
Abstract
Rh-hydride catalysis solves a synthetic challenge by affording the enantioselective reduction of allenes, thereby yielding access to motifs commonly used in medicinal chemistry. A designer Josiphos ligand promotes the generation of chiral benzylic isomers, when combined with a Hantzsch ester as the reductant. This semireduction proceeds chemoselectively in the presence of other functional groups, which are typically reduced using conventional hydrogenations. Isotopic labelling studies support a mechanism where the hydride is delivered to the branched position of a Rh-allyl intermediate.
Similar content being viewed by others
Introduction
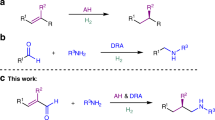
In nature, chemo- and stereocontrolled reduction of unsaturated bonds are catalysed by enzymes and mediated by cofactors such as nicotinamide adenine dinucleotide phosphate (NAD(P)H)1. Inspired by this cofactor, chemists have used Hantzsch esters as mild reagents to solve various challenges in asymmetric reductions2. It occurred to us that this cofactor mimic could be combined with Rh-hydride catalysis to enable a valuable strategy for reducing allenes to generate benzylic motifs, which are traditionally made by an allylic substitution between an allylic electrophile and an organometallic reagent3,4,5,6,7 or a hydride source8,9,10,11,12,13,14 (Fig. 1a). As allenes are readily accessible15, a method to access these motifs through a semireduction of allenes would avoid the pre-installation of a suitable leaving group. Allenes are challenging functional groups for reduction because of problems with chemo-, regio-, and stereoselectivity. Both π-bonds can be reduced to the corresponding alkane (Fig. 1b), or one π-bond can be reduced to afford one or a mixture of alkene isomers (Fig. 1c). Before studies in the regioselective semireduction of allenes have shown that the less substituted π-bond is typically reduced to afford the achiral internal alkene16,17,18. Existing methods that reduce the more substituted π-bond are limited to monosubstituted and symmetrical allenes, which give rise to achiral terminal alkenes19, 20.

Challenges in the selective reduction of allenes. a Traditional methods to access chiral allylic motifs. b Complete reduction affords alkanes. c Existing allene semireductions favour formation of the internal alkene. d Proposed strategy for regio- and enantioselective semireduction to afford the complementary terminal alkene
The generation of electrophilic metal-allyl species from allenes using iridium- and rhodium-hydrides is an emerging strategy in allene hydrofunctionalisation21, 22. These intermediates can undergo allylic substitution with various nucleophiles to afford branched allylated products. We envisioned that a Rh-hydride catalyst would transform an allene to an electrophilic Rh-allyl intermediate, which can then be trapped with a hydride nucleophile23,24,25,26. Given that allenes are known to isomerise to dienes in the presence of transition metal-hydrides27, we recognise that a key challenge would be identifying a catalyst that promotes semireduction over isomerisation.
Herein, we demonstrate an asymmetric semireduction of allenes enabled by Rh-hydride catalysis as a complementary approach to allylic alkylation and allylic reduction to generate chiral benzylic motifs. Using a designed Josiphos ligand and a Hantzsch ester reductant, various allenes are reduced to the corresponding chiral terminal alkenes with high selectivities.
Results
Reaction development
To test our hypothesis (Fig. 1d), we chose 1-methoxy-4-(3-phenylpenta-3,4-dien-1-yl)benzene (1a) as the model substrate for semireduction in the presence of [Rh(COD)Cl]2, (PhO)2P(O)(OH), and DPEphos (Fig. 2). Through a survey of achiral bidentate phosphine ligands, we found DPEphos to be the most promising scaffold for suppressing diene formation, in the presence of various reductants. Tsuji and Mandai8 demonstrated that formic acid and formates are competent reductants in the reduction of allylic carbonates. However, these reagents led to semireductions with little to no regiocontrol (50:50 to 67:33 2a:3a, Fig. 2, entries 1 and 2). Hayashi and Kawabata showed that a combination of formic acid and an amine base, such as 1,8-bis(dimethylamino)naphthalene, reduced allylic carbonates and esters9,10,11,12. In our system, this combination suppressed semireduction of the more substituted π-bond (Fig. 2, entry 3). NaBH4, a classical nucleophilic hydride source, gave trace reactivity (Fig. 2, entry 4), and a silanes13, 14 afforded unselective semireduction in low conversion (28%, 50:50 2a:3a, Fig. 2, entry 5). When Hantzsch ester 5a was used as the reductant (Fig. 2, entry 6), the reactivity increased (87% yield), and the desired terminal alkene was obtained as the major product (88:12 2a:3a).

Evaluation of reductants. Reaction conditions: 1a (0.050 mmol), reductant (0.10 mmol), [Rh(COD)Cl]2 (4 mol%), DPEphos (8 mol%), (PhO)2P(O)(OH) (8 mol%), CH2Cl2 (0.1 mL), 30 °C, 18 h. Yields and regioselectivities were determined by 1H NMR analysis of the unpurified reaction mixture using dimethyl terephthalate as an internal standard. aHCO2H (0.11 mmol), 1,8-bis(dimethylamino)naphthalene (0.060 mmol). bND, not determined
Next, we searched for a chiral ligand that could enable high enantio- and regioselectivities, in combination with Hantzsch ester 5a as the reductant (Fig. 3). Axially chiral bisphosphine ligands, such as (R)-BINAP (L1), afforded a mixture of alkenes 2a and 3a, as well as competitive isomerisation to diene 4a (1:3:2 2a:3a:4a). Ligands bearing point chirality, such as (R,R)-DIOP (L2), promoted semireduction over isomerisation, but with moderate regioselectivity (5:2 2a:3a). We discovered that the all-aryl substituted Josiphos ligand scaffold gave high selectivity for 2a. A significant increase in the reaction selectivity (20:1:1 2a:3a:4a) was observed when commercially available ligand L3 was employed. Josiphos ligand L4, where one phosphine is more electron-deficient, afforded an increase in the reaction rate, so the catalyst loading can be reduced two-fold. In addition, L4 further improved selectivity for 2a (>20:1:1 2a:3a:4a), but the enantioselectivity remained low (67:33 er). To improve the enantioselectivity, we replaced the 3,5-xylyl groups of L3 and L4 with the more electron-rich and sterically encumbered 3,5-di-tert-butyl-4-methoxyphenyl groups to afford new Josiphos ligands L5 and L6. With L5, the enantioselectivity increased (83:17 er), but low reactivity (27%) was observed. However, L6 afforded the desired terminal alkene in 85% yield and 95:5 er while maintaining the high selectivity for 2a (>20:1:1 2a:3a:4a).

Evaluation of chiral ligands. Reaction conditions: 1a (0.10 mmol), 5a (0.20 mmol), [Rh(COD)Cl]2 (4 mol%), L (8 mol%), (PhO)2P(O)(OH) (8 mol%), CH2Cl2 (0.2 mL), 30 °C, 18 h. Yields and product ratios were determined by 1H NMR analysis of the unpurified reaction mixture using dimethyl terephthalate as an internal standard. Enantioselectivities (er’s) were determined by chiral SFC analysis. aUsing [Rh(COD)Cl]2 (2 mol%), L (4 mol%), (PhO)2P(O)(OH) (4 mol%), CH2Cl2 (0.1 mL)
Reaction scope
With this protocol, we examined the generality of enantioselective semireduction using other allenes (Fig. 4). Generally, the terminal alkene was obtained as the sole product; no internal alkene or diene was observed. An allene with an ortho substituent (2b) on the phenyl group underwent semireduction with lower enantioselectivity (75%, 88:12 er). Substrates with meta (2c) and para (2d) substituents on the phenyl group reacted with similar efficiencies as the model substrate (88%, 96:4 er and 92%, 95:5 er, respectively). Allenes bearing electron-rich (2e) and electron-deficient (2f) substituents underwent semireduction (85–92%, 93:7–97:3 er). A benzyl ether is labile under typical hydrogenation conditions, but this protecting group was stable under our semireduction conditions (2e). Substrates bearing aryl halide bonds (2g and 2h) were tolerated (87–90%, 94:6–95:5 er). Extended aromatic systems, such as a naphthyl group (2i), reacted (91%, 94:6 er). The semireduction tolerates allenes with heteroaromatic moieties, such as an N-tosyl indole (2j, 70%, 94:6 er) and a thiophene (2k, 82%, 94:6 er). Chemoselective reduction occurred with substrates containing alkenes (2l), alkynes (2m), esters (2s) and nitriles (2s), affording the terminal alkenes selectively (67–81%, 89:11–95:5 er). 1-Aryl-1-propynes (2m) are reactive substrates towards isomerisation and hydrofunctionalisation22, but only the allene functionality reacted. Allenes bearing other alkyl groups were accommodated (2n–2s, 60–99%, 89:11–96:4 er). At last, the semireduction occurs chemoselectively in the presence of other nucleophiles, such as an alcohol (2q, 61%, 96:4 er). Allenes bearing dialkyl or diaryl substituents were unreactive under the present conditions. Notably, the semireduction tolerates acidic and electrophilic functionalities, such as an alcohol (2q), ester (2s), and nitrile (2s). Thus, this method to access benzylic motifs complements allylic substitutions using organometallic reagents. The absolute configuration of 2n was determined to be (S) by comparison of its optical rotation with literature data28.

Enantioselective semireduction of allenes. Reaction conditions: 1 (0.20 mmol), 5a (0.40 mmol), [Rh(COD)Cl]2 (2 mol%), L6 (4 mol%), (PhO)2P(O)(OH) (4 mol%), CH2Cl2 (0.2 mL), 30 °C, 18 h. Isolated yields. Product ratios were determined by 1H NMR analysis of the unpurified reaction mixture. Enantioselectivities (er’s) were determined by chiral SFC analysis. aReaction performed with 1,2-dichloroethane at 60 °C
Mechanistic studies
To shed light on the mechanism of this semireduction, we performed deuterium-labelling experiments using deuterated analogues of Hantzsch ester 5a. Semireduction of 1a with 5b afforded 2ab, where the deuterium label was completely transferred to the allylic carbon (Fig. 5a). In addition to its mechanistic significance, this experiment demonstrates a method to prepare chiral isotopically labelled stereogenic centres that complements allylic deuteration using formic acid-d 2 29, 30. Using 5c, 2ac was obtained, where the deuterium label was incorporated into the internal vinylic carbon (Fig. 5b). The remaining deuteriums were incorporated into the vinylic methyl groups of the Hantzsch ester 5c (31% D) and the pyridine byproduct (6% D) presumably as a statistical mixture of products31.

Deuterium-labelling studies. a Treatment of allene 1a with deuterated Hantzsch ester 5b afforded deuteration in the allylic position. b Analogous experiment with 5c gave deuteration in the vinylic position
On the basis of our observations and literature precedence, we propose the mechanism shown in Fig. 6. To initiate catalysis, the Rh(I) precursor undergoes oxidative addition to generate a Rh(III)-hydride species A. The insertion of allene 1 with A forms an electrophilic Rh(III)-allyl intermediate B, which undergoes allylic substitution with Hantzsch ester 5a to furnish the terminal alkene 2 and regenerate the catalyst.

Proposed mechanism. Mechanistic pathway of the allene semireduction
Discussion
As a complementary approach to allylic alkylation and allylic reduction, we have demonstrated a Rh-catalysed regio- and enantioselective semireduction of allenes as a strategy to generate chiral benzylic motifs. The high reaction selectivities are enabled by a designed Josiphos ligand and a Hantzsch ester reductant. Given the significance of deuterated pharmaceuticals32,33,34, new strategies for asymmetric hydride delivery are especially relevant. Our approach allows access to isotopically labelled stereogenic centres and occurs with excellent chemo- and stereocontrol in the presence of functional groups that are sensitive to conventional hydrogenations.
Methods
General procedure for the semireduction of allenes
In a N2-filled glovebox, [Rh(COD)Cl]2 (2.0 mg, 0.0040 mmol, 2 mol%), (PhO)2P(O)(OH) (2.0 mg, 0.0080 mmol, 4 mol%), Josiphos L6 (9.1 mg, 0.0080 mmol, 4 mol%), Hantzsch ester 5a (101.3 mg, 0.40 mmol, 2.0 equiv), allene 1 (0.20 mmol, 1 equiv), and anhydrous CH2Cl2 (0.20 mL, 1 M) were added to a 1 dram vial equipped with a magnetic stir bar. The vial was then sealed with a Teflon-lined screw cap and stirred at 30 °C for 18 h. The reaction mixture was cooled to rt and concentrated in vacuo. Regioselectivities were determined by 1H NMR analysis of the unpurified reaction mixture. The terminal alkene product 2 was purified by preparatory thin-layer chromatography. The enantioselectivities were determined by chiral SFC analysis. All new products were fully characterised. For the full experimental procedures and characterisation, including NMR spectra, of the new compounds in this article, see Supplementary Figs. 1–167.
Data availability
The authors declare that the data supporting the conclusions of this study are available within the article and its Supplementary Information file or from the authors upon reasonable request.
References
Alberts, B. et al. Molecular Biology of the Cell, 4th edn (Garland: New York & London, 2002).
Zheng, C. & You, S.-L. Transfer hydrogenation with Hantzsch esters and related organic hydride donors. Chem. Soc. Rev. 41, 2498–2518 (2012).
Langlois, J. B. & Alexakis, A. Copper-catalyzed enantioselective allylic substitution. Top. Organomet. Chem. 38, 235–268 (2012).
Hornillos, V., Gualtierotti, J.-B. & Feringa, B. L. Asymmetric allylic substitutions using organometallic reagents. Top. Organomet. Chem. 58, 1–39 (2016).
Luchaco-Cullis, C. A., Mizutani, H., Murphy, K. E. & Hoveyda, A. H. Modular pyridinyl peptide ligands in asymmetric catalysis: enantioselective synthesis of quaternary carbon atoms through copper-catalyzed allylic substitutions. Angew. Chem. Int. Ed. 40, 1456–1460 (2001).
Kacprzynski, M. A. & Hoveyda, A. H. Cu-catalyzed asymmetric allylic alkylations of aromatic and aliphatic phosphates with alkylzinc reagents. An effective method for enantioselective synthesis of tertiary and quaternary carbons. J. Am. Chem. Soc. 126, 10676–10681 (2004).
Larsen, A. O., Leu, W., Oberhuber, C. N., Campbell, J. E. & Hoveyda, A. H. Bidentate NHC-based chiral ligands for efficient Cu-catalyzed enantioselective allylic alkylations: structure and activity of an air-stable chiral Cu complex. J. Am. Chem. Soc. 126, 11130–11131 (2004).
Tsuji, J. & Mandai, T. Palladium-catalyzed hydrogenolysis of allylic and propargylic compounds with various hydrides. Synthesis 1996, 1–24 (1996).
Hayashi, T. et al. Catalytic asymmetric reduction of allylic esters with formic acid catalyzed by palladium-MOP complexes. J. Am. Chem. Soc. 116, 775–776 (1994).
Hayashi, T., Kawatsura, M., Iwamura, H., Yamaura, Y. & Uozumi, Y. Catalytic asymmetric synthesis of optically active alkenes by palladium-catalysed asymmetric reduction of racemic allylic esters with formic acid. Chem. Commun. 1996, 1767–1768 (1996).
Fuji, K., Sakurai, M., Kinoshita, T. & Kawabata, T. Palladium-catalyzed asymmetric reduction of allylic esters with a new chiral monodentate ligand, 8-diphenylphosphino-8’-methoxy-1,1′-binaphthyl. Tetrahedron. Lett. 39, 6323–6326 (1998).
Kawatsura, M., Uozumi, Y., Ogasawara, M. & Hayashi, T. Palladium-catalyzed asymmetric reduction of racemic allylic esters with formic acid: effects of phosphine ligands on isomerization of π-allylpalladium intermediates and enantioselectivity. Tetrahedron 56, 2247–2257 (2000).
Keinan, E. & Greenspoon, N. Highly chemoselective allylic reductions with silicon hydrides and palladium catalyst. Isr. J. Chem. 24, 82–87 (1984).
Nguyen, T. N. T., Thiel, N. O., Pape, F. & Teichert, J. F. Copper(I)-catalyzed allylic substitutions with a hydride nucleophile. Org. Lett. 18, 2455 (2016).
Yu, S. & Ma, S. How easy are the syntheses of allenes? Chem. Commun. 47, 5384–5418 (2011).
Bhagwat, M. M. & Devaprabhakara, D. Selective hydrogenation of allenes with chlorotris(triphenylphosphine) rhodium catalyst. Tetrahedron. Lett. 13, 1391–1392 (1972).
Guo, H. et al. [Pd(Ar-BIAN)(alkene)]-catalyzed highly chemo-, regio-, and stereoselective semihydrogenation of 1,2-allenylphosphonates and related compounds. Angew. Chem. Int. Ed. 45, 4997–5000 (2006).
Adler, P., Gomes, F., Fadel, A. & Rabasso, N. Selective reduction of amino allenephosphonates: preparation of α-amino vinylphosphonates. Eur. J. Org. Chem. 2013, 7546–7555 (2013).
Semba, K., Fujihara, T., Xu, T., Terao, J. & Tsuji, Y. Copper-catalyzed highly selective semihydrogenation of non-polar carbon-carbon multiple bonds using a silane and an alcohol. Adv. Synth. Catal. 354, 1542–1550 (2012).
Inés, B. et al. Metal-free hydrogenation of electron-poor allenes and alkenes. Angew. Chem. Int. Ed. 51, 12367–12369 (2012).
Kim, I. S. & Krische, M. J. Iridium-catalyzed hydrocarboxylation of 1,1-dimethylallene: byproduct-free reverse prenylation of carboxylic acids. Org. Lett. 10, 513 (2008).
Koschker, P. & Breit, B. Branching out: rhodium-catalyzed allylation with alkynes and allenes. Acc. Chem. Res. 49, 1524–1536 (2016).
Trost, B. M. & Van Vranken, D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 96, 395–422 (1996).
Hartwig, J. F. & Stanley, L. M. Mechanistically driven development of iridium catalysts for asymmetric allylic substitution. Acc. Chem. Res. 43, 1461–1475 (2010).
Kazmaier, U. (ed.). Transition metal catalyzed allylic substitution in organic synthesis (Springer, 2012).
Liu, Y., Han, S.-J., Liu, W.-B. & Stoltz, B. M. Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc. Chem. Res. 48, 740–751 (2015).
Al-Masum, M. & Yamamoto, Y. Palladium-catalyzed hydrocarboxylation of allenes. J. Am. Chem. Soc. 120, 3809–3810 (1998).
Jackowski, O. & Alexakis, A. Copper-free asymmetric allylic alkylation with Grignard reagents. Angew. Chem. Int. Ed. 49, 3346–3350 (2010).
Hayashi, T., Iwamura, H., Uozumi, Y., Matsumoto, Y. & Ozawa, F. A new optically active monodentate phosphine ligand, (R)-(+)-3-diphenylphosphino-3′-methoxy-4,4′-biphenanthryl (MOP-phen): preparation and use for palladium-catalyzed asymmetric reduction of allylic esters with formic acid. Synthesis 1994, 526–532 (1994).
Chau, A., Paquin, J.-F. & Lautens, M. Diastereoselective palladium-catalyzed formate reduction of allylic carbonates en route to polypropionate systems. J. Org. Chem. 71, 1924–1933 (2006).
Liu, M., Chen, X., Chen, T. & Yin, S.-F. A facile and general acid-catalyzed deuteration at methyl groups of N-heteroarylmethanes. Org. Biomol. Chem. 15, 2507–2511 (2017).
Gant, T. G. Using deuterium in drug discovery: leaving the label in the drug. J. Med. Chem. 57, 3595–3611 (2014).
Tung, R. D. Deuterium medicinal chemistry comes of age. Future Med. Chem. 8, 491–494 (2016).
Coppen, E. M. & Roos, R. A. C. Current pharmacological approaches to reduce chorea in Huntington’s disease. Drugs 77, 29 (2017).
Acknowledgements
Funding was provided by UC Irvine and the National Institutes of Health (GM105938). We thank Yannan Liu for preparing and evaluating allenes 1q and 1s.
Author information
Authors and Affiliations
Contributions
Z.C. and V.M.D. designed the project and wrote the manuscript. Z.C. performed the experiments and analysed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Dong, V.M. Enantioselective semireduction of allenes. Nat Commun 8, 784 (2017). https://doi.org/10.1038/s41467-017-00793-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-017-00793-0
This article is cited by
-
Allene reduction: Selectively stopping halfway
Nature Reviews Chemistry (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
