
Abstract
Gold-catalyzed intermolecular alkyne oxidation by an N–O bond oxidant has proven to be a powerful method in organic synthesis during the past decade, because this approach would enable readily available alkynes as precursors in generating α-oxo gold carbenes. Among those, gold-catalyzed oxidative cyclization of dialkynes has received particular attention as this chemistry offers great potential to build structurally complex cyclic molecules. However, these alkyne oxidations have been mostly limited to noble metal catalysts, and, to our knowledge, non-noble metal-catalyzed reactions such as diyne oxidations have not been reported. Herein, we disclose a copper-catalyzed oxidative diyne cyclization, allowing the facile synthesis of a wide range of valuable pyrrolo[3,4-c]quinolin-1-ones. Interestingly, by employing the same starting materials, the gold-catalyzed cascade cyclization leads to the divergent formation of synthetically useful pyrrolo[2,3-b]indoles. Furthermore, the proposed mechanistic rationale for these cascade reactions is strongly supported by both control experiments and theoretical calculations.
Similar content being viewed by others
Introduction
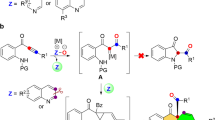
Highly efficient construction of N-heterocycle skeletons is one of the most important themes in organic synthesis. The structurally diverse and interesting family of tricyclic N-heterocycles, such as pyrrolo[3,4-c]quinolin-1-ones1,2,3,4,5,6,7 and pyrrolo[2,3-b]indoles8,9,10,11, are important structural motifs that can be frequently observed in bioactive molecules as well as in natural products (Fig. 1). It is surprising, however, that only a few preparative methods have been reported, with most employing the corresponding quinolines12,13,14 and indoles15,16,17 as precursors, respectively. Thus, new synthetic approaches for the direct construction of these skeletons are highly desired, especially those based on the assembly of structures directly from readily available and easily diversified building blocks.

Selected examples bearing the pyrrolo[3,4-c]quinolin-1-one and pyrrolo[2,3-b]indole core structure. Some of these molecules are synthesized in the next section
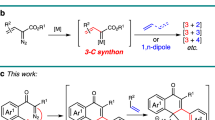
Gold-catalyzed intermolecular alkyne oxidation by an N–O bond oxidant, presumably via an α-oxo gold carbenoid intermediate, has attracted considerable interest during the past decade because this approach would enable readily available and safer alkynes to replace not easily accessible and hazardous α-diazo carbonyls as precursors in generating α-oxo metal carbenes18,19,20,21,22,23,24,25,26,27,28,29,30. Among those, gold-catalyzed oxidative cyclization of dialkynes has received particular attention because this chemistry offers great potential to build structurally complex cyclic molecules31,32,33,34,35. For example, Hashmi et al. reported an elegant protocol for the gold-catalyzed oxidative diyne cyclization via a presumable 1,6-carbene transfer (Fig. 2a)32. Notable is that haloalkynes are typically required for this strategy. Such a gold-catalyzed oxidative diyne cyclization has also been well exploited in the synthesis of various functionalized O-heterocycles by Zheng and Zhang33 and Ji et al.34 In addition, Tang et al. disclosed that rhodium could also catalyze this type of diyne oxidation (Fig. 2b)35. Despite these significant achievements, these alkyne oxidations have been mostly limited to noble metal catalysts, and, to our knowledge, non-noble metal-catalyzed such as diyne oxidation has not been reported.

Transition-metal-catalyzed oxidative diyne cyclization. a Au-catalyzed oxidative diyne cyclization (Hashmi). b Rh-catalyzed oxidative diyne cyclization (Tang). c Cu-catalyzed oxidative diyne cyclization and Au-catalyzed cascade cyclization (this work)
Inspired by our recent study on ynamide chemistry36,37,38,39,40,41,42,43, we envisioned that the synthesis of pyrrolo[3,4-c]quinolin-1-ones 2 might be accessed through such an oxidative cyclization of N-propargyl (azido)ynamides 1. However, realizing this cascade reaction is highly challenging because of two competing reactions. First, the generated vinyl metal carbene is highly reactive and often suffers the overoxidation by the same oxidant32, 35, 41, 42, in addition to many other side reactions. Second, the azido group would be expected to attack the ynamide directly to initiate the relevant alkyne amination via a presumable α-imino metal carbene pathway44,45,46,47,48,49,50,51,52,53,54,55. Herein, we describe the realization of a copper-catalyzed oxidative diyne cyclization protocol that allows the facile synthesis of a variety of valuable pyrrolo[3,4-c]quinolin-1-ones. Furthermore, by employing the same starting materials, the gold-catalyzed cascade cyclization leads to the divergent formation of pyrrolo[2,3-b]indoles. In addition, the mechanistic rationale for these cascade reactions, in particular accounting for the distinct selectivity, is also well supported by density functional theory (DFT) calculations.
Results
Optimization of reaction conditions
Table 1 shows the realization of the cascade cyclization of ynamide 1a in the presence of various transition metals (for more details see Supplementary Table 1, Supporting Information. To our delight, the tandem reaction indeed produced the desired pyrrolo[3,4-c]quinolin-1-one 2a under the previously optimized reaction conditions42, albeit in low yield (Table 1, entry 1). We then investigated other non-noble metals (Table 1, entries 2−6), and were pleased to find that Cu(CH3CN)4PF6 catalyzed the oxidative cyclization to produce the desired 2a in 41% yield (Table 1, entry 6). Of note, rhodium (Table 1, entry 7)35 and Brønsted acids56,57,58,59 such as TsOH and TfOH were not effective in promoting this reaction (for more details see Supplementary Table 1). Interestingly, pyrrolo[2,3-b]indole 3a was obtained as the main product in the presence of typical gold catalysts such as Ph3PAuNTf2 and IPrAuNTf2 (Table 1, entries 8 and 9). Further screening of oxidants revealed that the use of quinoline N-oxide 4b led to a significantly improved yield in the presence of Cu(CH3CN)4PF6 as catalyst (Table 1, entry 10, for more details see Supplementary Table 1), and 2a could be formed in 85% yield at 60 °C (Table 1, entry 11). In addition, condition optimization on the formation of 3a was also explored (for more details see Supplementary Table 1), and it was found that slightly improved yield was obtained by employing Ph3PAuNTf2 as catalyst in the absence of oxidant (Table 1, entry 12). Gratifyingly, 86% yield was achieved by using CH3NO2 as solvent (Table 1, entry 13), and similar yield was obtained when the catalyst loading was reduced to 2 mol% (Table 1, entry 14). Notably, no formation of 3a was observed under copper catalysis (for more details see Supplementary Table 1).
Synthesis of pyrrolo[3,4-c]quinolin-1-ones via Cu catalysis
With the optimal reaction conditions in hand (Table 1, entry 11), the reaction scope of the copper-catalyzed synthesis of pyrrolo[3,4-c]quinolin-1-ones was then explored (Fig. 3). The reaction proceeded smoothly with different aryl-substituted ynamides (R2 = Ar), affording the desired γ-lactam-fused quinolines 2a–h in generally good to excellent yields (Fig. 3, entries 1–8, 2a was confirmed by X-ray diffraction, for more details see Supplementary Table 2). In addition, heterocycle-substituted ynamide 1i was also a suitable substrate for this oxidative cyclization to produce the corresponding 2i in a serviceable yield (Fig. 3, entry 9), whereas none of the desired 2j was observed with alkyl-substituted ynamide 1j (Fig. 3, entry 10). The method worked efficiently for various aryl-substituted ynamides bearing both electron-donating and -withdrawing groups, and the desired 2k–o were obtained in 63−94% yields (Fig. 3, entries 11–15). Ynamides containing other protecting groups also reacted well to afford the tricyclic N-heterocycles in 68−85% yields (Fig. 3, entries 16–18). Importantly, no diketone formation via double oxidation by the same oxidant was observed in all cases32, 35.

Reaction scope for the formation of pyrrolo[3,4-c]quinolin-1-ones 2. Reaction conditions: [1] = 0.05 M; yields are those for the isolated products
This reaction was also extended to substituted N-propargyl ynamides and these chiral substrates could be readily prepared with excellent enantiomeric excesses by using Ellman′s tert-butylsulfinimine chemistry (for more details see Supplementary Fig. 87). Thus, the desired enantioenriched tricyclic N-heterocycles 2s–t were formed in good yields with well-maintained enantioselectivity by employing 8-isopropylquinoline N-oxide 4c as oxidant (Fig. 4).

Copper-catalyzed oxidative cyclization of chiral N-propargyl (azido)ynamides 1. Substrate scope of chiral N-propargyl ynamides 1
Synthesis of pyrrolo[2,3-b]indoles via Au catalysis
We also investigated the substrate scope for the gold-catalyzed synthesis of pyrrolo[2,3-b]indoles with the same ynamide substrates under the optimal reaction conditions (Table 1, entry 14). As shown in Fig. 5, this alkyne amination-initiated tandem reaction39, 44,45,46,47,48,49,50,51,52,53,54,55 proceeded very well and afforded the desired pyrrole-fused indoles 3a–h in mostly good to excellent yields (Fig. 5, entries 1–8, 3a was confirmed by X-ray diffraction, for more details see Supplementary Table 3). This chemistry could also be extended to heterocycle- or alkyl-substituted ynamides, leading to the corresponding 3i and 3j in 73% and 86% yields, respectively (Fig. 5, entries 9 and 10). Ynamides bearing different aryl groups and protecting groups were also suitable substrates for this gold catalysis to furnish the desired fused N-heterocycles in 56−86% yields (Fig. 5, entries 11–18).

Reaction scope for the formation of pyrrolo[2,3-b]indoles 3. Reaction conditions: [1] = 0.05 M; yields are those for the isolated products
Further synthetic transformations of the as-synthesized tricyclic N-heterocycles were also explored (Fig. 6). For example, the Ts group in γ-lactam-fused quinoline 2a, obtained on a gram scale in 77% yield, was easily removed by the treatment with H2SO4 to afford the corresponding 5a in 74% yield, which could be further transformed into DHODH inhibitor 5b 6. Alternatively, 5a could be converted into pyrrolo[3,4-c]quinoline-1,3-dione 5c, known for antibacterial activity against Gram-positive and Gram-negative bacteria2, via a facile K2CO3-mediated air oxidation60, 61 and metal-free oxidative arene imidation62. By using a similar strategy, the synthesis of caspase-3 inhibitor 5d was achieved starting from the corresponding ynamide 1u 5. In addition, pyrrole-fused indole 3a could be subjected to removal of the Ts group by NaOH or reduction of the carbonyl group by LiAlH4 to produce the desired 6a and 6b, respectively.

Synthetic applications. a Synthesis of bioactive molecules 5b and 5c. b Synthesis of caspase-3 inhibitor 5d. c Transformation of 3a into 6a and 6b
Mechanistic investigations
To understand the mechanism of these cyclizations, several control experiments were first conducted. As shown in Fig. 7, control experiments with H2 18O and18O2 isotopic labeling proved that the oxygen atom in the carbonyl group of 3a originates from water but not molecular oxygen. Of note, no incorporation of 18O into the 3a was observed when 3a was subjected to the reaction conditions with H2 18O (for more details see Supplementary Fig. 81).

Control experiments with 18O labeling study. a Reactions were run in the presence of 20 equiv of H2 18O. b Reactions were run in the presence of 18O2 atmosphere (1 atm)
In addition, when ynamide 1v was subjected to this copper-catalyzed cascade reaction, no 2v formation was observed, and the corresponding 2va was obtained in 66% yield instead (Fig. 8). These results suggested that vinyl copper carbene intermediate was presumably involved in such a diyne oxidation.

Trapping of the presumable vinyl copper carbene intermediate. Substrate scope of alkyl-substituted N-propargyl ynamide 1v
Based on the above experimental observations (for more details see Supplementary Figs. 80–85), previously published results32, 35, 44,45,46,47,48,49,50,51,52,53,54,55, and on DFT computations (for more details see Supplementary Figs. 75–79) plausible mechanisms for the divergent CuI/AuI-catalyzed synthesis of 2a and 3a are illustrated in Fig. 9. First, the catalytic [MI]-species is preferentially bound to the amide-neighbored, electron-richer triple bond of 1a, forming precursor A (for more details see Supplementary Fig. 75). In the oxidant-free cycle (for more details see Supplementary Fig. 76), intramolecular cyclization is thus triggered by nucleophilic attack of the proximal N atom of azide to form intermediate B, followed by elimination of N2 to form metal-carbenoid intermediate C, and a second cyclization to the enylium-cationic intermediate D. The latter can readily react with ambient H2O, leading eventually to product 3a (for more details see Supplementary Figs. 80, 81). The overall barrier height (OBH) (for more details see Supplementary Figs. 78, 79) is determined by the relative free energy of transition state TSc, which amounts up to 25.6 kcal/mol in CuI catalysis, 9.5 kcal/mol higher than that in AuI catalysis. This accounts well for the much higher efficiency of the AuI-catalyst in the oxidant-free synthesis of 3a. In the oxidant-involving cycle (for more details see Supplementary Fig. 77), precursor A subjects to nucleophilic attack of oxidant 4a to form vinyl metal intermediate B′. Upon N–O bond cleavage, B′ transforms into α-oxo metal-carbenoid intermediate C′ (for details, see the Supporting Information)63,64,65, leading smoothly to the final product 2a 66,67,68,69,70. It appears that the OBH (for more details see Supplementary Figs. 78, 79) of such oxidant-involving cycle is determined by the relative free energy of transition state TS B' , which amounts up to 18.0 and 23.4 kcal/mol in the AuI- and CuI-catalyses, respectively. Note that in the presence of oxidant, the oxidant-free cycle may even be favored over the oxidant-involving path, if the former has a lower OBH than the latter. This is true for the AuI-catalyst system, but not true for the CuI-catalyst system. Accordingly, the oxidant-involving CuI- and AuI-catalyst systems prefer to produce 2a and 3a, respectively (for details, see Supplementary Data 1).

Plausible mechanism accounting for the divergent CuI/AuI-catalyzed formation of 2a/3a. Relative free energies of key intermediates and transition states were computed at the SMD-M06/DZP level of theory in solvent (DCE for CuI catalysis and CH3NO2 for AuI catalysis) at 298 K. Data for AuI catalysis were given in parentheses
Discussion
In summary, we have developed a copper-catalyzed oxidative cyclization of azido-diynes, affording a wide range of functionalized pyrrolo[3,4-c]quinolin-1-ones in mostly good to excellent yields. Importantly, this protocol represents a non-noble metal-catalyzed diyne oxidation by an N–O bond oxidant. In addition, the gold-catalyzed cascade cyclization of the same substrates leads to the efficient formation of pyrrolo[2,3-b]indoles. Thus, this controllable cascade cyclization enables the efficient and divergent synthesis of two types of valuable tricyclic N-heterocycles from identical starting materials under exceptionally mild conditions. Moreover, the computational study provides further evidence on the feasibility of the proposed mechanism of these cascade reactions, especially for the distinct selectivity. Further studies on other controllable cascade cyclizations are currently underway.
Methods
Materials
Unless otherwise noted, materials were obtained commercially and used without further purification. All the solvents were treated according to general methods. Flash column chromatography was performed over silica gel (300–400 mesh). See Supplementary Methods for experimental details.
General methods
1H NMR spectra and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer and a Bruker AV-500 spectrometer in chloroform-d3. For 1H NMR spectra, chemical shifts are reported in ppm with the internal TMS signal at 0.0 ppm as a standard. For 13C NMR spectra, chemical shifts are reported in ppm with the internal chloroform signal at 77.0 ppm as a standard. Infrared spectra were recorded on a Nicolet AVATER FTIR330 spectrometer as thin film and are reported in reciprocal centimeter (cm−1). Mass spectra were recorded with Micromass QTOF2 Quadrupole/Time-of-Flight tandem mass spectrometer using electron spray ionization. 1H NMR, 13C NMR, and high-performance liquid chromatography (HPLC) spectra (for chiral compounds) are supplied for all compounds: see Supplementary Figs. 1–74. See Supplementary Methods for the characterization data of compounds not listed in this part.
General procedure for the synthesis of pyrrolo[3,4-c]quinolin-1-ones 2
Methylquinoline N-oxide (0.3 mmol, 47.7 mg) and Cu(CH3CN)4PF6 (0.02 mmol, 7.5 mg) were added in this order to the ynamide 1 (0.20 mmol) in DCE (4.0 mL) at room temperature. The reaction mixture was stirred at 60 °C and the progress of the reaction was monitored by TLC. The reaction typically took 2 h. Upon completion, the mixture was then concentrated and the residue was purified by chromatography on silica gel (eluent: petroleum ether/dichloromethane) to afford the desired pyrrolo[3,4-c]quinolin-1-one 2.
General procedure for the synthesis of pyrrolo[2,3-b]indoles 3
Ph3PAuNTf2 (0.004 mmol, 3.0 mg) was added in this order to the ynamide 1 (0.20 mmol) in CH3NO2 (4.0 mL) at room temperature. The reaction mixture was stirred at room temperature and the progress of the reaction was monitored by TLC. The reaction typically took 30 min. Upon completion, the mixture was then concentrated and the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate) to afford the desired pyrrolo[2,3-b]indole 3.
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1535333 (2a) and CCDC 1535335 (3a). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/ data_request/cif. Any further relevant data are available from the authors upon reasonable request.
References
Yu, F.-C. et al. Three-component synthesis of functionalized pyrrolo[3,4-c]. Tetrahedron 71, 1036–1044 (2015).
Xia, L., Idhayadhulla, A., Lee, Y. R., Kim, S. H. & Wee, Y. Microwave-assisted synthesis of diverse pyrrolo[3,4-c]quinoline-1,3-diones and their antibacterial activities. ACS Comb. Sci. 16, 333–341 (2014).
Xia, L. & Lee, Y. R. Efficient one-step synthesis of pyrrolo[3,4-c]quinoline-1,3-dione derivatives by organocatalytic cascade reactions of isatins and β-ketoamides. Org. Biomol. Chem. 11, 5254–5263 (2013).
Makki, M. S. T., Bakhotmah, D. A. & Abdel-Rahman, R. M. Highly efficient synthesis of novel fluorine bearing quinoline-4-carboxylic acid and the related compounds as amylolytic agents. Int. J. Org. Chem. 2, 49–55 (2012).
Kravchenko, D. V. et al. Synthesis and structure-activity relationship of 4-substituted 2-(2-acetyloxyethyl)-8-(morpholine-4-sulfonyl)pyrrolo[3,4-c]quinoline-1,3-diones as potent caspase-3 inhibitors. J. Med. Chem. 48, 3680–3683 (2005).
Boa, A. N. et al. Synthesis of brequinar analogue inhibitors of malaria parasite dihydroorotate dehydrogenase. Bioorg. Med. Chem. 13, 1945–1967 (2005).
Kravchenko, D. V. et al. Synthesis and caspase-3 inhibitory activity of 8-sulfonyl-1,3-dioxo-2,3-dihydro-1H-pyrrolo[3,4-c]quinolines. Farmaco 60, 804–809 (2005).
Handa, T., Singh, S. & Singh, I. P. Characterization of a new degradation product of nifedipine formed on catalysis by atenolol: a typical case of alteration of degradation pathway of one drug by another. J. Pharm. Biomed. Anal. 9, 6–17 (2014).
Prasad, B. et al. Conformationally restricted functionalized heteroaromatics: a direct access to novel indoloindoles via Pd-mediated reaction. Chem. Commun. 49, 3970–3972 (2013).
Wu, Q., Wu, Z., Qu, X. & Liu, W. Insights into pyrroindomycin biosynthesis reveal a uniform paradigm for tetramate/tetronate formation. J. Am. Chem. Soc. 134, 17342–17345 (2012).
Yin, W.-B., Yu, X., Xie, X.-L. & Li, S.-M. Preparation of pyrrolo[2,3-b]indoles carrying α β-configured reverse C3-dimethylallyl moiety by using a recombinant prenyltransferase CdpC3PT. Org. Biomol. Chem. 8, 2430–2438 (2010).
Zhao, Y., Duan, Q., Zhou, Y., Yao, Q. & Li, Y. Gold-catalyzed chemo- and diastereoselective C(sp2)–H functionalization of enaminones for the synthesis of pyrrolo[3,4-c]-quinolin-1-one derivatives. Org. Biomol. Chem. 14, 2177–2181 (2016).
Li, J. et al. Unexpected isocyanide-based cascade cycloaddition reaction with methyleneindolinone. Chem. Commun. 49, 10694–10696 (2013).
Cappelli, A. et al. Further studies on the interaction of the 5-hydroxytryptamine3 (5-HT3) receptor with arylpiperazine ligands. development of a new 5-HT3 receptor ligand showing potent acetylcholinesterase inhibitory properties. J. Med. Chem. 48, 3564–3575 (2005).
Kumar, S. et al. KOtBu-mediated aerobic transition-metal-free regioselective β-arylation of indoles: synthesis of β-(2-/4-nitroaryl)-indoles. Org. Lett. 17, 82–85 (2015).
Gao, H., Xu, Q.-L., Yousufuddin, M., Ess, D. H. & Kürti, L. Rapid synthesis of fused N-heterocycles by transition-metal-free electrophilic amination of arene C–H bonds. Angew. Chem. Int. Ed. 53, 2701–2705 (2014).
Yan, Q. et al. Oxidative cyclization of 2-aryl-3-arylamino-2-alkenenitriles to N-arylindole-3-carbonitriles mediated by NXS/Zn(OAc)2. J. Org. Chem. 76, 8690–8697 (2011).
Zheng, Z., Wang, Z., Wang, Y. & Zhang, L. Au-catalysed oxidative cyclisation. Chem. Soc. Rev. 45, 4448–4458 (2016).
Yeom, H.-S. & Shin, S. Catalytic access to α-oxo gold carbenes by N–O bond oxidants. Acc. Chem. Res. 47, 966–977 (2014).
Zhang, L. A non-diazo approach to α-oxo gold carbenes via gold-catalyzed alkyne oxidation. Acc. Chem. Res. 47, 877–888 (2014).
Xiao, J. & Li, X. Gold α-oxo carbenoids in catalysis: catalytic oxygen-atom transfer to alkynes. Angew. Chem. Int. Ed. 50, 7226–7236 (2011).
Zeng, X., Liu, S., Shi, Z., Liu, G. & Xu, B. Synthesis of α-fluoroketones by insertion of HF into a gold carbene. Angew. Chem. Int. Ed. 55, 10032–10036 (2016).
Zhang, Y., Xue, Y., Li, G., Yuan, H. & Luo, T. Enantioselective synthesis of Iboga alkaloids and vinblastine via rearrangements of quaternary ammoniums. Chem. Sci. 7, 5530–5536 (2016).
Wang, Y., Zheng, Z. & Zhang, L. Intramolecular insertions into unactivated C(sp3)-H bonds by oxidatively generated β-diketone-α-gold carbenes: synthesis of cyclopentanones. J. Am. Chem. Soc. 137, 5316–5319 (2015).
Chen, H. & Zhang, L. A desulfonylative approach in oxidative gold catalysis: regiospecific access to donor-substituted acyl gold carbenes. Angew. Chem. Int. Ed. 54, 11775–11779 (2015).
Ji, K., Zheng, Z., Wang, Z. & Zhang, L. Enantioselective oxidative gold catalysis enabled by a designed chiral P,N-bidentate ligand. Angew. Chem. Int. Ed. 54, 1245–1249 (2015).
Homs, A., Muratore, M. E. & Echavarren, A. M. Enantioselective total synthesis of (−)-nardoaristolone B via a gold(I)-catalyzed oxidative cyclization. Org. Lett. 17, 461–463 (2015).
Schulz, J., Jašíková, L., Škríba, A. & Roithová, J. Role of gold(I) α-oxo carbenes in the oxidation reactions of alkynes catalyzed by gold(I) complexes. J. Am. Chem. Soc. 136, 11513–11523 (2014).
Karad, S. N. & Liu, R.-S. Gold-catalyzed 1,2-oxoarylations of nitriles with pyridine-derived oxides. Angew. Chem. Int. Ed. 53, 5444–5448 (2014).
Wang, T. et al. Synthesis of highly substituted 3-formylfurans by a gold(I)-catalyzed oxidation/1,2-alkynyl migration/cyclization cascade. Angew. Chem. Int. Ed. 53, 3715–3719 (2014).
Asiri, A. M. & Hashmi, A. S. K. Gold-catalysed reactions of diynes. Chem. Soc. Rev. 45, 4471–4503 (2016).
Nösel, P. et al. 1,6-carbene transfer: gold-catalyzed oxidative diyne cyclizations. J. Am. Chem. Soc. 135, 15662–15666 (2013).
Zheng, Z. & Zhang, L. C–H insertions in oxidative gold catalysis: synthesis of polycyclic 2H-pyran-3(6H)-ones via a relay strategy. Org. Chem. Front. 2, 1556–1560 (2015).
Ji, K., Liu, X., Du, B., Yang, F. & Gao, J. Gold-catalyzed selective oxidation of 4-oxahepta-1,6-diynes to 2H-pyran-3(6H)-ones and chromen-3(4H)-ones via β-gold vinyl cation intermediates. Chem. Commun. 51, 10318–10321 (2015).
Liu, R. et al. Generation of rhodium(I) carbenes from ynamides and their reactions with alkynes and alkenes. J. Am. Chem. Soc. 135, 8201–8204 (2013).
Zhou, B. et al. Yttrium-catalyzed intramolecular hydroalkoxylation/claisen rearrangement sequence: efficient synthesis of medium-sized lactams. Angew. Chem. Int. Ed. 56, 4015–4019 (2017).
Shen, W.-B. et al. Highly site selective formal [5+2] and [4+2] annulations of isoxazoles with heterosubstituted alkynes by platinum catalysis: rapid access to functionalized 1,3-oxazepines and 2,5-dihydropyridines. Angew. Chem. Int. Ed. 56, 605–609 (2017).
Li, L. et al. Reversal of regioselectivity in catalytic arene-ynamide cyclization: direct synthesis of valuable azepino[4,5-b]indoles and β-carbolines and DFT calculations. ACS Catal. 7, 4004–4010 (2017).
Shu, C. et al. Generation of α-imino gold carbenes through gold-catalyzed intermolecular reaction of azides with ynamides. J. Am. Chem. Soc. 137, 9567–9570 (2015).
Zhou, A.-H. et al. Atom-economic generation of gold carbenes: gold-catalyzed formal [3+2] cycloaddition between ynamides and isoxazoles. Chem. Sci. 6, 1265–1271 (2015).
Li, L. et al. Generation of gold carbenes in water: efficient intermolecular trapping of the a-oxo gold carbenoids by indoles and anilines. Chem. Sci. 5, 4057–4064 (2014).
Li, L. et al. Zinc-catalyzed alkyne oxidation/C-H functionalization: highly siteselective synthesis of versatile isoquinolones and β-carbolines. Angew. Chem. Int. Ed. 54, 8245–8249 (2015).
Pan, F. et al. Catalytic ynamide oxidation strategy for the preparation of α-functionalized amides. ACS Catal. 6, 6055–6062 (2016).
Gorin, D. J., Davis, N. R. & Toste, F. D. Gold(I)-catalyzed intramolecular acetylenic Schmidt reaction. J. Am. Chem. Soc. 127, 11260–11261 (2005).
Wetzel, A. & Gagosz, F. Gold-catalyzed transformation of 2-alkynyl arylazides: efficient access to the valuable pseudoindoxyl and indolyl frameworks. Angew. Chem. Int. Ed. 50, 7354–7358 (2011).
Lu, B. et al. Umpolung reactivity of indole through gold catalysis. Angew. Chem. Int. Ed. 50, 8358–8362 (2011).
Xiao, Y. & Zhang, L. Synthesis of bicyclic imidazoles via [2+3] cycloaddition between nitriles and regioselectively generated α‑imino gold carbene intermediates. Org. Lett. 14, 4662–4665 (2012).
Yan, Z.-Y., Xiao, Y. & Zhang, L. Gold-catalyzed one-step construction of 2,3-dihydro-1H-pyrrolizines with an electron-withdrawing group in the 5-position: a formal synthesis of 7-methoxymitosene. Angew. Chem. Int. Ed. 51, 8624–8627 (2012).
Gronnier, C., Boissonnat, G. & Gagosz, F. Au-catalyzed formation of functionalized quinolines from 2-alkynyl arylazide derivatives. Org. Lett. 15, 4234–4237 (2013).
Tokimizu, Y., Oishi, S., Fujii, N. & Ohno, H. Gold-catalyzed cascade cyclization of (azido)ynamides: an efficient strategy for the construction of indoloquinolines. Org. Lett. 16, 3138–3141 (2014).
Pawar, S. K., Sahani, R. L. & Liu, R.-S. Diversity in gold-catalyzed formal cycloadditions of ynamides with azidoalkenes or 2H-azirines: [3+2] versus [4+3] cycloadditions. Chem. Eur. J. 21, 10843–10850 (2015).
Li, N., Wang, T.-Y., Gong, L.-Z. & Zhang, L. Gold-catalyzed multiple cascade reaction of 2-alkynylphenylazides with propargyl alcohols. Chem. Eur. J. 21, 3585–3588 (2015).
Li, N. et al. Gold-catalyzed direct assembly of aryl-annulated carbazoles from 2-alkynyl arylazides and alkynes. Org. Lett. 18, 4178–4181 (2016).
Pan, Y., Chen, G.-W., Shen, C.-H., He, W. & Ye, L.-W. Synthesis of fused isoquinolines via gold-catalyzed tandem alkyne amination/intramolecular O–H insertion. Org. Chem. Front. 3, 491–495 (2016).
Matsuoka, J., Matsuda, Y., Kawada, Y., Oishi, S. & Ohno, H. Total synthesis of dictyodendrins by the gold-catalyzed cascade cyclization of conjugated diynes with pyrroles. Angew. Chem. Int. Ed. 56, 7444–7448 (2017).
Patil, D. V. et al. Brønsted acid catalyzed oxygenative bimolecular friedel–crafts-type coupling of ynamides. Angew. Chem. Int. Ed. 56, 3670–3674 (2017).
Graf, K., Rühl, C. L., Rudolph, M., Rominger, F. & Hashmi, A. S. K. Metal-free oxidative cyclization of alkynyl aryl ethers to benzofuranones. Angew. Chem. Int. Ed. 52, 12727–12731 (2013).
Chen, D.-F., Han, Z.-Y., He, Y.-P., Yu, J. & Gong, L.-Z. Metal-free oxidation/C(sp3)–H functionalization of unactivated alkynes using pyridine-N-oxide as the external oxidant. Angew. Chem. Int. Ed. 51, 12307–12310 (2012).
Chen, X., Ruider, S. A., Hartmann, R. W., González, L. & Maulide, N. Metal-free meta-selective alkyne oxyarylation with pyridine N-oxides: rapid assembly of metyrapone analogues. Angew. Chem. Int. Ed. 55, 15424–15428 (2016).
Lee, J. H. et al. 18F-labeled isoindol-1-one and isoindol-1,3-dione derivatives as potential PET imaging agents for detection of β-amyloid fibrils. Bioorg. Med. Chem. Lett. 18, 5701–5704 (2008).
Shah, J. H. et al. PCT Int. Appl. WO 2003014315A2 (2003).
Kim, H. J., Kim, J., Cho, S. H. & Chang, S. Intermolecular oxidative C-N bond formation under metal-free conditions: control of chemoselectivity between aryl sp2 and benzylic sp3 C-H bond imidation. J. Am. Chem. Soc. 133, 16382–16385 (2011).
Chen, M. et al. Gold-catalyzed oxidative ring expansion of 2-alkynyl-1,2-dihydropyridines or -quinolines: highly efficient synthesis of functionalized azepine or benzazepine scaffolds. Angew. Chem. Int. Ed. 54, 1200–1204 (2015).
Qian, D. et al. Gold(I)-catalyzed highly diastereo- and enantioselective alkyne oxidation/cyclopropanation of 1,6-enynes. Angew. Chem. Int. Ed. 53, 13751–13755 (2014).
Henrion, G., Chavas, T. E. J., Le Goff, X. & Gagosz, F. Biarylphosphonite gold(I) complexes as superior catalysts for oxidative cyclization of propynyl arenes into indan-2-ones. Angew. Chem. Int. Ed. 52, 6277–6282 (2013).
Yao, R., Rong, G., Yan, B., Qiu, L. & Xu, X. Dual-functionalization of alkynes via copper-catalyzed carbene/alkyne metathesis: a direct access to the 4-carboxyl quinolines. ACS Catal. 6, 1024–1027 (2016).
Day, D. P. & Chan, P. W. H. Gold-catalyzed cycloisomerizations of 1,n-diyne carbonates and esters. Adv. Synth. Catal. 358, 1368–1384 (2016).
Yan, J., Tay, G. L., Neo, C., Lee, B. R. & Chan, P. W. H. Gold-catalyzed cycloisomerization and Diels–Alder reaction of 1,6-diyne esters with alkenes and diazenes to hydronaphthalenes and -cinnolines. Org. Lett. 17, 4176–4179 (2015).
Rao, W. & Chan, P. W. H. Gold-catalyzed [2+2+1] cycloaddition of 1,6-diyne carbonates and esters with aldehydes to 4-(cyclohexa-1,3-dienyl)-1,3-dioxolanes. Chem. Eur. J. 20, 713–718 (2014).
Rao, W., Koh, M. J., Li, D., Hirao, H. & Chan, P. W. H. Gold-catalyzed cycloisomerization of 1,6-diyne carbonates and esters to 2,4a-dihydro-1H-fluorenes. J. Am. Chem. Soc. 135, 7926–7932 (2013).
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21572186, 21622204, and 91545105), the Natural Science Foundation of Fujian Province for Distinguished Young Scholars (2015J06003), the President Research Funds from Xiamen University (20720150045), XMU Training Program of Innovation and Enterpreneurship for Undergraduates (2016G10384076), and NFFTBS (J1310024). We also thank Professor Dr Nanfeng Zheng from Xiamen University for assistance with X-ray crystallographic analysis.
Author information
Authors and Affiliations
Contributions
W.-B.S., L.L., X.L. and B.Z. performed experiments. Q.S. and X.L. performed DFT calculations. X.L. revised the paper. L.-W.Y. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, WB., Sun, Q., Li, L. et al. Divergent synthesis of N-heterocycles via controllable cyclization of azido-diynes catalyzed by copper and gold. Nat Commun 8, 1748 (2017). https://doi.org/10.1038/s41467-017-01853-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-01853-1
This article is cited by
-
Copper-catalyzed alkyne oxidation/Büchner-type ring-expansion to access benzo[6,7]azepino[2,3-b]quinolines and pyridine-based diones
Communications Chemistry (2023)
-
Regioselective access to polycyclic N-heterocycles via homogeneous copper-catalyzed cascade cyclization of allenynes
Communications Chemistry (2023)
-
Catalytic hydrative cyclization of aldehyde-ynamides with water for synthesis of medium-sized lactams
Science China Chemistry (2021)
-
Stereoselective synthesis of medium lactams enabled by metal-free hydroalkoxylation/stereospecific [1,3]-rearrangement
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
