst. Inset d: high-resolution XPS spectrum of Pd 3d peaks
Electroreduction of N2 to NH3 on the Pd/C catalyst
The electrochemical measurements were performed using a gas-tight two-compartment electrochemical cell separated by a piece of Nafion 115 membrane (Supplementary Fig. 2). A piece of Pt gauze and Ag/AgCl electrode (filled with saturated KCl solution) were used as counter electrode and reference electrode, respectively. The working electrode was prepared by dispersing the Pd/C catalyst on a carbon paper or a glassy carbon substrate, as specified below. N2 gas was delivered into the cathodic compartment by N2 gas bubbling. The N2RR activities of the electrodes were evaluated using controlled potential electrolysis with N2-saturated electrolyte for 3 h. All potentials were iR-compensated and converted to the RHE scale via calibration (Supplementary Fig. 3). The gas-phase product (H2) was quantified by periodic gas chromatography of the headspace. The produced NH3 in the solution phase was quantified at the end of each electrolysis using the calibration curves established by the indophenol blue method38 (Supplementary Fig. 4). Another possible solution-phase product, N2H4, was also determined using a spectrophotometric method developed by Watt and Chrisp39 (Supplementary Fig. 5), whereas no N2H4 was detected in our studies within the detection limit of the method.
To boost the selectivity for N2RR, we need to find an electrolyte that can effectively suppress the competing HER. We compared the HER activities of the Pd/C catalyst in three Ar-saturated electrolytes: 0.05 M H2SO4 (pH = 1.2), 0.1 M PBS (pH = 7.2), and 0.1 M NaOH (pH = 12.9). Linear sweep voltammograms (LSV) of the Pd/C catalyst in the three electrolytes show that the current densities measured in H2SO4 and NaOH are both several times higher than those in PBS in a wide potential range (see Fig. 2a and Supplementary Fig. 6a), indicating an effective suppression of the HER activity in the neutral PBS electrolyte. The less favorable kinetics of HER in PBS is because of its higher barrier for mass- and charge-transfer40, as evidenced by the electrochemical impedance spectra in Supplementary Fig. 6b. Similarly, the controlled potential electrolysis in N2-saturated PBS at −0.05 V vs. RHE produces a current density of about 0.3 mA cm−2 (Fig. 2b), which is much lower than that in H2SO4 (~8 mA cm−2) and NaOH (~3.5 mA cm−2). However, the NH3 yield rate in PBS reaches 4.9 μg mg−1Pd h−1 (Fig. 2c), which is around two times of that in H2SO4 (2.5 μg mg−1Pd h−1) and that in NaOH (2.1 μg mg−1Pd h−1). More strikingly, a Faradaic efficiency of 2.4% is achieved in PBS, whereas both H2SO4 and NaOH electrolytes give rise to a Faradaic efficiency lower than 0.1%. These results clearly indicate that PBS is a promising electrolyte for electrochemical N2RR due to its effective suppression of the HER activity. Therefore, all the following N2RR experiments were performed with 0.1 M PBS electrolyte.

Electroreduction of N2 to NH3 on the Pd/C catalyst. a Linear sweep voltammetric curves of the Pd/C catalyst on the glassy-carbon support measured in the three Ar-saturated electrolytes. b Chronoamperometric curves of the Pd/C catalyst on the carbon-paper support measured at −0.05 V vs. RHE in the three N2-saturated electrolytes, and c corresponding NH3 yield rates and Faradaic efficiencies in the three electrolytes. d Chronoamperometric curves of the Pd/C catalyst on the carbon-paper support in N2-saturated 0.1 M PBS at various potentials, and e corresponding NH3 yield rates and Faradaic efficiencies at the potentials. The error bars correspond to the standard deviations of measurements over three separately prepared samples under the same conditions. f UV-vis absorption spectra of the electrolytes after electrolysis at −0.05 V vs. RHE for 3 h under different conditions. No apparent NH3 was detected for the control experiments with Ar-saturated electrolyte (CP-Pd/C-Ar) or without Pd catalyst (CP-N2), indicating that NH3 was produced by Pd-catalyzed electroreduction of N2
Subsequently, the activities of the Pd/C catalyst for N2RR were systematically investigated in N2-saturated PBS at various potentials with separately prepared electrodes. As shown in Fig. 2d, the total current density increases from ~0.05 to more than 1.2 mA cm−2, as the potential shifts from 0.1 to −0.2 V. Interestingly, the NH3 yield rate remains similar within this potential range, fluctuating around 4.5 μg mg−1Pd h−1 (Fig. 2e). Strikingly, the Faradaic efficiency for NH3 production reaches a maximum value of 8.2% at 0.1 V, which corresponds to a low overpotential of 56 mV, given the equilibrium potential of 0.156 V for N2 reduction to NH3 under our experimental conditions (see Methods section for the calculations). The Faradaic efficiency decreases gradually at more negative potentials, which is mainly caused by the rapid rising of the HER activity (Supplementary Fig. 7). To the best of our knowledge, the Pd/C catalyst achieves an NH3 yield rate and Faradaic efficiency that are comparable to the recently reported catalysts for N2RR under ambient conditions (Supplementary Table 1), but uses an overpotential lower by at least 300 mV, making it one of the most active and selective electrocatalysts for ambient NH3 synthesis.
In addition, we have carefully examined the N source of the produced NH3. First, control experiments with Ar-saturated electrolyte or without Pd catalyst were performed. As shown in Fig. 2f, no apparent NH3 was detected using the indophenol blue method when the bubbled N2 gas was replaced by Ar or when a carbon paper electrode without Pd was used, indicating that the NH3 was produced by N2 reduction in the presence of Pd catalyst. Furthermore, 15N isotopic labeling experiment was performed as an alternative method to verify the N source of the produced NH3 in 0.1 M PBS electrolyte. A triplet coupling for 14NH4+ and a doublet coupling for 15NH4+ in the 1H nuclear magnetic resonance (1H NMR) spectra are used to distinguish them25. As shown in Supplementary Fig. 8, only 15NH4+ was observed in the electrolyte when 15N2 was supplied as the feeding gas, and no NH4+ was detected when Ar was supplied, which are consistent with the control experiments and confirm that the NH3 was produced by Pd-catalyzed electroreduction of N2.
The stability of the Pd/C catalyst for electrochemical N2RR was assessed by consecutive recycling electrolysis at −0.05 V vs. RHE. After five consecutive cycles, only a slight decline in the total current density was observed, as shown in Supplementary Fig. 9. However, the NH3 yield rate and Faradaic efficiency decreased to 2.4 μg mg−1Pd h−1 and 1.2% after five cycles, indicating a loss of the N2RR activity by 50% after the 15 h operation. The decrease in the N2RR activity is due to the loss of active Pd surface area caused by the aggregation of Pd nanoparticles on the carbon support, as evidenced by the TEM images of the Pd/C catalyst after the recycling test (Supplementary Fig. 9). Further improvement in the dispersion of Pd nanoparticles on the support and interaction between them will be beneficial for the long-term durability of the catalyst.
Discussion
The Pd/C catalyst exhibits high activity and selectivity for the N2RR at a low overpotential of 56 mV. To explore the underlying mechanism and to see whether it is exclusive to Pd, we prepared Au/C and Pt/C catalysts with identical metal loading using the same method, and compared their N2RR catalytic performance with that of the Pd/C catalyst (Fig. 3). The structural and compositional characterizations of the Au/C and Pt/C catalysts, including TEM, XRD, and XPS (Supplementary Fig. 10), confirm the successful synthesis of the metal nanoparticles with similar sizes as that of the Pd/C catalyst. At −0.05 V vs. RHE, the Au/C catalyst exhibits a current density (<0.04 mA cm−2) much lower than that of the Pd/C catalyst, whereas the Pt/C catalyst shows a slightly higher current density (see inset of Fig. 3). Both the Au/C and Pt/C catalysts produce NH3 at a rate of about 0.3 μg mg−1metal h−1, which is lower than that of the Pd/C catalyst by more than one order of magnitude. The Au/C catalyst achieves a Faradaic efficiency of 1.2% for N2RR, due to its low activity for the HER. In contrast, the Faradaic efficiency for N2RR on the Pt/C catalyst is only 0.2%, much lower than that of the Au/C catalyst, because Pt is intrinsically the most active catalyst for HER (Supplementary Fig. 11). In comparison, such a significant difference in both N2RR activity and selectivity clearly indicates that Pd is a unique catalyst for N2RR. Actually, Pd can readily absorb H atoms in its lattice, forming Pd hydrides under operating conditions41. The cathodic current observed at 0.10 and 0.05 V is similar to the data in a previous study42, and the unaccounted current at the two potentials (see Supplementary Fig. 7b) may be similarly attributed to the dynamic hydrogen adsorption and absorption on Pd42, in addition to the capacitance of the carbon support. It has been reported that Pd catalyzes the electroreduction of CO2 to formate with high activity and selectivity at low overpotentials43, 44, and both experimental and computational studies have confirmed that it is attributed to a hydrogenation mechanism through in situ formed PdHx phase45. Interestingly, a recent study of N2RR on commercial Pd/C catalysts in acidic and alkaline electrolytes showed Faradaic efficiencies lower than 0.1% at −0.2 V vs. RHE31, which are consistent with our results under similar conditions (Fig. 2c), and highlight the critical roles of the HER suppression in PBS and the hydrogenation via hydride transfer pathway at low overpotentials (vide infra).

Comparison of the Pd/C catalyst with Au/C and Pt/C catalysts for N2RR. NH3 yield rates and Faradaic efficiencies of the Au/C, Pd/C, and Pt/C catalysts on the carbon-paper support measured in N2-saturated 0.1 M PBS electrolyte at −0.05 V vs. RHE. The error bars correspond to the standard deviations of measurements over three separately prepared samples under the same conditions. Inset: chronoamperometric curves obtained for each catalyst
To understand the unique activity of N2RR on Pd/C, we performed density functional theory (DFT) calculations for the energetics of HER and N2RR steps on the (211) surfaces of Au, Pt, and Pd hydride (see Supplementary Note 1 for the computational details). As employed previously, the undercoordinated step atoms are assumed to be the catalytic site for activating the N≡N bond46. For Pd, we have two subsurface *H (*Hsub) at the octahedral sites underneath the Pd edge atoms to simulate the α-phase Pd hydride (α-PdH), which is the stable phase under operating potentials45. According to the differential adsorption free energies of *H on the (211) surfaces (Supplementary Fig. 12), we adopted models with 2/3 monolayer of *H for α-PdH and Pt, on which the step/terrace sites are occupied by *H and the bottom-of-the-edge sites are free, while a clean surface for Au was used (geometric structures shown in Fig. 4). In the inset of Fig. 4, we can see that the HER on Pt is facile with the free formation energy of *H close to zero47. In contrast, the process is limited by *H desorption on α-PdH and by *H adsorption on Au. Interestingly, the free energy cost in creating a *H vacancy (*H-v) necessary for N2 collision at step sites is much lower on α-PdH (0.18 eV) than that on Pt (0.41 eV). Furthermore, the hydrogenation of N2 by a surface *H to form *N2H on α-PdH (1.18 eV) is thermodynamically more favorable than that on Pt (1.37 eV) and Au (2.21 eV). On α-PdH, the surface hydrogenation is accompanied by the transfer of *Hsub to the surface site, analogous to the Grotthuss-like proton-hopping mechanism in a water network48. The reaction-free energy of N2 hydrogenation on Pd without the hydride transfer is less favorable by 0.3 eV (see Supplementary Fig. 13 for a direct comparison). The nature of this rate-determining step, i.e., surface chemical hydrogenation instead of the proton-coupled electron transfer, supports the observed weak potential dependence of the NH3 yield rate at low overpotentials. DFT-calculated free energies of hydrogenation of N2 vs. hydrogen evolution across metal surfaces (Au, Pt, and α-PdH) rationalize the activity and selectivity trends in Fig. 3.

Computational studies. DFT-calculated free energy pathways of HER (inset) and the relevant steps of N2RR on the (211) surfaces of Au, Pt, and α-PdH at surface potential of 0 V vs. RHE under 298.15 K (atomic structures shown in the insets). The first step denotes the formation of *H on the Au surface and *H vacancy (*H-v) on Pt and α-PdH step sites. The second step is the direct surface hydrogenation of N2(g) forming *N2H on Au and Pt, and the Grotthuss-like hydride transfer pathway on α-PdH
In summary, we have discovered an efficient electrohydrogenation of N2 to NH3 on Pd/C catalyst at an overpotential as low as 56 mV for the electrochemical NH3 synthesis under ambient conditions. The Pd/C catalyst exhibits high activity and selectivity for N2RR in a PBS electrolyte, achieving an NH3 yield rate of about 4.5 μg mg−1Pd h−1 and a Faradaic efficiency of 8.2% at 0.1 V vs. RHE. Comparative experiments indicate an effective suppression of the HER in the neutral PBS electrolyte, and a significantly higher N2RR activity of Pd than other catalysts including Au and Pt. The DFT calculations suggest that the in situ formed α-PdH allows the activation of N2 via a Grotthuss-like hydride transfer pathway that is thermodynamically more favorable than direct surface hydrogenation or proton-coupled electron transfer steps. Our findings open up an avenue to develop efficient electrocatalysts for not only the electroreduction of N2 to NH3, but also other challenging electrocatalytic reactions for renewable energy conversions.
Methods
Synthesis of carbon black-supported metal catalysts
The Pd/C catalyst was synthesized using polyol reduction method. First, the Pd precursor (K2PdCl4) solution was prepared by dissolving PdCl2 in water in the presence of KCl. Typically, 70 mg of carbon black was dispersed in 100 mL of ethylene glycol, followed by sonication for 30 min. Then, 5 mL of K2PdCl4 solution (Pd 6 mg mL−1) was added into the mixture. After stirring for another 30 min, the mixture was heated to 130 °C and kept at this temperature for 2 h. The catalyst slurry was filtered and washed with water. The resulting Pd/C catalyst was dried at 60 °C overnight, with a Pd loading of 30 wt%. Comparative samples of Au/C and Pt/C catalysts with a metal loading of 30 wt% were prepared using the same procedure, except with different metal precursors.
Physical characterizations
TEM images were acquired using a FEI Tecnai F30 Transmission Electron Microscope, with a field emission gun operated at 200 kV. The XRD pattern was collected using a PANalytical Empyrean diffractometer, with a 1.8 KW copper X-ray tube. XPS was performed using a Physical Electronics 5400 ESCA photoelectron spectrometer. Gas products were analyzed by a gas chromatograph (SRI Multiple Gas Analyzer #5) equipped with molecular sieve 5 A and HayeSep D columns, with Ar as the carrier gas.
Preparations of the working electrodes
First, 8 mg of carbon black-supported metal catalyst was dispersed in diluted Nafion alcohol solution containing 1.5 mL ethanol and 60 μL Nafion, which formed a homogeneous suspension after sonication for 1 h. Two types of substrates were used to prepare electrodes in this study: one was carbon paper, and the other was glassy carbon. The carbon-paper electrodes were prepared by drop-casting the suspension on a piece of carbon paper (1 × 1 cm2), with a total mass loading of 1 mg (of which 30 wt% is Pd), which were used for all controlled potential electrolyses. The glassy-carbon electrodes were prepared by drop-casting the suspension on a round glassy carbon (diameter = 3 mm), with a total mass loading of 0.435 mg cm−2 (of which 30 wt% is Pd), which were used for all linear sweep voltammetry measurements, except Supplementary Fig. 6a.
Electrochemical measurements
Prior to N2RR tests, Nafion 115 membranes were heat-treated in 5% H2O2, 0.5 M H2SO4, and water for 1 h, respectively. After being rinsed in water thoroughly, the membranes were immersed in deionized water for future use. Electrochemical measurements were performed using a CH Instruments 760E Potentiostat, with a gas-tight two-compartment electrochemical cell separated by a piece of Nafion 115 membrane at room temperature (Supplementary Fig. 2). A piece of Pt gauze and Ag/AgCl/sat. KCl were used as counter electrode and reference electrode, respectively. The linear sweep voltammetry was scanned at a rate of 5 mV s−1. The N2RR activity of an electrode was evaluated using controlled potential electrolysis in an electrolyte for 3 h at room temperature (~293 K). Prior to each electrolysis, the electrolyte was presaturated with N2 by N2 gas bubbling for 30 min. During each electrolysis, the electrolyte was continuously bubbled with N2 at a flow rate of 10 sccm, and was agitated with a stirring bar at a stirring rate of ~800 rpm. No in-line acid trap was used to capture NH3 that might escape from the electrolyte in our study, as no apparent NH3 was detected in the acid trap under our experimental conditions. The applied potentials were iR-compensated, and the reported current densities were normalized to geometric surface areas.
Calibration of the reference electrodes
All potentials in this study were converted to the RHE scale via calibration (Supplementary Fig. 3). The calibration was performed using Pt gauze as both working electrode and counter electrode in H2-saturated electrolyte. Cyclic voltammograms were acquired at a scan rate of 1 mV s−1. The two potentials at which the current equaled zero were averaged and used as the thermodynamic potential for the hydrogen electrode reactions.
Ammonia quantification
The produced NH3 was quantitatively determined using the indophenol blue method38. Typically, 2 mL of the sample solution was first pipetted from the post-electrolysis electrolyte. Afterwards, 2 mL of a 1 M NaOH solution containing salicylic acid (5 wt%) and sodium citrate (5 wt%) was added, and 1 mL of NaClO solution (0.05 M) and 0.2 mL of sodium nitroferricyanide solution (1 wt%) were added subsequently. After 2 h, the absorption spectra of the resulting solution were acquired with an ultraviolet-visible (UV-vis) spectrophotometer (BioTek Synergy H1 Hybrid Multi-Mode Reader). The formed indophenol blue was measured by absorbance at λ = 653 nm. In order to quantify the produced NH3, the calibration curves were built using standard NH4Cl solutions in the presence of 0.05 M H2SO4, 0.1 M PBS, and 0.1 M NaOH, respectively (Supplementary Fig. 4), to take into account the possible influence of different pH values38. The measurements with the background solutions (no NH3) were performed for all experiments, and the background peak was subtracted from the measured peaks of N2RR experiments to calculate the NH3 concentrations and the Faradaic efficiencies.
Hydrazine quantification
The yellow color developed upon the addition of p-dimethylaminobenzaldehyde (PDAB) to solutions of N2H4 in dilute hydrochloric acid solution was used as the basis for the spectrophotometric method to quantify the N2H4 concentration39. Typically, 5 mL of the electrolyte solution was taken out and then mixed with 5 mL of the coloring solution (4 g of PDAB dissolved in 20 mL of concentrated hydrochloric acid and 200 mL of ethanol). After 15 min, the absorption spectra of the resulting solution were acquired using a UV-vis spectrophotometer (BioTek Synergy H1 Hybrid Multi-Mode Reader). The solutions of N2H4 with known concentrations in 0.1 M PBS were used as calibration standards, and the absorbance at λ = 458 nm was used to plot the calibration curves (Supplementary Fig. 5).
Calculation of the equilibrium potential
The standard potential for the half reaction of N2 reduction to NH4OH was calculated according to the standard molar Gibbs energy of formation at 298.15 K49. The equilibrium potential under our experimental conditions is calculated using the Nernst equation, assuming 1 atm of N2 and a NH4OH concentration of 0.01 mM in the solution.
E° = −ΔG°/nF = 0.058 V, where n = 6 is the number of electrons transferred in the reaction and F is the Faraday constant.
The equilibrium potential under our experimental conditions is calculated using the Nernst equation, assuming 1 atm of N2 and a 0.01 mM concentration of NH4OH in the solution.
Calculation of the Faradaic efficiency and the yield rate
The Faradaic efficiency was estimated from the charge consumed for NH3 production and the total charge passed through the electrode:
The yield rate of NH3 can be calculated as follows:
where F is the Faraday constant (96,485 C mol−1), \(c_{{\mathrm{NH}}_{\mathrm{3}}}\) is the measured NH3 concentration, V is the volume of the electrolyte, Q is the total charge passed through the electrode, t is the electrolysis time (3 h), and m is the metal mass of the catalyst (typically 0.3 mg). The reported NH3 yield rate, Faradaic efficiency, and error bars were determined based on the measurements of three separately prepared samples under the same conditions.
15N isotope labeling experiment
The isotopic labeling experiment was carried out using 15N2 as the feeding gas (Sigma-Aldrich, 98 atom % 15N) with 0.1 M PBS electrolyte. After electrolysis at −0.05 V vs. RHE for 10 h, 10 mL of the electrolyte was taken out and acidized to pH ~3 by adding 0.5 M H2SO4, and then concentrated to 2 mL by heating at 70 °C. Afterwards, 0.9 mL of the resulting solution was taken out and mixed with 0.1 mL D2O containing 100 ppm dimethyl sulphoxide (Sigma-Aldrich, 99.99%) as an internal standard for 1H nuclear magnetic resonance measurement (1H NMR, Bruker Avance III 400 MHz).
Computational studies
DFT calculations were performed using the plane-wave-based PWSCF (Quantum-ESPRESSO) program and the Atomic Simulation Environment (ASE). The ultrasoft Vanderbilt pseudopotential method with Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional was adopted. More calculation details and relevant references are provided in the Supplementary Note 1, Supplementary Table 2, and Supplementary References.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information file or are available from the corresponding authors upon reasonable request.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production (MIT Press, Cambridge, 2004).
Jennings, J. R. Catalytic Ammonia Synthesis: Fundamentals and Practice (Springer Science & Business Media, New York, 2013).
Gruber, N. & Galloway, J. N. An Earth-system perspective of the global nitrogen cycle. Nature 451, 293–296 (2008).
Erisman, J. W., Sutton, M. A., Galloway, J., Klimont, Z. & Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 1, 636–639 (2008).
Apodaca, L. E. In Mineral Commodity Summaries 2016: U.S. Geological Survey (ed. Kimball, S. M.) 118–119 (Government Publishing Office, Washington, DC, 2016).
Kitano, M. et al. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat. Chem. 4, 934–940 (2012).
Service, R. F. New recipe produces ammonia from air, water, and sunlight. Science 345, 610 (2014).
van der Ham, C. J. M., Koper, M. T. M. & Hetterscheid, D. G. H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 43, 5183–5191 (2014).
Ali, M. et al. Nanostructured photoelectrochemical solar cell for nitrogen reduction using plasmon-enhanced black silicon. Nat. Commun. 7, 11335 (2016).
Brown, K. A. et al. Light-driven dinitrogen reduction catalyzed by a CdS:nitrogenase MoFe protein biohybrid. Science 352, 448–450 (2016).
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209–2244 (2009).
Renner, J. N., Greenlee, L. F., Ayres, K. E. & Herring, A. M. Electrochemical synthesis of ammonia: a low pressure, low temperature approach. Electrochem. Soc. Interface 24, 51–57 (2015).
Kyriakou, V., Garagounis, I., Vasileiou, E., Vourros, A. & Stoukides, M. Progress in the electrochemical synthesis of ammonia. Catal. Today 286, 2–13 (2017).
Shipman, M. A. & Symes, M. D. Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal. Today 286, 57–68 (2017).
Zamfirescu, C. & Dincer, I. Using ammonia as a sustainable fuel. J. Power Sources 185, 459–465 (2008).
Klerke, A., Christensen, C. H., Nørskov, J. K. & Vegge, T. Ammonia for hydrogen storage: challenges and opportunities. J. Mater. Chem. 18, 2304–2310 (2008).
Soloveichik, G. Liquid fuel cells. Beilstein J. Nanotechnol. 5, 1399–1418 (2014).
Licht, S. et al. Ammonia synthesis by N2 and steam electrolysis in molten hydroxide suspensions of nanoscale Fe2O3. Science 345, 637–640 (2014).
Bao, D. et al. Electrochemical reduction of N2 under ambient conditions for artificial N2 fixation and renewable energy storage using N2/NH3 Cycle. Adv. Mater. 29, 1604799 (2017).
Shi, M. M. et al. Au sub-nanoclusters on TiO2 toward highly efficient and selective electrocatalyst for N2 conversion to NH3 at ambient conditions. Adv. Mater. 29, 1606550 (2017).
Li, S. J. et al. Amorphizing of Au nanoparticles by CeOx–RGO hybrid support towards highly efficient electrocatalyst for N2 reduction under ambient conditions. Adv. Mater. 29, 1700001 (2017).
Ma, J. L., Bao, D., Shi, M. M., Yan, J. M. & Zhang, X. B. Reversible nitrogen fixation based on a rechargeable lithium-nitrogen battery for energy storage. Chem 2, 525–532 (2017).
McEnaney, J. M. et al. Ammonia synthesis from N2 and H2O using a lithium cycling electrification strategy at atmospheric pressure. Energy Environ. Sci. 10, 1621–1630 (2017).
Chen, G. F. et al. Ammonia electrosynthesis with high selectivity under ambient conditions via a Li+ incorporation strategy. J. Am. Chem. Soc. 139, 9771–9774 (2017).
Kordali, V., Kyriacou, G. & Lambrou, C. Electrochemical synthesis of ammonia at atmospheric pressure and low temperature in a solid polymer electrolyte cell. Chem. Commun. (https://doi.org/10.1039/B004885M (2000).
Giddey, S., Badwal, S. P. S. & Kulkarni, A. Review of electrochemical ammonia production technologies and materials. Int. J. Hydrog. Energy 38, 14576–14594 (2013).
Lan, R., Irvine, J. T. S. & Tao, S. Synthesis of ammonia directly from air and water at ambient temperature and pressure. Sci. Rep. 3, 1145 (2013).
Kim, K. et al. Electrochemical reduction of nitrogen to ammonia in 2-propanol under ambient temperature and pressure. J. Electrochem. Soc. 163, F610–F612 (2016).
Chen, S. et al. Electrocatalytic synthesis of ammonia at room temperature and atmospheric pressure from water and nitrogen on a carbon-nanotube-based electrocatalyst. Angew. Chem. Int. Ed. 56, 2699–2703 (2017).
Nash, J. et al. Electrochemical nitrogen reduction reaction on noble metal catalysts in proton and hydroxide exchange membrane electrolyzers. J. Electrochem. Soc. 164, F1712–F1716 (2017).
Montoya, J. H., Tsai, C., Vojvodic, A. & Nørskov, J. K. The challenge of electrochemical ammonia synthesis: a new perspective on the role of nitrogen scaling relations. ChemSusChem 8, 2180–2186 (2015).
Singh, A. R. et al. Electrochemical ammonia synthesis—the selectivity challenge. ACS Catal. 7, 706–709 (2017).
Zhu, D., Zhang, L., Ruther, R. E. & Hamers, R. J. Photo-illuminated diamond as a solid-state source of solvated electrons in water for nitrogen reduction. Nat. Mater. 12, 836–841 (2013).
Akagi, F., Matsuo, T. & Kawaguchi, H. Dinitrogen cleavage by a diniobium tetrahydride complex: formation of a nitride and its conversion into imide species. Angew. Chem. Int. Ed. 46, 8778–8781 (2007).
Shima, T. et al. Dinitrogen cleavage and hydrogenation by a trinuclear titanium polyhydride complex. Science 340, 1549–1552 (2013).
Wang, P. et al. Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation. Nat. Chem. 9, 64–70 (2017).
Searle, P. L. The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. A review. Analyst 109, 549–568 (1984).
Watt, G. W. & Chrisp, J. D. A spectrophotometric method for the determination of hydrazine. Anal. Chem. 24, 2006–2008 (1952).
Strmcnik, D., Lopes, P. P., Genorio, B., Stamenkovic, V. R. & Markovic, N. M. Design principles for hydrogen evolution reaction catalyst materials. Nano Energy 29, 29–36 (2016).
Wickman, B. et al. Depth probing of the hydride formation process in thin Pd films by combined electrochemistry and fiber optics-based in situ UV/vis spectroscopy. Phys. Chem. Chem. Phys. 17, 18953–18960 (2015).
Hara, M., Linke, U. & Wandlowski, T. Preparation and electrochemical characterization of palladium single crystal electrodes in 0.1 M H2SO4 and HClO4: Part I. Low-index phases. Electrochim. Acta 52, 5733–5748 (2007).
Min, X. & Kanan, M. W. Pd-catalyzed electrohydrogenation of carbon dioxide to formate: high mass activity at low overpotential and identification of the deactivation pathway. J. Am. Chem. Soc. 137, 4701–4708 (2015).
Klinkova, A. et al. Rational design of efficient palladium catalysts for electroreduction of carbon dioxide to formate. ACS Catal. 6, 8115–8120 (2016).
Gao, D. F. et al. Switchable CO2 electroreduction via engineering active phases of Pd nanoparticles. Nano Res. 10, 2181–2191 (2017).
Honkala, K. et al. Ammonia synthesis from first-principles calculations. Science 307, 555–558 (2005).
Hinnemann, B. et al. Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 127, 5308–5309 (2005).
Agmon, N. The Grotthuss mechanism. Chem. Phys. Lett. 244, 456–462 (1995).
Rumble, J. CRC Handbook of Chemistry and Physics 98th edn (CRC Press, Boca Raton, FL, 2017).
Acknowledgements
This work is supported by the Startup Fund from the University of Central Florida (UCF). X.F. is a member of the Energy Conversion and Propulsion Cluster at UCF. J.W. gratefully acknowledges the Preeminent Postdoctoral Program (P3) award from UCF. L.Y. and H.X. acknowledge the financial support from the American Chemical Society Petroleum Research Fund (ACS PRF 55581-DNI5) and the NSF CBET Catalysis Program (CBET-1604984).
Author information
Authors and Affiliations
Contributions
J.W. and X.F. conceived and designed the experiments. J.W. synthesized the materials and carried out the experimental work. L.Y. and H.X. performed the computational work. L.H. assisted in the experimental work. G.C. contributed to NH3 quantification. X.F. and H.X. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Yu, L., Hu, L. et al. Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential. Nat Commun 9, 1795 (2018). https://doi.org/10.1038/s41467-018-04213-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-018-04213-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
