
Abstract
Photocatalytic formation of hydrocarbons using solar energy via artificial photosynthesis is a highly desirable renewable-energy source for replacing conventional fossil fuels. Using an l-cysteine-based hydrothermal process, here we synthesize a carbon-doped SnS2 (SnS2-C) metal dichalcogenide nanostructure, which exhibits a highly active and selective photocatalytic conversion of CO2 to hydrocarbons under visible-light. The interstitial carbon doping induced microstrain in the SnS2 lattice, resulting in different photophysical properties as compared with undoped SnS2. This SnS2-C photocatalyst significantly enhances the CO2 reduction activity under visible light, attaining a photochemical quantum efficiency of above 0.7%. The SnS2-C photocatalyst represents an important contribution towards high quantum efficiency artificial photosynthesis based on gas phase photocatalytic CO2 reduction under visible light, where the in situ carbon-doped SnS2 nanostructure improves the stability and the light harvesting and charge separation efficiency, and significantly enhances the photocatalytic activity.
Similar content being viewed by others
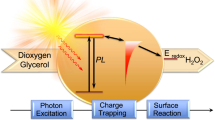


Introduction
Artificial photosynthesis is one of the future energy sources that promises an environmentally friendly alternative to fossil fuels1,2,3,4. In this process, photocatalysts can directly harvest energy from solar light and simultaneously convert CO2 to hydrocarbons, tackling both energy and global environmental problems. Photocatalytic CO2 reduction to hydrocarbon fuels is a solar energy based process that requires highly efficient and stable catalytic materials5, 6. In the past decades, following the pioneering discovery by Inoue et al.7 of photoelectrochemical CO2 reduction in aqueous semiconductor suspensions, various semiconductor materials, in particular TiO2, ZnO, NiO, WO3, and Bi2WO3 have been tested as catalysts for the photocatalytic CO2 reduction reaction8, 9. However, most of these semiconductor materials have a bandgap with energy in the ultraviolet range, resulting in low conversion efficiencies owing to their large band gaps and high charge-carrier recombination rates. To overcome these limitations and to improve photocatalytic CO2 reduction efficiency, semiconductors have been modified by several strategies: nanostructuring, band gap engineering by doping and modification with metal nanoparticles, and hybridization with carbonaceous materials such as Pt/TiO2, GO, g-C3N4, g-C3N4/Bi2WO6, Cu/GO10,11,12,13,14,15,16. Although these hybrid heterogeneous photocatalysts improved the catalytic performance significantly, the overall catalytic selectivity and quantum efficiency are far from the commercial requirements. Recently, an enzyme and semiconductor-hybrid system has been demonstrated to have a high photocatalytic CO2 reduction efficiency of ~ 3–4%17. However, this photocatalyst system suffered from poor enzyme stability. Moreover, the most used narrow band gap semiconductor, CdS, poses another challenge due to its toxicity problem18. As far as photocatalytic CO2 reduction is concerned, we need to develop an environmentally friendly nanostructured hybrid semiconductor, which can take us one step forward from CdS. Apart from the critical narrow band gap and high absorption coefficient to utilize maximum solar energy, efficient charge separation is the other important factor for high activity of a photocatalyst system. Therefore, controlling the carrier diffusion pathway and controlling the defects in the bulk or at the interfaces and surface of a nanostructure semiconductor are the key factors for designing a highly efficient photocatalyst system19.
Since the discovery of graphene, two-dimensional (2D) layered transition metal dichalcogenides and metal sulfide nanostructures are playing an important role in catalysis owing to their wide range of optical and electronic properties20, 21. Moreover, the high surface area and low charge recombination characteristics of 2D materials can potentially enhance the photocatalyst activity22. Among various metal sulfides, SnS2 is a naturally occurring bronze-colored n-type narrow band gap (2.2–2.4 eV) semiconductor known as mosaic gold. During the last few years, it has been demonstrated to be a promising photocatalyst for dye degradation processes23, 24. Recently, it has proven attractive for its potential applications as a light absorber layer for dye-sensitized solar cells25, optoelectronics26, gas sensing27, and energy storage and conversion28. The narrow band gap with around 0.19 µm average photocarriers diffusion length29 and the high quantum yield of SnS2 thus give two advantages for a good photocatalyst under visible light. Sun et al.30 first explored the freestanding single layer SnS2 as an efficient visible-light photocatalyst for water-splitting. Recently, Sun et al.31 have reported the photocatalytic CO2 reduction to CO using SnS2. However, the overall photocatalytic performance is far lower than the practical requirement due to fast recombination of the photogenerated charge carriers. To overcome this problem, it is necessary to synthesize this semiconductor nanostructure doped with metals or non-metals to control the carrier diffusion pathway and charge-carrier recombination. Semiconductor doping with non-metal carbonaceous materials is very popular in photocatalyst systems due to their wide range of light absorption and low photo corrosion as compared with metals. These doped carbon sites act as excellent electron acceptor centers and suppress the charge recombination in the electron transfer process due to electronic interaction between doped carbon and semiconductor. Huang and co-workers32 introduced novel carbon-doped h-BN nanosheets as a sustainable and stable visible photocatalyst system with high efficiency . In 2012, Lin et al.33 reported enhanced photocatalytic water-splitting based on carbon-doped porous ZnO nanoarchitecture. Moreover, the most commonly reported carbon-doped photocatalysts are based on wide band gap semiconductors. Interestingly, doping can create microstrain in the crystal, which affects the electronic and optical properties. Recently, a simulation study has shown a strain-induced indirect-to direct band gap transition in bulk SnS234. This strain induction and non-metal doping studied in semiconductor material promises that there are possibilities of improving the photocatalytic CO2 reduction activity with tuning the optoelectronic property and enhancing separation of photo induced electron-hole pairs by introducing carbon as a doping element into the semiconductor. In this work, we propose a carbon-doped SnS2 nanostructure system with limited average lifetime of photogenerated electrons and holes by shortening the diffusion time so that they can reach the reaction sites before losing their energy. In the hybrid system the conductive carbon incorporated into SnS2 provides the opportunities for fast charge transport in the nanostructure with an interconnected planar structure, thus shortening the diffusion time from semiconductor interior to surface reaction sites. Here, we performed the photocatalytic CO2 reduction using this carbon-doped SnS2 nanostructure (hereafter, referred as SnS2-C) and demonstrated enhanced photocatalytic performance compared with the undoped SnS2 nanoplate (hereafter referred as SnS2). A theoretical study of the CO2 adsorption and dissociation activity for C doped SnS2 has been performed to support the experimental observation. Our results indicate that the carbon-doped nanostructure of SnS2 has a key role in enhancing the visible light photocatalytic activity of the CO2 reduction to solar fuels.
Results
Photocatalyst synthesis
The SnS2-C and SnS2 photocatalyst materials were synthesized by a simple hydrothermal method, using a 1:5 stoichiometric mixture of SnCl4.5H2O and an S source (l-cysteine or thiourea) at 180 oC. The two different sulfur sources were chosen to obtain different nanostructures resulting from their different nucleation processes and pH values during the hydrothermal synthesis. Apparently, both l-cysteine and thiourea provide a controlled condition for anisotropic growth of SnS2 nanostructures and nanoplates respectively as schematically shown in Supplementary Figure 1. The detailed synthesis process is described in the Methods.
Crystal structure analysis
The crystal structure of the as-prepared SnS2-C and SnS2 were characterized by powder X-ray diffraction (PXRD) as shown in Fig. 1a. The PXRD patterns of SnS2-C and SnS2 match well with that of polycrystalline hexagonal SnS2 berndtite (JCPDS no. 01–075–0367) and berndtite-2T (JCPDS no. 00–023–0677), respectively. The facets of SnS2 show quite sharp strong intensities, which demonstrates that thiourea helps for large and thick crystal growth. The PXRD peaks of SnS2-C are broader than the peaks of the SnS2. The peak broadening of SnS2-C implies the nanocrystal and amorphous nature of hexagonal SnS2 after carbon doping. Compared with SnS2, the diffraction peaks of SnS2-C are slightly shifted towards lower angle. The characteristic (001) peak is quite broad and shifts from 15.12 ° to 14.66°, indicating that (001) plane growth of the SnS2-C crystals is greatly inhibited and only few-layered SnS2 is formed, along with lattice expansion, resulting in an enlarged d-spacing owing to the carbon doping during the hydrothermal synthesis in presence of l-cysteine. It is interesting to note that carbon doping occurred in the l-cysteine-assisted hydrothermal process, but not in the thiourea-assisted counterpart. The d-spacing of the (001) plane of SnS2-C is calculated to be 0.604 nm, which is slightly larger than that of SnS2 (0.585 nm). The decreased number of layers and the enlarged inter-layer spacing of SnS2-C could be attributed to the structural strain generated by the expansion of the crystal lattice after interstitial incorporation of carbon. The crystallite size, microstrain and lattice d-spacing of SnS2-C and SnS2 are summarized in Supplementary Table 1. Interestingly, we observed that the crystallite size in SnS2-C is smaller than that in SnS2 and the corresponding microstrain significantly enhanced around 3.6, 3.3 and 1.8 times based on (001), (101), and (110) planes after interstitial C doping into SnS2. Elemental analysis is adopted to identify the elemental compositions of the SnS2-C and SnS2 samples as depicted in Fig. 1b, which shows that the SnS2-C sample contains around 20.78 atomic % C. Although SnS2 shows ~ 2.79 atomic % of C, however, this low C level is very close to the error limit. The calculated atomic ratio of S to Sn is ~ 1.92 and 1.86 for SnS2-C and SnS2, respectively.

Crystal structure and optical properties of SnS2-C and SnS2. a XRD patterns of SnS2-C, SnS2 and commercial SnS2. b Chemical compositions, c UV-vis diffuse reflectance and (insets) Tauc plots with both direct and indirect fittings, and d Raman spectra, of the SnS2-C and SnS2
Optical properties
The optical absorption measurement was performed, followed by a tauc plot to estimate the band gap for the as-prepared SnS2-C and SnS2 (Fig. 1c). The observed direct band gaps of SnS2-C and SnS2 are 2.54 and 2.43 eV, respectively. In addition, SnS2-C shows an absorption band edge towards longer wavelength ~ 2.34 eV, indicating a decreased band gap in SnS2-C as compared with undoped SnS2. It is also worth noting that the SnS2-C exhibits a significantly higher absorption ranging from a visible-light wavelength of 530 nm, which is the most intense region in the solar spectrum, towards longer wavelength. In addition, SnS2-C exhibits an indirect band gap 1.75 eV that is smaller than its direct band gap. For SnS2, on other hand its indirect band gap (2.05 eV) is nearly close to its direct band gap. Presumably, the interstitial C doping creating microstrain on SnS2-C, as compared with SnS2, affects the electronic character of the valance band and conduction band edges. This is closely similar with the recently strain-induced band gap transition on bulk SnS2 simulation study34. Thus, we expect that the indirect band gap and additional band edge in SnS2-C, whereas maximize photon absorption, will also affect the electron-hole pair’s lifetime, which is beneficial for charge carrier to participate in the surface photocatalytic application.
To further quantify the doped carbon content in SnS2-C, we have performed solid-state NMR spectroscopic characterizations as shown in Supplementary Figure 2 and 3. The 13C Cross-Polarization Magic Angle Spinning (CPMAS) NMR spectra of SnS2-C shows two broad resonances at 23.8 ppm and at 133.3 ppm, respectively. The observed chemical shifts do not match those of pristine l-cysteine or the related decomposition compound (pyruvic acid) as shown in Supplementary Table 2. The observed resonance in the 13C CPMAS NMR spectrum of SnS2-C remains unchanged after the light activated CO2 reduction reaction. Thus, we believe that the observed chemical shifts in the 13C CPMAS NMR spectra of SnS2-C should be related to the carbon containing molecules doped inside the SnS2 layer during hydrothermal synthesis.
The samples were further investigated by Raman spectral analysis as shown in Fig. 1d. The spectra of both samples illustrate the strong characteristic peaks at 312 and 314.2 cm−1, respectively, of the SnS2-C and SnS2 sample, which are assigned to the A1g mode of SnS2. This observed Raman in plane mode of the atomic vibration shift around 2 cm−1 is strongly related to the significant changes to the inter-layer covalent interaction of SnS in SnS2-C after interstitial carbon doping and is well supported with the reported simulation study34. SnS2 shows another weak peak ~ 206.1 cm−1, resulting from Eg symmetry transition owing to out of plane atomic vibration in the 2H polytype of SnS2. However, for SnS2-C, instead of a single Eg peak in the 2H, we observed a broad peak from 190 to 225 cm−1 corresponding to the 4H and 18R polytypes of the SnS2 phase. Overall, the broadening and softening of A1g peak observed in the SnS2-C, in comparison with SnS2, can be attributed to the interstitial doping into the SnS2-C layer structure and formation of different SnS2 polytypes35. It is also worth noting that excessive incorporation of carbon may lead to formation of carbonaceous matters. As shown in Supplementary Figure 4 the Raman spectra of SnS2-C reveal additional peaks ~ 1186.9, 1336.6, and 1470.1 cm−1, which match the characteristic vibrational modes of 7A1, 6A1g, and 6E2g, respectively, in doped amorphous carbon with pentatomic and heptatomic rings36. The presence of the carbonaceous matters in the SnS2-C may introduce heterogeneous interfaces favorable for carrier separation, as will be discussed later.
Morphology and microstructure analysis
The morphology of the as-prepared SnS2 samples was characterized by field emission scanning electron microscopy (FESEM). Figure 2(a, b) shows the typical SEM images of SnS2-C and SnS2, respectively, and their insets are the corresponding high-resolution transmission electron microscopy (HRTEM) images. The SnS2-C samples exhibit flower type morphology composed of a number of nanosheets having uniform sheet dimension ~ 300–400 nm; however, these aggregated nanosheets have rough surface. Typical thickness of the SnS2-C nanosheets is ~ 30–60 nm, well underneath the photogenerated carriers diffusion length of SnS2 crystal, which is more favorable for the carrier diffusion process during photocatalytic reaction. For the SnS2 samples, we observed plate-like nanostructures, where the nanoplates are thicker and bigger than those nanosheets in SnS2-C. The nanoplates are ~ 0.5–1 micron in size and 150–250 nm in thickness, whilst showing a smooth surface morphology. Figure 2(c, d) shows the HRTEM lattice fringes of the SnS2-C and SnS2, respectively, and their insets are the corresponding selective area electron diffraction (SAED) patterns. The SAED of SnS2-C reveals the polycrystalline nature and dominant 001, 100, 101, and 110 diffraction planes with other planes, whereas SnS2 shows single-crystal diffraction along the [001] axis. It shall be noted that the HRTEM image analysis indicates the interplaner spacing of SnS2-C is larger than that of SnS2. This result is in good agreement with the XRD analysis results. Figure 2e shows the high-angle annular dark field (HAADF) image and energy dispersive x-ray spectroscopy (EDX) elemental maps of the SnS2-C samples, signifying that the Sn, S and C are evenly distributed within the SnS2-C nanostructure. In Fig. 2f the HAADF-EDX elemental mapping of SnS2 clearly shows Sn and S are well distributed without any elemental carbons. This is consistent with the previous elemental analysis data.

Morphology and microstructure analysis of SnS2-C and SnS2. a, b SEM images of SnS2-C and SnS2, respectively, and (inset) corresponding HRTEM images. c, d HRTEM lattice fringes and (inset) corresponding SAED patterns of SnS2-C and SnS2. e, f High-angle annular dark-field (HAADF) image and EDX elemental mapping of C, S, Sn from selected area for SnS2-C and SnS2
Chemical composition and photoluminescence study
Figure 3(a, b) presents the comparison of high-resolution XPS spectra of Sn 3d and S 2p of the as-prepared SnS2-C and SnS2 samples. In Fig. 3a, the measured binding energies of SnS2-C as compared with SnS2, corresponding to Sn 3d5/2 and Sn 3d3/2, are higher binding energy shifted and ~ 486.7 and 495.2 eV, respectively; these binding energies indicate Sn4+ ions in the SnS2 samples. This shift is induced by the distortion of the SnS2 lattice after carbon doping. A difference of around 8.4 eV between the two strong Sn 3d peaks is characteristic of tetravalent Sn 3d states. Furthermore, in Fig. 3b, the high-resolution S 2p core level analysis of SnS2-C at binding energies of ~ 161.6 and 162.8 eV corresponds to S 2p3/2 and S 2p1/2, which are good typical values for a metal sulfide with a doublet separation of around 1.2 eV. The observed S 2p3/2 and S 2p1/2 values of SnS2-C are higher binding energy shifted as compared with SnS2. The observed XPS-binding energies of Sn 3d and S 2p spectra confirmed the Sn4+ and S2+ characters of the as-prepared SnS2 samples. Interestingly, in SnS2-C, we observed two extra resolvable peaks around 163.4 and 164.6 eV, which revealed the corresponding S 2p3/2 and S 2p1/2 states of polysulfide. The XPS results are well consistent with the reported value37, 38. To study the transfer and exciton separation behavior of the photogenerated electrons and holes of the as-prepared SnS2 we carried out the photoluminescence (PL) and time-resolved photoluminescence (TRPL) measurements as shown in Fig. 3c (see Supplementary Fig. 5). Two PL peaks around 493 and 548 nm were observed for SnS2; in contrast, these peaks become weak in case of SnS2-C, revealing that the recombination of photo-induced charge carriers is reduced greatly, presumably by the enhanced interfacial charge transfer between the carbonaceous matters and SnS2. The normalized PL spectrum of SnS2-C shows a nearly threefold lower PL intensity as compared with that of SnS2, revealing that carbon doping lowers the recombination rate. To understand the exciton separation behavior, we measured TRPL spectroscopy at 493 and 548 nm emissions to estimate the lifetime of the electron-hole pair. The emission decay data of SnS2 and SnS2-C were fitted biexponentially (see Supplementary Fig. 5) and the calculated slow decay time τ1, fast decay time τ2 and average lifetime τavg are summarized in Fig. 3c. The observed average lifetimes for SnS2-C are 4.88 and 0.33 ns, which are much less than the 12.98 and 24.53 ns for SnS2 at 493 and 548 nm, respectively. This shortening of the lifetime in SnS2-C indicates the emergence of a nonradiative pathway, that is, the delocalization of electrons from SnS2 to C and hence effective carrier separation. Therefore, the lower recombination of photogenerated electrons in the SnS2-C allows them to reach the surface and consequently enhance the photoreduction process. Supplementary Figure 6 shows the impedance spectroscopy data for the SnS2-C and SnS2 on FTO electrodes at an applied potential of 1.2 V (vs NHE) with amplitude of 10 mV and a frequency ranging from 0.01 to 105 Hz in 0.1 M Na2SO4. The Nyquist plot reveals ideal semiconductor behavior of both SnS2-C and SnS2. However, the Nyquist plot for the SnS2-C in the high-frequency domain shows a smaller semicircle with a low RC (270.9 Ω/cm2) compared with that of the SnS2 (398.7 Ω/cm2), suggesting that the presence of carbon in the SnS2-C not only improves the charge transfer behavior but also offers more conducting pathway.

Electronic structure analysis of SnS2-C and SnS2. a High-resolution XPS Sn 3d spectra of SnS2-C and SnS2. b High-resolution XPS S 2p spectra of SnS2-C and SnS2. c Normalized PL spectra of SnS2-C and SnS2 and the summary table of TRPL slow, fast, and average lifetime calculated at both 493 and 548 nm emissions
Adsorption study
Nitrogen adsorption–desorption isotherm measurements were carried out to determine the surface area of the as-prepared SnS2 samples. Supplementary Figure 7 shows the corresponding N2 adsorption–desorption isotherms for both SnS2 and SnS2-C architectures. The shape of the curve is typical for a type II isotherm, indicating the presence of a macroporous structure for both samples. In addition, the hysteresis loops of type H3 are observed, reflecting the presence of non-rigid aggregates of plate-like particles with macropores network. Using the Brunauer–Emmett–Teller (BET) method, the specific surface area of the SnS2-C was measured as about 26.56 m2/g, which was larger than that of the SnS2 (10.73 m2/g). The two times higher surface area of SnS2-C could offer more active sites exposed for CO2 adsorption, thus more favorable for the photocatalytic CO2 reduction to solar fuels products.
Density functional theory calculation
Photocatalytic CO2 reduction activity has been shown to be primarily dependent on the adsorption energy of the CO2 molecule to the photocatalyst surface and corresponding CO2 dissociation energy. To understand the photocatalytic CO2 reduction activity for carbon-doped SnS2 surface, theoretical calculations were performed with density functional theory (DFT) plane-wave method utilizing the Vienna ab initio simulation package to predict the CO2 adsorption energy and conversion into CO on the carbon-doped SnS2. We considered two possible ways of introducing carbon-doping atom into the two-dimensional SnS2 2H polytype. The first was an S atom substituted with a C atom (hereafter denoted as CS-S) (~ 12.5% atomic C doping); and the second was a C doping in an interstitial position (hereafter, denoted as Cint) (~ 25% atomic C doping). We calculated the formation energies (Ef) of a dopant atom in substitutional and interstitial configurations to characterize the stability of the doped SnS2, as shown in Supplementary Table 3. The supercell model and partial geometries from the structurally optimized C-doped SnS2 shown in Supplementary Figures 8 are explained in detail in Supplementary Method. The calculated energy results indicate that interstitial C doping is of lower formation energy than that of the S-substituted one as shown in Supplementary Table 3. We compared the energies of CO2 adsorption and their dissociation energies on the two different carbon-doped SnS2, C-SnS2 (Cs-s) and 2C-SnS2 (Cs-s: Cint), the resulting energies are presented in Fig. 4. First, CO2(g) can undergo adsorption on the C doped SnS2 [C-SnS2(Cs-s)] forming CO2–C-SnS2(a) with an exothermicity of 25.0 kcal mol−1. The dissociation of CO2–C-SnS2(a) yielding CO–C-SnS2(a) has to overcome a rather low energy barrier of 19.5 kcal mol−1 at TS1, with an exothermicity of 30.8 kcal mol−1. The CO2 adsorption energy on the interstitial C-doped SnS2 [2C-SnS2(Cs-s: Cint)] has a binding energy of 10.2 kcal mol−1, which is 15 kcal mol−1 smaller than that in the C-SnS2(Cs-s) case. The CO2–2C-SnS2(a) dissociation barrier at TS2 is only 13.3 kcal mol−1, which is readily accessible at room temperature. In the former case, the deeper adsorption well will help accommodate more CO2 than the latter case, which has, however, a lower dissociation barrier. We therefore expect that both cases are competitive in practice. To confirm the CO2 adsorption characteristic on the as-prepared photocatalyst surface, we performed the CO2 adsorption study at lower temperature (195 K) as shown in Supplementary Figure 9, which could reveal more structural information of porosity instead. The total pore volume of SnS2-C analyzed by CO2 adsorption (P/Po = 0.96) at 195 K is 1.5-fold higher than the one of SnS2 and this aforementioned comparison suggested that SnS2-C might possess higher pore volume with narrow porosity (<0.4 nm) than SnS2 while physically adsorbing CO2. It should be mentioned that the present data is consistently comparable with the total pore volumes analyzed by nitrogen sorption (P/Po = 0.97) performed at 77 K previously, in which it was observed that the total pore volume of SnS2-C is 2.1-fold higher than the corresponding one of SnS2. Moreover, the overall CO2 adsorption isotherm study is comparable with the theoretical prediction. Thus we believe that SnS2-C can offer more active sites and different surface energy exposed for CO2 adsorption.

Theoretical energy calculation by DFT. a, b Potential comparative free energy of CO2 adsorption, and dissociation energy on carbon-doped SnS2[C-SnS2 (Cs-s) and 2C-SnS2 (Cs-s: Cint)] with their corresponding model structure (Unit: kcal mol−1)
Photocatalytic CO2 reduction study
Photoreaction characteristics of the as-prepared SnS2-C and SnS2 nanostructures were determined through reaction between CO2 and water in the gas phase (see Supplementary Figure 10). Figure 5a illustrates the cumulative acetaldehyde production yield after 14 h for the SnS2-C and SnS2 nanostructure photocatalysts. The observed maximum cumulative acetaldehyde yields after 13 h are around 125.66 µmole/100 mgcat and 0.55 µmole/100 mgcat for the SnS2-C and SnS2 photocatalysts, respectively. It can be seen that the prepared SnS2-C nanostructure photocatalyst exhibited prominent photocatalytic CO2 reduction activity under visible light and selectively produced acetaldehyde as a major product through multi-electron reduction. SnS2-C photocatalyst performance started slow decay after 12 h performance as shown in Supplementary Figure 11. The maximum photocatalytic performance was observed at 9 h, after that slow decay started and showed ~ 6 % deterioration after 5 h. We believe that this is owing to the absorbed product on the catalyst surface during continuous photocatalytic reaction. To avoid this problem, instead of continuous stability study we performed 8 h consecutive cycle reaction using AM 1.5 light source. After each reaction cycle, we discontinued the reaction and cleaned the reactor together with degassing the catalyst to start a new cycle. The photocatalytic stability by consecutive cycles is shown in Supplementary Figure 11 (inset). The consecutive cycle stability results revealed that the SnS2-C retained its stable catalytic performances for the CO2 reduction. In addition, Fig. 5b shows a comparison of visible light photocatalytic CO2 reduction to solar fuel yields of SnS2-C and SnS2. The maximum solar fuel formation yields for the SnS2-C and SnS2 photocatalysts are ~ 13.98 and 0.055 µmole/100 mgcat-hr, respectively. The photocatalytic solar fuel formation yield for SnS2-C is almost 250 times higher than that for SnS2. The calculated solar fuel photochemical quantum efficiency (PCQE) for SnS2-C is ~ 0.72% (see Supplementary Table 4). To further characterize the photocatalytic behavior a control experiment in the absence of CO2 and another one without light irradiation were performed. The absence of acetaldehyde detection in the control experiments confirmed photocatalytic acetaldehyde generation from CO2 under visible light. In addition, to confirm the acetaldehyde formation from CO2 reduction, we have conducted an isotope tracer experiment under 13CO2 atmosphere. Analysis of the gas chromatography-mass spectrometry (GC-MS), as shown in Supplementary Figure 12, revealed the reaction product to be 13CH313CHO (m/z=46), without any product containing 12C, confirming that acetaldehyde was indeed produced from the photocatalytic reduction of 13CO2 (m/z = 45). To understand better visible-light absorption and photocatalytic CO2 reduction performance of SnS2-C, we performed the wavelength dependence photocatalytic reaction and summarized the PCQE in Supplementary Table 5. SnS2-C showed different PCQE of 1.64, 1.04, and 0.32% at 400 ± 25, 500 ± 25, and 600 ± 25 nm wavelength respectively using specific band pass filters. The 13C CPMAS NMR spectra of SnS2-C before and after light irradiation also confirmed that the doped C species inside SnS2-C were unaltered after the CO2 reduction study under light irradiation, as shown in Supplementary Figure 3. Additional XPS analysis of SnS2-C after 14 h photocatalytic performance shows that Sn 3d and S 2p peaks are unchanged (see Supplementary Fig. 13). To confirm the chemical stability of the SnS2-C photocatalyst we conducted XPS analysis after photocatalytic reaction 2, 6, 10, and 14 h, respectively as shown in Supplementary Figure 14. It shall be noted that the S 2p spectra were deconvoluted into four peaks (F1, F2, F3 and F4), as depicted in Supplementary Figure 13b and Supplementary Figure 14 f-j. The XPS spectra of SnS2-C show nearly unchanged in the Sn (3d) and S (2p) deconvoluted peaks as a result of irradiation. The correlation between the relative peak intensity of the deconvoluted Sn(3d) and S(2p) components, as summarized in Supplementary Table 6, shows a small change in S(2p) peaks after 2 h photocatalytic reaction as compared with the pristine sample before irradiation. Moreover, after 2 h photocatalytic reaction the S (2p) components remained unchanged and stable during photoirradiation. We believe that during the initial 2 h irradiation, chemisorptions of CO2 molecule and the catalyst surface occurred, causing the catalyst surface’s little change, and however, after that it remained stable.

Comparative photocatalytic CO2 reduction activity of SnS2-C and SnS2. a Cumulative acetaldehyde formation yield of SnS2-C and SnS2. b Comparative solar fuel formation rate and quantum efficiency of SnS2-C, SnS2, and commercial SnS2 under a visible light source (300 W halogen lamp)
CO2 reduction mechanism
It is well accepted that the photocatalytic CO2 reduction is a multi-electron reduction. In the initial step, direct photon absorption by SnS2 generates electron-hole pairs. Specifically, carbon-doped SnS2-C significantly extends the absorption band of the materials into longer wavelength range (near 530 nm and above) as compared with undoped SnS2. The carbon doping also promotes the CO2 molecule adsorption on the surface with a relatively small dissociation barrier, as shown in simulation studies. Moreover, carbon-doped SnS2-C containing smaller nanosheets with only a few atomic layers can shorten the charge diffusion time as compared with SnS2. The band edge positions of the photocatalysts directly influence the photocatalytic reduction and oxidation reactions at the catalyst surface. To understand the details of the electronic state and band energy alignment of SnS2-C and SnS2, we performed the ultraviolet photoemission spectroscopy (UPS) study shown in Supplementary Figure 15. The work functions of SnS2-C and SnS2 were calculated to be 4.4 and 4.16 eV (vs vacuum level) (see Supplementary Table 7), from which the corresponding Fermi levels can be deduced. Based on the calculated Fermi levels, conduction band and valence band maxima of SnS2-C and SnS2, we have drawn the electronic band diagram as shown in Fig. 6. A corresponding hypothetical photoreduction mechanism has been proposed. The electronic band diagram clearly shows that the position of the frontier orbitals of CO2 with respect to the conduction band position in both SnS2 and SnS2-C would make multi-electron reduction process feasible. However, in SnS2-C, interstitially doped carbon introduced somewhat longer band tail owing to the microstrain induced new electronic state penetration into the bulk. The doped carbon helps the electrons to migrate faster to the surface of SnS2-C for the reduction reaction. Besides electrons, the photogenerated holes may react with the water molecules to generate oxygen, hydrogen peroxide or hydroxide radicals. The conduction band position of SnS2-C with respect to the onset reduction potential energy of CO2 favors ten-electron reduction on the surface of the photocatalyst. The ten-electron reduction processes are involved in the production of acetaldehyde in our experiment. The overall reactions can be described in the following equations.

Band edge positions and photocatalytic reaction mechanism: Comparative band diagram of SnS2-C and SnS2, together with a proposed electron-hole separation of photo-excited electron-hole pairs in SnS2-C
Discussion
The overall photocatalytic multi-electron CO2 reduction mechanism is more complex than the single electron water-splitting reaction. However, in the photocatalytic CO2 reduction process, the photogenerated holes move around to the surface and cannot be excluded. The S 2p XPS analysis of SnS2-C reveals that the two extra deconvoluted peaks are owing to polysulfides, which may act as scavenging agents to eliminate the photogenerated holes, resulting in more efficient separation of the photogenerated electrons and holes. Moreover, the generation of O2 is suppressed, whereas the yield of acetaldehyde is enhanced. On the other hand, the excess polysulfide oxidizes to elemental sulfur via redox process and suppresses the corrosion of SnS2 during the photocatalytic reaction. Overall, the enhanced photocatalytic reaction may result from combined favorable situations, including band edges tuning by induced microstrain together with high surface area, reduced photocarriers diffusion length and improved charge separation process in carbon-doped SnS2. It is worth to mention that carbon-doped SnS2-C can be synthesized by a simple l-cysteine assisted hydrothermal method and is an effective way to improve the photocatalytic CO2 reduction activity under visible light. Nevertheless, more studies are needed to better understand the mechanism and to further enhance the photocatalytic activity and selectivity of hydrocarbon formation.
In conclusion, carbon-doped SnS2-C was successfully synthesized by an l-cysteine assisted hydrothermal process and was demonstrated to be a highly efficient photocatalyst for CO2 reduction under visible light. The synthesized SnS2-C photocatalyst shows selective photocatalytic CO2 reduction to acetaldehyde with moderately high PCQE above 0.7%. Based on various structural analyses, the C doping is mainly incorporated as interstitials, which introduce micro strains and affect electronic band structures as well as the optical properties. Moreover, DFT calculations suggest that carbon doping also promotes the CO2 molecule adsorption on the surface with a relatively small dissociation barrier in C doped SnS2-C. All these factors lead to significantly enhanced photocatalytic reduction of CO2. We believe that carbon doping in the narrow-band gap of dichalcogenides and other metal sulfides is a promising approach to develop high quantum efficiency photocatalysts for CO2 reduction to solar fuels.
Methods
Synthesis
The carbon-doped SnS2 nanoflower (SnS2-C) and SnS2 nanoplate (SnS2) were prepared by hydrothermal process. All the reagents used in the experiment were of analytical grade and used without further purification. In a typical procedure, 1 mM of tin (IV) chloride pentahydrate (SnCl4, 5H2O) and 5 mM l-cysteine (C3H7NO2S) were added to a 60 ml of distilled water and gradually dispersed to form a homogeneous solution by vigorous magnetic stirring for 1 h at room temperature. Finally, the resulting solution was transferred into a Teflon-lined stainless autoclave. The autoclave was sealed and heated at 180 °C for 24 h. After hydrothermal reaction, the sample was cooled to room temperature naturally. The resulting product was collected by centrifugation at 8000 rpm for 10 min and washed several times with distilled water. Finally, the collected yellow SnS2-C powder was vacuum-dried at 80 °C overnight. In a similar procedure using 5 mM thiourea (CH4N2S) as an S source, SnS2 nanoplate (SnS2) was synthesized at 180 °C for 12 h and vacuum-dried at 80 °C. The overall SnS2-C and SnS2 synthesis process is schematically presented in the Supplementary Figure 1.
Characterization
The ultraviolet-visible absorption spectrum of powder samples was measured with a Jasco V-670 spectrophotometer using an integrated sphere. The crystal structures were determined by XRD using CuKα radiation (Bruker, D2 PHASER with XFlash). The surface morphology of all samples was characterized by FESEM (JEOL, 6700F). The HRTEM (JEOL-2100) studies with SAED and EDX were also performed to determine morphology, crystal phase and elemental compositions. The Raman spectra were measured using Jobin-Yvon LabRAM HR800 with laser source of 633 nm. X-ray photoelectron spectroscopy (XPS) analysis was performed on a theta probe ESCA VG Scientific (2002) using a monochromatic AlKα as the exciting source. The peak positions of the XPS were calibrated carefully with respect to the Au 4f peak. Finally, all the XPS spectra were deconvoluted by Voigt fitting function after a Shirley background subtraction procedure. Excitation-dependent PL measurements were performed using a spectrofluorometer (Horiba Jobin-Yvon FluoroMax-4). TRPL techniques were carried out using time-correlated single-photon counting. A pulsed laser with a wavelength of 375 nm, duration of 250 fs, and repetition frequency of 20 MHz was used as the excitation source for the steady state PL and TRPL studies. The collected PL was dispersed by a 0.75 m spectrometer and detected by the photomultiplier tube. For work function and valence band maxima measurement, UPS was performed using Perkin-Elmer phi 5400 system under vacuum with Fermi energy (Ef) calibration using in situ deposited gold. For UPS measurement, the samples were uniformly dispersed on the gold-coated ITO. BET surface area was determined by recording nitrogen adsorption and desorption isotherms using Micromeritics ASAP 2010 Accelerated Surface Area and Porosimetry System. The total volume was calculated from the amount of nitrogen adsorbed at P/Po=0.97, assuming that adsorption on the external surface was negligible in comparison to the adsorption in pores. For the microporosity study, the adsorption data were acquired at relative pressure P/Po between 0 and 0.01 with little incremental dose at liquid nitrogen temperature (77 K), using a Micromeritics 3Flex analyzer, which was used for carbon dioxide gas (CO2, purity of 99.9992%) adsorption measurements as well. All samples (50 mg each) were initially degassed at 423 K for 12 h under a 1 × 10−2 mmHg vacuum level, by Micromeritics Smart VacPrep degasser. The CO2 sorption was analyzed at relative pressure (P/P0) between 0.0003 and 0.96 for the P0 of 789.5 mmHg at 195 K. During the analysis, the temperatures were maintained by the slurry of the combination of dry ice and acetone (wt/wt = 0.86) in Dewar. After the analysis, the free space of the sample tube was determined by using Helium gas (purity of 99.9992%). The photocatalytic CO2 reduction products were analyzed by gas chromatography (GC). The GC analyses were performed on a GC-FID-CHINA CHROMATOGRAPHY 9800 system using glass column Porapak Q (80–100 mesh), at injection temperature of 50 °C, FID temperature of 150 °C and oven temperature of 80 °C. GC-MS analysis was performed on GC (HP6890)/MS(5973) system (column-Agilent J&W 122–7032 DB-WAX, inj. Temp. 250 °C and oven temperature of 35 °C utilizing 13CO2 source (Cambridge Isotope laboratories, Inc. USA).
Microstructural parameters analysis of the SnS2-C and SnS2
The average crystallite size of the sample estimated using Scherrer’s formula, i.e., (D) = Kλ/βcosθ, where K = 0.89 is the shape factor, λ is the X-ray wavelength of X-ray radiation, θ is Bragg’s angle, and β is the full width at half maximum of the respective peak. The microstrain is calculated using the following relation: ε = βcosθ/4. Supplementary Table 1 shows the average crystal size, microstarin, and lattice d-spacing calculation.
Solid-state NMR analysis of SnS2-C
The 119Sn MAS NMR spectrum of SnS2-C and commercial SnS2 (MKN-SnS2-900 purchased from M K Implex Corp. Canada) are shown in Supplementary Figure 2. SnS2-NS synthesized from cysteine shows a major chemical shift at −76 ppm, which is consistent with 119Sn MAS NMR of commercial SnS2. The observed line width broadening may be a result of greater distribution of crystal grain size. The additional small peak at around −605 ppm, could be attributed to different stacking of 4H and 18R polytype of SnS2 as supported by our Raman spectroscopy. The 13C CPMAS NMR spectra of SnS2-C before and after CO2 reduction reaction are shown in Supplementary Figure 3. For comparison, 13C NMR chemical shift of l-cysteine and pyruvic acid are summarized in Supplementary Table 2. For other details of experimental procedures, please refer to the Supplementary Methods.
Data availability
The authors declare that data supporting the findings of this study are available within the paper and the supplementary information file.
References
Abelson, P. H. Energy and climate. Science 197, 941–941 (1977).
Lewis, N. S. & Nocera, D. G. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl Acad. Sci. 103, 15729–15735 (2006).
Alfano, O. M., Bahnemann, D., Cassano, A. E., Dillert, R. & Goslich, R. Photocatalysis in water environments using artificial and solar light. Catal. Today 58, 199–230 (2000).
Hoffmann, M. R., Martin, S. T., Choi, W. & Bahnemann, D. W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 95, 69–96 (1995).
Cowan, A. J. & Durrant, J. R. Long-lived charge separated states in nanostructured semiconductor photoelectrodes for the production of solar fuels. Chem. Soc. Rev. 42, 2281–2293 (2013).
Tran, P. D., Wong, L. H., Barber, J. & Loo, J. S. C. Recent advances in hybrid photocatalysts for solar fuel production. Energy Environ. Sci. 5, 5902–5918 (2012).
Inoue, T., Fujishima, A., Konishi, S. & Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 277, 637–638 (1979).
Habisreutinger, S. N., Schmidt-Mende, L. & Stolarczyk, J. K. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 52, 7372–7408 (2013).
Das, S. & Wan Daud, W. M. A. A review on advances in photocatalysts towards CO2 conversion. RSC Adv. 4, 20856–20893 (2014).
Wang, W.-N. et al. Size and structure matter: enhanced CO2 photoreduction efficiency by size-resolved ultrafine pt nanoparticles on TiO2 single crystals. J. Am. Chem. Soc. 134, 11276–11281 (2012).
Ong, W.-J., Tan, L.-L., Ng, Y. H., Yong, S.-T. & Chai, S.-P. Graphitic carbon nitride (g-C3N4)-Based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 116, 7159–7329 (2016).
Shown, I. et al. Highly efficient visible light photocatalytic reduction of CO2 to hydrocarbon fuels by Cu-Nanoparticle decorated graphene oxide. Nano. Lett. 14, 6097–6103 (2014).
Hsu, H.-C. et al. Graphene oxide as a promising photocatalyst for CO2 to methanol conversion. Nanoscale 5, 262–268 (2013).
Li, M. et al. Highly selective CO2 photoreduction to CO over g-C3N4/Bi2WO6 composites under visible light. J. Mater. Chem. A 3, 5189–5196 (2015).
Neaţu, Ş., Maciá-Agulló, J. A., Concepción, P. & Garcia, H. Gold–Copper nanoalloys supported on TiO2 as photocatalysts for CO2 reduction by water. J. Am. Chem. Soc. 136, 15969–15976 (2014).
Low, J., Cheng, B., Yu, J. & Jaroniec, M. Carbon-based two-dimensional layered materials for photocatalytic CO2 reduction to solar fuels. Energy Storage Mater. 3, 24–35 (2016).
Sakimoto, K. K., Wong, A. B. & Yang, P. Self-photosensitization of nonphotosynthetic bacteria for solar-to-chemical production. Science 351, 74–77 (2016).
Hossain, S. T. & Mukherjee, S. K. Toxicity of cadmium sulfide (CdS) nanoparticles against Escherichia coli and HeLa cells. J. Hazard. Mater. 260, 1073–1082 (2013).
Osterloh, F. E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 42, 2294–2320 (2013).
Voiry, D., Yang, J. & Chhowalla, M. Recent strategies for improving the catalytic activity of 2D TMD nanosheets toward the hydrogen evolution reaction. Adv. Mater. 28, 6197–6206 (2016).
Chhowalla, M. et al. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. Chem. 5, 263–275 (2013).
Singh, A. K., Mathew, K., Zhuang, H. L. & Hennig, R. G. Computational screening of 2D materials for photocatalysis. J. Phys. Chem. Lett. 6, 1087–1098 (2015).
Lei, Y., Song, S., Fan, W., Xing, Y. & Zhang, H. Facile synthesis and assemblies of flowerlike SnS2 and In3+-Doped SnS2: Hierarchical structures and their enhanced photocatalytic property. J. Phys. Chem. C. 113, 1280–1285 (2009).
Zhang, Y. C., Du, Z. N., Li, K. W., Zhang, M. & Dionysiou, D. D. High-performance visible-light-driven SnS2/SnO2 nanocomposite photocatalyst prepared via in situ hydrothermal oxidation of SnS2 nanoparticles. ACS Appl. Mater. Interfaces 3, 1528–1537 (2011).
Yang, B. et al. SnS2 as low-cost counter-electrode materials for dye-sensitized solar cells. Mater. Lett. 133, 197–199 (2014).
Zhou, X., Zhang, Q., Gan, L., Li, H. & Zhai, T. Large-size growth of ultrathin SnS2 nanosheets and high performance for phototransistors. Adv. Funct. Mater. 26, 4405–4413 (2016).
Ou, J. Z. et al. Physisorption-based charge transfer in Two-Dimensional SnS2 for selective and reversible NO2 gas sensing. ACS Nano 9, 10313–10323 (2015).
Ma, C. et al. Investigating the energy storage mechanism of SnS2-rGO composite anode for advanced Na-Ion batteries. Chem. Mater. 27, 5633–5640 (2015).
Bryushinin, M. A., Dubrovsky, G. B. & Sokolov, I. A. Non-steady-state photocurrents in SnS2crystals. Appl. Phys. B 68, 871–875 (1999).
Sun, Y. et al. Freestanding tin disulfide single-layers realizing efficient visible-light water splitting. Angew. Chem. Int. Ed. 51, 8727–8731 (2012).
Sun, Y., Li, G., Xu, J. & Sun, Z. Visible-light photocatalytic reduction of carbon dioxide over SnS2. Mater. Lett. 174, 238–241 (2016).
Huang, C. et al. Carbon-doped BN nanosheets for metal-free photoredox catalysis. Nat. Commun. 6, 7698 (2015).
Lin, Y.-G. et al. Visible-light-driven photocatalytic carbon-doped porous ZnO nanoarchitectures for solar water-splitting. Nanoscale 4, 6515–6519 (2012).
Ram, B. & Singh, A. K. Strain-induced indirect-to-direct band-gap transition in bulk SnS2. Phys. Rev. B 95, 075134 (2017).
Nakashima, S., Katahama, H. & Mitsuishi, A. The effect of polytypism on the vibrational properties of SnS2. Phys. B+C. 105, 343–346 (1981).
Doyle, T. E. & Dennison, J. R. Vibrational dynamics and structure of graphitic amorphous carbon modeled using an embedded-ring approach. Phys. Rev. B 51, 196–200 (1995).
Chen, Q., Lu, F., Xia, Y., Wang, H. & Kuang, X. Interlayer expansion of few-layered Mo-doped SnS2 nanosheets grown on carbon cloth with excellent lithium storage performance for lithium ion batteries. J. Mater. Chem. A 5, 4075–4083 (2017).
Chia, X., Lazar, P., Sofer, Z., Luxa, J. & Pumera, M. Layered SnS versus SnS2: Valence and structural implications on electrochemistry and clean energy electrocatalysis. J. Phys. Chem. C. 120, 24098–24111 (2016).
Acknowledgements
We thank the Ministry of Science and Technology (MOST, especially 103–2745-M-002–006-ASP, 104–2745-M-002–004-ASP), Academia Sinica, National Taiwan University, the Ministry of Education (MOE), Taiwan for financial support. Technical support from Nano-Core, the Core facilities for nanoscience and nanotechnology at Academia Sinica in Taiwan, is acknowledged.
Author information
Authors and Affiliations
Contributions
I.S. proposed and performed the synthesis, UV-Vis, XRD, Raman, experiments; S.S. performed the GC and SEM measurements, Y.-C.C. performed GC calibration and photocatalyst experimental setup; R.P. performed DFT calculations directed by M.C.L., I.S. and A.S. performed the GC-MS isotope tracer analysis, F.-Y.F. performed PL and TRPL measurement, T.-Y.Y. performed the solid-state NMR measurement; P.-H.C. performed the UPS measurements; P.-W.C. performed CO2 adsorption analysis; I.S., W.-F.C., P.-W.C., C.-I.W., M.-C.L., K.-H.C. and L.-C.C. discussed and analyzed the results; I.S., K.-H.C. and L.-C.C. co-wrote the manuscript; K.-H.C. and L.-C.C. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shown, I., Samireddi, S., Chang, YC. et al. Carbon-doped SnS2 nanostructure as a high-efficiency solar fuel catalyst under visible light. Nat Commun 9, 169 (2018). https://doi.org/10.1038/s41467-017-02547-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-02547-4
This article is cited by
-
Photocatalytic CO2 reduction
Nature Reviews Methods Primers (2023)
-
Fabrication of g-C3N4/SnS2 type-II heterojunction for efficient photocatalytic conversion of CO2
Journal of Materials Science: Materials in Electronics (2023)
-
Nanohybrid embedded ferroelectric polymer blend for bipolar memristive application
Applied Physics A (2023)
-
Ellipsometry study of optical properties and dielectric response of SnS2 and GaS crystals
Journal of Materials Science: Materials in Electronics (2023)
-
Construction of hierarchical SnO2@SnS2 nanostructures on carbon cloth as advanced microwave absorbers
Journal of Materials Science: Materials in Electronics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
