
Abstract
Coronavirus disease-2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has affected more than seven million people worldwide, contributing to 0.4 million deaths as of June 2020. The fact that the virus uses angiotensin-converting enzyme (ACE)-2 as the cell entry receptor and that hypertension as well as cardiovascular disorders frequently coexist with COVID-19 have generated considerable discussion on the management of patients with hypertension. In addition, the COVID-19 pandemic necessitates the development of and adaptation to a “New Normal” lifestyle, which will have a profound impact not only on communicable diseases but also on noncommunicable diseases, including hypertension. Summarizing what is known and what requires further investigation in this field may help to address the challenges we face. In the present review, we critically evaluate the existing evidence for the epidemiological association between COVID-19 and hypertension. We also summarize the current knowledge regarding the pathophysiology of SARS-CoV-2 infection with an emphasis on ACE2, the cardiovascular system, and the kidney. Finally, we review evidence on the use of antihypertensive medication, namely, ACE inhibitors and angiotensin receptor blockers, in patients with COVID-19.
Similar content being viewed by others

Coronavirus disease-2019 (COVID-19) and hypertension: premonition of convergence of communicable diseases and noncommunicable diseases
In recent years, it has been well recognized that noncommunicable diseases (NCDs), including hypertension, are the main health issue, since ~70% of the causes of death in the world (57 million deaths/year) are attributed to NCDs. However, the sudden emergence of COVID-19, a communicable disease (CD), has changed our concept toward health and diseases. Indeed, we are harshly reminded that CDs can seriously threaten all aspects of life, including health, economics, education, and human relations. Furthermore, we must change our concept of a normal life to a “New Normal” one [1]. It is predicted that the pandemic of COVID-19 will create another pandemic of NCDs [2], and this prediction is plausible considering the similar situations manifested in the evacuation performed in the Great East Japan Earthquake on March 11, 2011. Even two years after the disaster, the blood pressure (BP) of the evacuees remained significantly elevated by an average of ~4~5 mmHg [3]. In this context, it is deeply concerning that a new disease entity is emerging, that is, the convergence of CDs and NCDs without borders. Thus far, we have been attempting to understand NCDs as lifestyle-related diseases, focusing upon each person’s specific lifestyle in personalized medicine. However, from now on, we should also dissect out the pathophysiology of convergence of CDs and NCDs from each person’s lifestyle in society where he/she is living, and should take care of life-environment as life-environment-related diseases. We also should pay more attention to virus-infected spaces of life as pressing matters, as well as global warming, which has been gradually recognized as an urgent issue.
To cope with this new disease entity, it is necessary to know the FACTS with regard to COVID-19 and NCDs. This is especially true for hypertension because severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel virus that causes COVID-19, utilizes angiotensin-converting enzyme (ACE)-2, one of the components of the renin-angiotensin system (RAS), for its entry into the body and because hypertension-associated diseases have been revealed to be factors contributing to the severity of COVID-19. We should also realize what is known and not yet known, and what requires further clarification. Here, we review recent literature regarding hypertension and COVID-19 available as of June 7th, 2020.
Epidemiological findings on the association between hypertension and COVID-19
Prevalence of hypertension among COVID-19 patients
Although emerging reports have demonstrated a high prevalence of hypertension among patients with COVID-19 [4,5,6], evidence is still insufficient (Table 1). For example, 58 of 191 Chinese patients with COVID-19 (30.4%; median age, 56.0 years) [4] and 509 of 1043 Italian patients with available data (48.8%, 95% confidence intervals, 46-52%; mean age among all 1591 study patients, 63 years) [5] had hypertension, and investigators in these studies [4, 5] reported a high prevalence of hypertension among patients with COVID-19. However, a Chinese nationwide survey showed that 44.6% of the population aged 55–64 years had hypertension [7], and 45.2% of the Italian population aged 60–69 years had hypertension according to an Italian inclusive general practitioner database [8]. Furthermore, in a US case series including 5700 sequentially hospitalized COVID-19 patients (median age, 63 years), 3026 (56.6%) had hypertension [6], with the prevalence of hypertension among COVID-19 patients also being lower than the US general population (estimated to be 63–77% of the population aged 55–64 years [9]). Furthermore, it should be noted that demographic data among patients, including comorbidities, were collected by different methods (by electronic medical records [4, 6] or by phone [5]). Most importantly, none of the currently available epidemiological studies regarding COVID-19 and hypertension (Table 1) clearly state the diagnostic methodology used or the criteria for hypertension, e.g., BP values based on office or out-of-office measurement, as well as self-reported or testimony of a family in serious situations. These variations would make the comparison of reported incidence with that in the general population inaccurate. Taking the high proportion of individuals with hypertension who are not aware of their condition [10] into account, it would be a reasonable assumption that the prevalence of hypertension in these reports [4,5,6] may be underestimated. Nevertheless, no supporting report was found that shows a higher rate of hypertension among patients with COVID-19, and there have been no reliable reports demonstrating an increased risk of SARS-CoV-2 infection in the presence of hypertension.
Impact of hypertension on the severity and mortality of COVID-19
Comparisons of COVID-19 patients with mild and severe clinical symptoms can be used to evaluate whether hypertension is a risk factor for aggravation of the disease. According to a retrospective study consisting of 487 COVID-19 patients in Zhejiang Province of China, the prevalence of hypertension was higher in the 49 severe cases than in the 438 mild cases (53.1% vs. 16.7%, p < 0.0001) [11]. Further multivariable-adjusted analysis revealed that male sex, age ≥50 years old, and hypertension were independent factors for COVID-19 severity on admission (odds ratio, 2.71; 95% confidence intervals 1.32–5.59) [11]. In another study involving 548 inpatients in Wuhan, China, where the first COVID-19 outbreak occurred in December 2019 [12], the prevalence of hypertension was significantly higher in patients with severe COVID-19 than in nonsevere cases (38.7% vs. 22.2%, p < 0.001). In a logistic model with adjustment for age, high lactate dehydrogenase (LDH), and D-dimer, hypertension was independently associated with the severity of COVID-19 on admission (odds ratio, 2.01; 95% confidence intervals, 1.27–3.17) [12].
However, it should be noted that hypertension is commonly accompanied by many comorbidities that are major determinant factors for the severity of COVID-19. The study in Wuhan [12] also reported that male sex, age ≥65 years old, high white blood cell count, LDH, cardiac injury, hyperglycemia, and high dose corticosteroid were independent predictive factors for death in a multivariable-adjusted Cox proportional hazard model. Nonetheless, hypertension was not included as a candidate explanatory factor in the multivariate analysis. A French single-center study [13] reported that hypertension was not significantly associated with progression of COVID-19, as defined as the requirement of invasive mechanical ventilation during hospitalization (OR, 2.29; 95% CI, 0.89–5.84; p = 0.08), even though the association was significant in a univariate model. In these and other studies, hypertension was not selected as an independent factor for COVID-19 severity based on multivariable-adjusted analysis, despite being identified as a risk factor by univariate [4, 12,13,14] or bivariate [15] survival analysis. Although some studies report that hypertension can be an independent risk factor for severe COVID-19 [11, 12], it would be plausible to interpret that the high prevalence of hypertension among patients with severe and fatal COVID-19 may be attributed to the vulnerability of older individuals to SARS-CoV-2 infection. At present, there is no clear epidemiological evidence supporting that hypertension itself is an independent risk factor for developing severe disease in patients with COVID-19. We, therefore, agree with the conclusion of the Centers for Disease Control and Prevention (CDC), which does not include hypertension in the list of risk factors for COVID-19 severity [16].
Key comorbidities of hypertension in relation to COVID-19
As mentioned above, age is the essential risk factor for increased severity and mortality in COVID-19. In Japan, the mortality rates among COVID-19 patients aged 70–79 and ≥80 years were 6.8% and 14.8%, respectively, whereas COVID-19 mortality in all age groups was 2.6% as of 7 May 2020 [17]. In addition to age, available reports have suggested a variety of underlying medical conditions to be associated with COVID-19 disease severity and mortality [4,5,6, 11, 12, 15, 16]. Furthermore, the majority of these risk factors are observed in patients with hypertension [9, 18]. In clinical practice, one may not always have enough time to assess comorbidities in acute-phase patients with COVID-19. In such situations, hypertension diagnosed by various methods might be regarded as a marker of the severity of the disease.
Obesity is another crucial issue that needs to be discussed since it is often accompanied by hypertension and is recognized as a novel risk factor for COVID-19 [13]. It may also be a determinant of disease severity, independent of age and hypertension [14, 19]. Obesity might worsen the clinical course of COVID-19 by decreasing expiratory reserve volume, impeding diaphragm excursion, and restricting ventilation [20]. Furthermore, obesity, especially abdominal obesity, increases inflammatory cytokines and oxidative stress and causes hypertension, diabetes, and dyslipidemia [21]. These comorbidities accelerate atherosclerosis, which leads to cardiovascular complications and may increase the severity and mortality of COVID-19. Recently, patients with both obesity and hypertension have been increasing in Japan [22], and these patients are at particularly high risk for severe disease in COVID-19 and thus require careful observation and intensive treatment. Similar to hypertension, the definition of obesity is not consistent among studies/countries, which needs to be considered.
COVID-19 and ACE2
ACE2 as the entry receptor for SARS-CoV-2
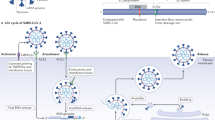
The first step of SARS-CoV-2 infection in humans is contact of the virus with cell-surface ACE2 (Fig. 1). ACE2 interacts with external SARS-CoV-2 by binding to the receptor-binding domain (RBD) of the viral spike protein [23]. This process is followed by proteolytic cleavage of the spike protein, which allows fusion to cells, and transmembrane protease serine 2 (TMPRSS2) has been identified as a protease responsible for the reaction (Fig. 1) [24]. Although SARS-CoV, which was responsible for SARS outbreaks in 2002–2004, uses the same ACE2 and TMPRSS2 for entry, our understanding of the differences between SARS-CoV and SARS-CoV-2 with regard to their cell entry mechanism has been rapidly developed by recent studies, including structural analysis of these viruses [25,26,27,28,29]. Interestingly, ACE2 binds with higher affinity to the RBD of SARS-CoV-2 than that of SARS-CoV. Paradoxically, however, the affinity of ACE2 for the entire SARS-CoV-2 spike protein is comparable to that of SARS-CoV, suggesting that the SARS-CoV-2 RBD is less exposed than is the SARS-CoV RBD [26].

Possible mechanism of SARS-CoV-2-induced vascular complications. ACE2 angiotensin-converting enzyme 2, ADAM17 a disintegrin and metalloprotease 17, AII angiotensin II, A1-7 angiotensin 1-7, ARDS acute respiratory distress syndrome, EC endothelial cell, EpC epithelial cell, IFN interferon, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, TLRs Toll-like receptors, TMPRSS2 transmembrane serine protease 2, RAS renin-angiotensin system
Organs responsible for SARS-CoV-2 entry
In the human lung, type II alveolar epithelial cells coexpress ACE2 and TMPRSS2 and are considered to be primarily responsible for virus entry in both SARS and COVID-19 [30, 31]. It was recently reported that ACE2 and TMPRSS2 are highly coexpressed in nasal and corneal epithelial cells [31]. Enterocytes also coexpress ACE2 and TMPRSS2 [31], and SARS-CoV-2 as well as SARS-CoV rapidly infect human small intestinal organoids [31]. These findings suggest alternative pathways of viral entry via the upper airways, eyes and intestinal organs. A recent report by Bunyavanich et al. found that ACE2 mRNA expression in the nasal epithelium increased with age when data were stratified into groups of younger children (<10 years), older children (10–17 years), young adults (18–24 years), and adults (≥25 years), which might help to explain the relatively low prevalence of COVID-19 in children [32].
The role of ACE2 in COVID-19
The argument about the role of ACE2 in the pathogenesis of COVID-19 has been complicated by the multifunctionality of ACE2 in addition to serving as the entry receptor for SARS-CoV-2. ACE2 was identified in 2000, and accumulating evidence has established diverse roles of ACE2, including as a negative regulator of the RAS, interacting protein with apelin peptides, and as a chaperone protein for the amino acid transporter B0AT1 (SLC6A19) [33]. Similar to SARS-CoV [34], infection with SARS-CoV-2 may downregulate cell-surface ACE2, leading to the reduced activity of ACE2 in infected organs. Moreover, the binding of ACE2 to SARS-CoV, and probably to SARS-CoV-2, increases the activity of disintegrin and metalloproteinase domain-containing protein 17 (ADAM17), also named tumor necrosis factor-α convertase (TACE) [35]. ADAM17 induces ectodomain shedding of ACE2 and produces circulating soluble ACE2, which can be detected in blood tests (Fig. 1) [36]. This mechanism could also reduce the cell-surface ACE2 [35]. The decrease in cell-surface ACE2 by these pathways appears to reduce the chance of further invasion by the virus. However, it might also attenuate the inhibition of RAS by ACE2 in infected organs. It was reported that depletion of ACE2 worsened acute lung inflammation induced by acid aspiration, sepsis, or endotoxin in mice [37]. In that study, severe lung inflammation induced by acid aspiration was alleviated in mice genetically lacking ACE or angiotensin II type 1a receptor, and also by losartan, an angiotensin II type 1 receptor antagonist, suggesting that local activation of RAS contributes to lung inflammation in pneumonia [37]. Moreover, a research group reported that the SARS spike protein binds to pulmonary ACE2, exacerbating acid-induced pneumonia accompanied by an increase in angiotensin II concentration, and that the pathologic alterations were rescued by losartan in wild-type mice [34]. Given the evidence that angiotensin II is a key mediator of tissue inflammation [38, 39], these findings suggest that downregulation of ACE2 in response to the binding of SARS-CoV, and probably SARS-CoV2, may serve as a mechanism to counteract viral infection at the expense of an increase in angiotensin II.
Interestingly, Ziegler et al. recently reported that ACE2 is upregulated by stimulation by interferon, an antiviral cytokine, in SARS-CoV-2 target cells such as in the lung, nose, and small intestine [40]. Given the abovementioned roles of ACE2 in the respiratory system, upregulation of ACE2 appears to be involved in the series of tissue-protective responses induced by interferon. However, SARS-CoV-2 may exploit this innate immune mechanism to enhance infection [40], and the process would eventually lead to a decrease in cell-surface ACE2. Although this proposed mechanism requires further validation, including in vivo experiments, these data indicate that the pathophysiology of COVID-19 depends on the critical balance between the SARS-CoV-2 viral load and host defense mechanism.
The potential influence of RAS inhibitors on COVID-19
In the early phase of the COVID-19 pandemic, it was proposed that antihypertensive treatment using ACE inhibitors or angiotensin receptor blockers (ARBs) may contribute to adverse outcomes in patients with hypertension and COVID-19 [41]. This hypothesis was primarily based on previous experimental studies showing that RAS inhibitors could alter tissue activity or expression of ACE2. While these studies were mainly conducted using heart [42,43,44,45,46,47,48,49,50,51], arterial [52,53,54,55], and kidney [43, 45, 46, 56,57,58,59] tissues, some papers showed that changes in ACE2 expression also occurred in the lung [60,61,62] (Table 2). There are also several reports indicating that mineralocorticoid receptor antagonists (MRAs) increase the expression or activity of ACE2 in the heart and kidney [48, 49, 63,64,65] (Table 2). Most of these studies have suggested that these antihypertensive drugs might increase tissue expression and/or activity of ACE2, though some have reported no alternation or a decrease of ACE2 in response to the drug [48,49,50, 59] (Table 2). Nevertheless, increased membrane expression of ACE2 induced by these drugs can theoretically increase the chance of virus entry into organs, but it is also conceivable that RAS inhibition contributes to organ protection against respiratory infection, as mentioned above [34, 37]. In addition, cellular entry of SARS-CoV-2 depends not only on ACE2 but also on proteolytic cleavage primarily by TMPRSS2. Therefore, while the increase in ACE2 by these drugs can be harmful at least in terms of viral invasion in organs that coexpress ACE2 and TMPRSS2, it remains unknown how increased tissue levels of ACE2 would alter the severity of COVID-19 in organs that lack proteases mediating fusion of the virus. Finally, there is little evidence that the therapeutic dose of RAS inhibitors influences tissue expression or activity of ACE2 in humans. Further investigation is required to clarify whether the influence of RAS inhibitors on COVID-19 is beneficial, neutral, or harmful.
Circulating ACE2 as a biomarker of SARS-CoV-2 infection?
Given the essential role of ACE2 in SARS-CoV-2 infection, it has been postulated that the circulating plasma concentration of ACE2 can serve as a biomarker to predict susceptibility to COVID-19 or disease severity. Circulating ACE2 levels are theoretically modulated by the activity of ADAM17, which cleaves cellular ACE2 in the cardiovascular system, as well as ACE2 abundance in each organ. Given the previous in vitro finding that ACE2 binding to SARS-CoV increases the truncated form of ACE2 by activating ADAM17 [35], it is theoretically conceivable that SARS-CoV and SARS-CoV-2 infection can alter circulating ACE2 levels. Nevertheless, it remains unknown whether circulating ACE2 levels have any relationship with ACE2 abundance in the respiratory system or intestinal tissues. Moreover, circulating ACE2 levels are increased in patients with cardiovascular diseases (CVDs), including heart failure [66, 67] and arterial fibrillation [68], chronic kidney disease (CKD) [69], atherosclerosis [70], and stroke [71]. In addition, circulating ACE2 levels are reported to be higher in male than in female patients with heart failure [72]. These data indicate that circulating levels of ACE2 can largely be affected by cardiovascular comorbidities or other characteristics. Further studies are needed to clarify whether circulating ACE2 is indeed associated with susceptibility to or disease severity of COVID-19.
ACE2 in COVID-19 from a therapeutic point of view
From a therapeutic point of view, supplementation with soluble exogenous ACE2 can theoretically be favorable for protection against COVID-19, as it can inhibit interaction of the virus with endogenous ACE2. In fact, it was recently reported that human recombinant soluble ACE2 can inhibit infection of SARS-CoV-2 in human blood vessel organoids and human kidney organoids (Fig. 1) [73].
Cardiovascular and cerebrovascular complications in COVID-19
COVID-19 and thromboembolic complications
The risk of venous and arterial thromboembolic complications has been reported to be higher in patients with COVID-19. Klok et al. demonstrated the cumulative incidence of venous thromboembolism (VTE) in 27% and ischemic stroke in 3.7% of patients with COVID-19 pneumonia [74]. Lodigiani et al. also reported that among 388 COVID-19 inpatients, the ratio of thromboembolic events, including VTE, ischemic stroke, and ischemic heart disease, was higher in intensive care unit (ICU) patients (27.6%) than in patients in the general ward (6.6%) [75]. Regarding stroke, patients with severe infection exhibited neurologic manifestations such as acute cerebrovascular diseases (5.7% in severe vs 0.8% in nonsevere, respectively) [76]. In SARS, a case of VTE in multiple organs was described [77], but there are very few reports on SARS-induced thrombotic complications. Large-artery ischemic strokes occurred in 0.7% of Taiwanese [78] and 2% of Singaporean [79] SARS patients. For cases in Singapore, the authors considered that stroke occurred as a side effect of intensive treatment, such as intravenous immunoglobulin; thus, the incidence of VTE and ischemic stroke in COVID-19 patients appears to be remarkably higher than that in SARS patients. According to several reports of “stroke cases” with COVID-19 [80,81,82,83,84], almost all showed elevated plasma D-dimer levels. Additionally, higher D-dimer levels on admission effectively predicted in-hospital mortality in patients with COVID-19 [4, 85, 86]. Among thromboembolic complications, VTE was a common complication in hospitalized patients (observed in 20%) with COVID-19 and was associated with death (adjusted hazard ratio [HR]: 2.4) [87]. In particular, lower extremity deep-vein thrombosis (DVT) was detected in 85.4% of critically ill COVID-19 patients [88]. These findings indicate that abnormalities in the coagulation cascade result in VTE and stroke after SARS-CoV-2 infection. Therefore, systemic anticoagulation therapy may improve outcomes in COVID-19 patients [89].
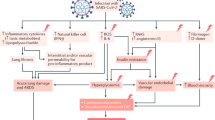
Possible mechanisms of SARS-CoV-2-induced endothelial injury
SARS-CoV-2-induced ischemic organ damage appears to be associated with a hyperinflammatory state, cytokine storm, vascular endothelial damage or fibrinogen consumption coagulopathy (Fig. 1) [84]. Yang et al. clearly reviewed the underlying mechanisms of the dysfunctional coagulatory response in the pathogenesis of influenza A virus [90]. Toll-like receptors (TLRs) have a central role in innate immunity. Viral pathogen-associated molecular patterns such as viral proteins, double-stranded RNA, and single-stranded RNA initially activate the innate immune system [91]. Using in silico studies on the interaction of the SARS-CoV-2 spike glycoprotein with human TLRs, Choudhury et al. demonstrated that TLR4 is most likely to be involved in recognizing molecular patterns from SARS-CoV-2 to induce inflammatory responses [92]. Other TLRs, such as TLR5, TLR7, and TLR8, have been also reported to also be involved in SARS-CoV-2 infection [93, 94]. TLRs activate a common signaling pathway via MyD88, leading to the production of proinflammatory cytokines [95]. Further activation of the innate immune response to eradicate the virus induces overproduction of pro-inflammatory cytokines, resulting in a “cytokine storm” [96] of overactivated neutrophils, monocytes, and lymphocytes. Coagulation is a highly organized process that involves endothelial cells (ECs), platelets and coagulation factors in the sequential action of primary and secondary hemostasis and fibrinolysis [97]. Inflammatory cytokines and leukocyte activation lead to EC activation and endothelial dysfunction via multiple mechanisms, including direct damage, loss of tight junctions, and hyperpermeability induced by inflammatory factors [90]. Activated or injured ECs initiate coagulation by activating platelets and expression of coagulation components (Fig. 1). Visseren et al. found that respiratory virus-infected ECs exhibit procoagulant activity associated with the induction of tissue factor (TF) expression [98]. The coagulation cascade is initiated after exposure of TF to the blood. Recently, Varga et al. reported the pathology of EC dysfunction in COVID-19 based on postmortem analysis of three cases [99]. As ECs express ACE2 [30], viral inclusion structures in ECs and diffuse endothelial inflammation, such as endotheliitis, has been observed. These results suggest that SARS-CoV-2 virus infection directly and indirectly injures ECs and may activate the coagulation pathway. Very recently, Ackermann et al. performed morphologic and molecular analyses in the peripheral lung of seven cases of COVID-19 at autopsy [100], and severe EC injury associated with the presence of intracellular virus, disrupted cell membranes, widespread thrombosis with microangiopathy, and new vessel growth were observed. Ma et al. reported that human umbilical vein endothelial cells (HUVECs) express both ACE2 and TMPRSS2 mRNAs [101]. Human dermal microvascular endothelial cells are also known to express TMPRSS2 [102]. However, whether SARS-CoV-2 induces EC injury directly or indirectly remains unclear. An increase in vascular permeability induced by EC injury plays a pathogenic role in the development of pulmonary fibrosis [103], and acute respiratory distress syndrome (ARDS) is characterized by acute respiratory failure, bilateral pulmonary infiltrates, and noncardiogenic pulmonary edema resulting from vascular hyperpermeability [104]. Thus, EC injury can lead to respiratory dysfunction and may be a key factor that determines the clinical outcome of COVID-19 patients.
COVID-19 and Kawasaki disease
Verdoni et al. surprisingly reported a 30-fold increase in the incidence of Kawasaki-like disease (KLD) during the SARS-CoV-2 epidemic [105]. In that study, 10 patients were diagnosed with KLD between February 18, 2020 and April 20, 2020, compared with 19 patients in the five years before the beginning of the COVID-19 pandemic [105]. Two of 10 patients were positive for SARS-CoV-2 by reverse-transcriptase quantitative PCR assay, and eight of 10 patients diagnosed with KLD were positive for SARS-CoV-2 IgG and/or IgM antibodies. Kawasaki disease (KD) is an acute systemic vasculitis with coronary artery abnormalities that predominantly affects young children [106]. Necrotizing arteritis, subacute chronic vasculitis, and luminal myofibroblastic proliferation are three linked processes associated with KD [107]. Although a correlation between viral infections and KD was reported [108, 109], it is not clear whether KD-associated infections are causal or incidental. KD might also be associated with a dysregulated innate immune response;[110] however, a link between SARS-CoV-2 infection and KLD has not been demonstrated.
Myocardial injury associated with COVID-19
Clinical manifestations of cardiovascular disorders associated with COVID-19 are diverse, with heart failure, arrhythmia, cardiogenic shock, acute myocardial infarction (AMI), and myocarditis having been reported [4, 85, 111,112,113,114]. In an initial report from Wuhan, China, Huang et al. documented that acute cardiac injury, as defined by the elevation of cardiac biomarkers (e.g., troponin I) or new abnormalities in electrocardiography and echocardiography, was present in 12.2% of 41 inpatients with COVID-19 [115]. In another study, acute cardiac injury (7.2%; using the same definition as above) and arrhythmia (16.7%) were common complications among 138 hospitalized patients with COVID-19 [85]. Cardiac complications in COVID-19 are associated with a poor clinical outcome [116, 117]. In one study, 19.7% of 416 hospitalized patients with COVID-19 had abnormally high levels of troponin I, and those with elevated troponin I levels had higher mortality (51.2%) than did those with lower levels (4.5%) [117]. Another study showed that elevation in troponin T levels is associated with the occurrence of malignant arrhythmias and fatal outcomes [116].
As described above, SARS-CoV-2 infection promotes vascular endothelial injury and thromboembolism, which can result in ischemic heart disease and stroke [84, 112]. However, cases of myocardial injury with no evidence of obstructive coronary disease have also been reported [114, 118, 119], suggesting that COVID-19 interacts with the cardiovascular system through multiple mechanisms.
Possible mechanisms of cardiovascular injury in COVID-19
Although the molecular causes of myocardial injury in COVID-19 have remained elusive, several mechanisms can be considered. Severe viral infection triggers systemic inflammatory response syndrome, which increases the risk of plaque rupture and thrombus formation, resulting in the occurrence of atherosclerotic diseases. For example, there is a significant association between the incidence of AMI and influenza [120]. In addition, several papers have reported the occurrence of AMI in COVID-19 patients [112, 121], though more studies are necessary to determine the actual prevalence. Cytokine storms can both cause plaque instability and promote cardiovascular inflammation and myocardial depression [122, 123], which may be involved in myocarditis and stress-induced cardiomyopathy in COVID-19 patients [114, 124].
Furthermore, recent data suggest that a substantial portion of patients with COVID-19 have risk factors for atherosclerotic disease, which include hypertension, obesity, smoking, and diabetes mellitus [6, 85, 111, 115]. Because severe respiratory viral infection induces hypoxemia and abnormal hemodynamic changes, enhanced BP variability may also trigger cardiovascular events in COVID-19 patients with atherosclerotic risk factors. These mechanisms likely act in parallel to cause cardiac damage, resulting in severe clinical manifestations such as AMI, ventricular arrhythmias, and congestive heart failure.
It is currently unknown whether SARS-CoV-2 infects cardiomyocytes. Autopsy studies have reported the detection of SARS-CoV-2 RNA in the heart [125]. However, the cell types in which viral RNA was detected were not defined. Endomyocardial biopsy in a case of COVID-19 with cardiogenic shock revealed viral particles in interstitial macrophages but not in cardiomyocytes [126]. Although ACE2 is expressed in human cardiomyocytes [127], there appears to be little expression of TMPRSS2 in these cells [31]. Therefore, the direct pathogenic role of SARS-CoV-2 in cardiomyocytes needs further investigation.
Acute kidney injury (AKI) in COVID-19: incidence and clinical significance
In addition to cardiac complications, accumulating data indicate that renal abnormalities frequently accompany COVID-19, though there does seem to be a geographic difference in the occurrence of AKI. Several single-center studies from Wuhan report that AKI developed in 3–7% of patients with COVID-19 [85, 115, 128], and the reported incidence of AKI in a multicenter study involving 1099 in-hospital patients in 30 provinces across mainland China was 0.5% [129]. Studies in the US found AKI incidence to be higher than that in the aforementioned reports. For instance, the incidence of AKI was 19.1% among critically ill patients in a small single-center study in Seattle [111], and in a multicenter study conducted in the largest academic health system in New York [6, 130], AKI occurred in 36.6% of 5449 patients admitted with COVID-19, of whom 14.3% required renal replacement therapy (RRT). This geographic difference in the occurrence of AKI might be explained by several factors, including disease severity, ethnicity, and comorbid conditions (such as diabetes and coronary artery disease), all of which are reported to be independent risk factors for AKI in COVID-19 [130].
AKI in COVID-19 is highly associated with respiratory failure. In one study, as many as 89.7% of mechanically ventilated patients developed AKI, whereas AKI occurred in 21.7% of nonventilated patients [130]. The demand for RRT may significantly increase during the COVID-19 pandemic, and clinicians should be aware of the possibility of facing challenges in delivering RRT to COVID-19 patients [131]. Moreover, in line with findings for SARS [132], the mortality rate seems much higher among COVID-19 patients with AKI than among those without AKI. In a report by Zhou et al. [4], AKI was observed only in 0.7% of survivors, whereas 50.0% of nonsurvivors had AKI. Consistently, a dose-dependent relationship between mortality and the severity of AKI was observed in a different cohort, with stage 3 AKI being associated with a fourfold increase in mortality risk [133]. A poor prognosis for COVID-19 patients with AKI was also confirmed in a US cohort [130]. In addition, renal abnormalities associated with COVID-19 may not necessarily be limited to AKI. Pei et al. reported that proteinuria and hematuria were seen in 65.7% and 41.7% of 333 patients with COVID-19 pneumonia, respectively [134]. Glomerulopathy associated with COVID-19 has also been reported [135, 136].
Mechanisms of kidney injury associated with COVID-19
Multiple mechanisms explaining the occurrence of kidney injury in COVID-19 have been proposed, which include but are not limited to the following: hemodynamic instability and renal ischemia, ARDS and the cytokine storm, rhabdomyolysis, hypercoagulability and thrombosis, cardiac failure and kidney congestion, and direct renal infection of SARS-CoV-2. In one study, AKI was more common in patients with cardiac injury than in those without cardiac injury, suggesting a cardio-renal interaction [117].
In autopsy data for 26 cases, diffuse proximal tubule injury was prominent, along with erythrocyte aggregates in peritubular capillaries, ischemic changes with fibrin thrombi in glomeruli, and pigmented casts indicative of rhabdomyolysis [137]. Of note, this study identified virus particles with crown-like morphology in electron microscopy within renal tubules and podocytes, which was accompanied by degenerative changes such as vacuolization and necrotic epithelia. Another study also identified abundant viral forms in the area of vacuolated tubules in a case of COVID-19 at autopsy [138]. These reports were followed by the demonstration of SARS-CoV-2 RNA in the kidney [125, 139]. Such histopathological analyses of cases postmortem indicate that SARS-CoV-2 may have tropism for the kidney, especially in severe cases, though the pathogenic role of direct infection remains undetermined. ACE2 is abundant in renal proximal tubules [31, 140], and its levels are altered in disease states such as diabetes mellitus [140,141,142]. However, TMPRSS2 is highly expressed in the more distal portion of the renal tubules [31]. At present, it is unclear how SARS-CoV-2 enters renal cells and whether comorbid conditions affect the cellular tropism of the virus in the kidney. It also remains to be determined whether renal infection with SARS-CoV-2 indeed contributes to kidney injury.
COVID-19 in advanced CKD
Several lines of evidence suggest that CKD patients, especially those at advanced stages, are vulnerable to SARS-CoV-2 infection. In a multicenter study of 5700 patients hospitalized with COVID-19, end-stage kidney disease (ESKD) was present in 3.5% of all cases [6]. Conversely, the prevalence of laboratory-confirmed COVID-19 was 2.1% in 7154 patients undergoing hemodialysis in Wuhan, which was apparently higher than the morbidity of the general population in that area (~0.5% as of March 2020) [143]. In a cross-sectional study of 3802 cases with SARS-CoV-2 test results, CKD was associated with a positive SARS-CoV-2 result after adjustment for potential confounding variables [144]. Moreover, a higher baseline serum creatinine level is an independent risk factor for in-hospital death in COVID-19 [133], suggesting that ESKD patients are at high risk of developing severe disease. In recent reports, the overall mortality rate for ESKD patients with COVID-19 was 29% (out of 94 cases) in Italy [145] and 31% (of 59 cases) in New York [146]. The basis for such vulnerability is likely multifactorial, and both medical (older age, immune cell dysfunction, cardiovascular and pulmonary comorbidities) and environmental factors need to be considered [147, 148]. Currently, several guiding principles have been proposed to mitigate the risk of COVID-19 in ESKD patients [149,150,151].
Antihypertensive agents during the COVID-19 pandemic
Use of ACE inhibitors and ARBs in patients with COVID-19
Because ACE inhibitors and ARBs may increase the amount of ACE2, whether these drugs should be discontinued during the COVID-19 pandemic has been a topic of discussion [152, 153]. The role of ACE2 in the pathophysiology of COVID-19 as well as experimental evidence for ACE2 and RAS inhibitors are reviewed in detail in the previous sections. In the following sections, we discuss the clinical evidence for COVID-19 and antihypertensive agents.
Although several reports in an early phase of the COVID-19 pandemic have suggested the relationship between COVID-19 and ACE inhibitors or ARBs [154, 155], it is possible that the association between COVID-19 and hypertensive medication results from reverse causality because older patients, who are at the highest risk for COVID-19, tend to have multiple comorbidities, including hypertension and CVD. Moreover, adjustments for age and other possible confounding factors were not performed in most of the early studies [156,157,158].
Several recent reports analyze the effect of ACE inhibitors and ARBs on clinical outcomes in patients with COVID-19 and hypertension [159, 160]. A retrospective, single-site, cohort study from Wuhan compared clinical outcomes among 126 COVID-19 patients with pre-existing hypertension (43 of whom were taking either ACE inhibitors or ARBs; 83 of whom were not taking these agents) and 125 age- and sex-matched COVID-19 control patients without hypertension [159]. In that study, it was found that ACE inhibitors or ARBs did not increase the risk of morbidity or mortality in patients with SARS-CoV-2 infection. Moreover, the study showed a nonsignificant trend toward marginally lower critical illness and death rates in patients taking ACE inhibitors or ARBs compared to those taking other antihypertensive agents. In a study from Spain, a case-population study (1139 cases and 11,390 population controls) showed that users of ACE inhibitors or ARBs had an adjusted odds ratio for COVID-19 requiring admission to hospital of 0.94 (95% CI 0.77–1.15) compared to users of other antihypertensive drugs, with no increased risk for ACE inhibitors or ARBs [160]. This study concluded that ACE inhibitors or ARBs do not increase the risk of COVID-19 requiring admission to the hospital, including fatal cases and those admitted to ICU, and that these agents should not be discontinued to prevent the development of severe COVID-19 [160].
Two other retrospective cohort studies from China comparing disease severity and mortality rates between hypertensive patients taking ACE inhibitors or ARBs and those not taking these agents are available [161, 162]. One, a single-center study, showed that the percentage of patients taking ACE inhibitors and ARBs did not differ between those with severe and nonsevere infections or between survivors and nonsurvivors [161]. In the other, a multicenter cohort study, hypertensive patients with COVID-19 who were taking ACE inhibitors or ARBs were compared with those who were taking antihypertensive drugs other than ACE inhibitors and ARBs as well as patients with COVID-19 without hypertension [162]. The risk for 28-day all-cause mortality was lower in the ACE inhibitor/ARB group than in the control group (adjusted HR 0.42, 95% CI 0.19–0.92; P = 0.03) and in matched subgroup analysis (adjusted HR 0.30, 95% CI 0.12–0.70; P = 0.01).
In addition, two clinical studies from northern Italy and from New York provide further evidence on the association between antihypertensive medication and COVID-19 [163, 164]. In the case-control study in Italy, which included 6272 patients with COVID-19 and 30,759 controls matched for age, sex, and municipality of residence, after adjustment for drugs and coexisting conditions, the odds ratios for the use of ACE inhibitors and ARBs were 0.96 (95% CI 0.87–1.07) and 0.95 (95% CI 0.86–1.05), respectively, among all patients and 0.91 (95% CI 0.69–1.21) and 0.83 (95% CI 0.63–1.10), respectively, among patients who had a severe or fatal course of the disease [163]. In the New York study, none of the major classes of antihypertensive drugs, including ACE inhibitors and ARBs, were associated with a positive SARS-CoV-2 test or disease severity [164].
Taken together, the data from these six studies, although retrospective, from different countries [159,160,161,162,163,164] provide evidence for continuing treatment with ACE inhibitors or ARBs in patients with hypertension during the COVID-19 pandemic. Moreover, a recent meta-analysis showed the potential benefit of ACE inhibitors or ARBs in patients with hypertension [165]. In nine studies comprising 3936 patients with hypertension and COVID-19, ACE inhibitors or ARB treatment was not associated with disease severity but was related to lower mortality from COVID-19 compared with other antihypertensive drugs. Although future well-designed randomized controlled trials are needed, these results suggest that treatment with ACE inhibitors or ARBs should be continued in COVID-19 patients with hypertension [165,166,167].
Use of other antihypertensive agents in patients with COVID-19
The aforementioned two studies on RAS inhibitors also address the association of COVID-19 with other classes of antihypertensive agents [163, 164]. In one study, the adjusted odds ratios for COVID-19 associated with the use of calcium-channel blockers, beta-blockers, thiazide diuretics, loop diuretics, and MRA were 1.03 (95% CI 0.95–1.12), 0.99 (95% CI 0.91–1.08), 1.03 (95% CI 0.86–1.23), 1.46 (95% CI 1.23–1.73), and 0.90 (95% CI 0.75–1.07), respectively [163]. Thus, none of the antihypertensive agents except loop diuretics were associated with an increased risk of COVID-19 in multivariate analysis. In the study population, loop diuretics were used more frequently in patients with COVID-19 than controls (13.9% versus 7.8%; relative difference, 43.6%) [163], and their use may reflect the existence of severe comorbidities such as heart failure and renal dysfunction, the severities of which were not appropriately quantified [163]. In another study, calcium-channel blockers, beta-blockers, and thiazide diuretics were not associated with an increased likelihood of a positive SARS-CoV-2 test [164].
Statements on the use of antihypertensive agents from hypertensive societies and associations worldwide
The International Society of Hypertension [168], the European Society of Hypertension [169], the European Society of Cardiology [170], and the American Heart Association/Heart Failure Society of America/American College of Cardiology [171] have already made a statement on the use of RAS inhibitors during the COVID-19 pandemic. the Japanese Society of Hypertension [172] and the Japanese Circulation Society [173, 174] have also provided statements regarding the management of cardiovascular diseases during the COVID-19 pandemic. All these statements indicate that there is no good evidence to change or discontinue ACE inhibitors or ARBs to avoid or manage SARS-CoV-2 infection.
Current conclusion regarding the use of antihypertensive agents during the COVID-19 pandemic
In the early phase of the COVID-19 pandemic, there was considerable confusion regarding whether ACE inhibitors or ARBs may have adverse effects on COVID-19 patient morbidity and mortality, which was based on the speculation that ACE2 can be upregulated by ACE inhibitors or ARBs. However, the evidence of ACE2 upregulation is limited to experimental studies. Furthermore, there have been no clinical studies supporting the hypothesis that ACE inhibitors and ARBs augment susceptibility to infection and worsen all-cause mortality and cardiovascular outcomes in COVID-19 patients. Thus, treatment with ACE inhibitors and ARBs should be continued in high-risk patients who have received guideline-directed medical therapy, and hypertension should be managed in accordance with the Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH2019) [18].
Areas of uncertainty and future perspectives
In this article, we review recent studies on COVID-19 in the context of hypertension and related diseases. As discussed in detail, there are no reliable reports on whether SARS-CoV-2 infection risk is increased in patients with hypertension. Hypertension is known to be associated with endothelial injury, especially in the elderly [175], and recent evidence suggests that thromboembolism triggered by endothelial injury is one of the important complications that influence disease outcome in COVID-19. At this time, it is unclear whether pre-existing endothelial injury increases the severity of COVID-19; however, hypertensive patients with atherosclerotic diseases may need to be carefully monitored for the occurrence of new-onset CVDs during SARS-CoV-2 infection.
Given that hypertension is the leading contributor to the development of cardiovascular and kidney diseases and given that myocardial injury and advanced CKD are associated with an increased risk of severe disease following SARS-CoV-2 infection, optimal management of hypertension can contribute to a better prognosis of COVID-19 by mitigating the progression of these disorders. Regardless, a major challenge is to achieve target BP control in the “New Normal” lifestyle, in which health care workers may have a reduced opportunity for in-person clinical examination of patients. In the Great East Japan Earthquake in 2011, medical care for patients with hypertension was compromised (“disaster hypertension”) [176]. After the recognition of BP increases following the disaster, which may have contributed to the increased cardiovascular events [176, 177], the remote BP monitoring system using information and communication technology (ICT) was introduced and was indeed useful in achieving target BP control [178, 179]. Therefore, in the post-COVID-19 era, medical practice using ICT may need to be widely implemented for the management of hypertension. In addition, the COVID-19 pandemic may increase the risk of mental disorders—owing to, for example, anxiety, economic issues, and decreased physical activity—all of which can potentially compromise BP control. It is currently unknown whether the COVID-19 pandemic will affect BP control and the development of CVDs in the long term; nonetheless, it is necessary to carefully monitor each patient’s BP.
Last, in addition to the development of effective treatment, vaccination for SARS-CoV-2 will of course be helpful. In the case of influenza infection, several epidemiological studies and randomized control trials have clearly shown that there is a strong inverse relationship between influenza vaccination and the risk of cardiovascular events [180].
References
Itoh H. A new normal for hypertension medicine with coronavirus disease-2019 (COVID-19): proposal from president of Japanese Society of Hypertension. Hypertension Res. 2020. https://doi.org/10.1038/s41440-020-0497-y.
Hall G, Laddu DR, Phillips SA, Lavie CJ, Arena R. A tale of two pandemics: how will COVID-19 and global trends in physical inactivity and sedentary behavior affect one another? Prog Cardiovasc Dis. 2020. https://doi.org/10.1016/j.pcad.2020.04.005.
Ohira T, Hosoya M, Yasumura S, Satoh H, Suzuki H, Sakai A, et al. Evacuation and risk of hypertension after the Great East Japan Earthquake: The Fukushima Health Management Survey. Hypertension. 2016;68:558–64.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.
Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323:1574–81.
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York city area. JAMA. 2020;323:2052–59.
Wang Z, Chen Z, Zhang L, Wang X, Hao G, Zhang Z, et al. Status of Hypertension in China: results from the China Hypertension Survey, 2012–2015. Circulation. 2018;137:2344–56.
Tocci G, Nati G, Cricelli C, Parretti D, Lapi F, Ferrucci A, et al. Prevalence and control of hypertension in the general practice in Italy: updated analysis of a large database. J Hum Hypertens. 2017;31:258–62.
Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71:1269–324.
Satoh A, Arima H, Ohkubo T, Nishi N, Okuda N, Ae R, et al. Associations of socioeconomic status with prevalence, awareness, treatment, and control of hypertension in a general Japanese population: NIPPON DATA2010. J Hypertens. 2017;35:401–8.
Shi Y, Yu X, Zhao H, Wang H, Zhao R, Sheng J. Host susceptibility to severe COVID-19 and establishment of a host risk score: findings of 487 cases outside Wuhan. Crit Care. 2020;24:108.
Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. 2020. https://doi.org/10.1016/j.jaci.2020.04.006.
Simonnet A, Chetboun M, Poissy J, Raverdy V, Noulette J, Duhamel A, et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity. 2020;28:1195–9.
Cai Q, Chen F, Wang T, Luo F, Liu X, Wu Q, et al. Obesity and COVID-19 severity in a designated hospital in Shenzhen, China. Diabetes Care. 2020;43:1392–98.
Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Internal Medicine. 2020;180:934–43.
Centers for Disease Control and Prevention. People who are at higher risk for severe illness. https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-at-higher-risk.html. Accessed 25 May 2020.
Ministry of Health Labour and Welfare. Updates on COVID-19 in Japan. https://www.mhlw.go.jp/content/10900000/000628617.pdf. Accessed 31 May 2020.
Umemura S, Arima H, Arima S, Asayama K, Dohi Y, Hirooka Y, et al. The Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2019). Hypertens Res. 2019;42:1235–481.
Zheng KI, Gao F, Wang XB, Sun QF, Pan KH, Wang TY, et al. Letter to the Editor: Obesity as a risk factor for greater severity of COVID-19 in patients with metabolic associated fatty liver disease. Metabolism. 2020;108:154244.
Kass DA, Duggal P, Cingolani O. Obesity could shift severe COVID-19 disease to younger ages. Lancet. 2020;395:1544–5.
Tanaka M, Itoh H. Hypertension as a metabolic disorder and the novel role of the gut. Curr Hypertens Rep. 2019;21:63.
Nagai M, Ohkubo T, Murakami Y, Takashima N, Kadota A, Miyagawa N, et al. Secular trends of the impact of overweight and obesity on hypertension in Japan, 1980–2010. Hypertens Res. 2015;38:790–5.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280 e278.
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620.
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. 2020. https://doi.org/10.1073/pnas.2003138117.
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–4.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e286.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7.
Sungnak W, Huang N, Becavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26:681–7.
Bunyavanich S, Do A, Vicencio A Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA. 2020. https://doi.org/10.1001/jama.2020.8707.
Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circulation Res. 2020;126:1456–74.
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9.
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci USA. 2008;105:7809–14.
Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167–76.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6.
Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900.
Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247–57.
Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020. https://doi.org/10.1016/j.cell.2020.04.035.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respiratory Med. 2020;8:e21.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–10.
Jessup JA, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell MC, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol. 2006;291:H2166–2172.
Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–6.
Agata J, Ura N, Yoshida H, Shinshi Y, Sasaki H, Hyakkoku M, et al. Olmesartan is an angiotensin II receptor blocker with an inhibitory effect on angiotensin-converting enzyme. Hypertens Res. 2006;29:865–74.
Iwanami J, Mogi M, Tsukuda K, Wang XL, Nakaoka H, Ohshima K, et al. Role of angiotensin-converting enzyme 2/ angiotensin-(1-7)/Mas axis in the hypotensive effect of azilsartan. Hypertens Res. 2014;37:616–20.
Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, Ma M, et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensinconverting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci. 2011;7:1077–92.
Takeda Y, Zhu A, Yoneda T, Usukura M, Takata H, Yamagishi M. Effects of aldosterone and angiotensin II receptor blockade on cardiac angiotensinogen and angiotensin-converting enzyme 2 expression in Dahl salt-sensitive hypertensive rats. Am J Hypertens. 2007;20:1119–24.
Karram T, Abbasi A, Keidar S, Golomb E, Hochberg I, Winaver J, et al. Effects of spironolactone and eprosartan on cardiac remodeling and angiotensin-converting enzyme isoforms in rats with experimental heart failure. Am J Physiol Heart Circ Physiol. 2005;289:H1351–1358.
Burrell LM, Risvanis J, Kubota E, Dean RG, MacDonald PS, Lu S, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26:369–75. discussion 322–364
Ocaranza MP, Godoy I, Jalil JE, Varas M, Collantes P, Pinto M, et al. Enalapril attenuates downregulation of Angiotensinconverting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension. 2006;48:572–8.
Zhang YH, Hao QQ, Wang XY, Chen X, Wang N, Zhu L, et al. ACE2 activity was increased in atherosclerotic plaque by losartan: Possible relation to anti-atherosclerosis. J Renin Angiotensin Aldosterone Syst. 2015;16:292–300.
Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H1013–1019.
Zhong JC, Ye JY, Jin HY, Yu X, Yu HM, Zhu DL, et al. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul Pept. 2011;166:90–97.
Jin HY, Song B, Oudit GY, Davidge ST, Yu HM, Jiang YY, et al. ACE2 deficiency enhances angiotensin II-mediated aortic profilin-1 expression, inflammation and peroxynitrite production. PLoS ONE. 2012;7:e38502. https://doi.org/10.1371/journal.pone.0038502
Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Ann Tallant E, et al. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005;68:2189–96.
Soler MJ, Ye M, Wysocki J, William J, Lloveras J, Batlle D. Localization of ACE2 in the renal vasculature: amplification by angiotensin II type 1 receptor blockade using telmisartan. Am J Physiol Ren Physiol. 2009;296:F398–405.
Velkoska E, Dean RG, Burchill L, Levidiotis V, Burrell LM. Reduction in renal ACE2 expression in subtotal nephrectomy in rats is ameliorated with ACE inhibition. Clin Sci (Lond). 2010;118:269–79.
Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–25.
Wosten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der LoosCM, et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J Pathol. 2011;225:618–27.
Han SX, He GM, Wang T, Chen L, Ning YY, Luo F, et al. Losartan attenuates chronic cigarette smoke exposure-induced pulmonary arterial hypertension in rats: possible involvement of angiotensin-converting enzyme-2. Toxicol Appl Pharmacol. 2010;245:100–7.
Li Y, Zeng Z, Li Y, Huang W, Zhou M, Zhang X, et al. Angiotensin-converting enzyme inhibition attenuates lipopolysaccharide-induced lung injury by regulating the balance between angiotensin-converting enzyme and angiotensinconverting enzyme 2 and inhibiting mitogen-activated protein kinase activation. Shock. 2015;43:395–404.
Kong EL, Zhang JM, An N, Tao Y, Yu WF, Wu FX. Spironolactone rescues renal dysfunction in obstructive jaundice rats by upregulating ACE2 expression. J Cell Commun Signal. 2019;13:17–26.
Mulder P, Mellin V, Favre J, Vercauteren M, Remy-Jouet I, Monteil C, et al. Aldosterone synthase inhibition improves cardiovascular function and structure in rats with heart failure: a comparison with spironolactone. Eur Heart J. 2008;29:2171–9.
Keidar S, Gamliel-Lazarovich A, Kaplan M, Pavlotzky E, Hamoud S, Hayek T, et al. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circulation Res. 2005;97:946–53.
Epelman S, Tang WH, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol. 2008;52:750–4.
Epelman S, Shrestha K, Troughton RW, Francis GS, Sen S, Klein AL, et al. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail. 2009;15:565–71.
Walters TE, Kalman JM, Patel SK, Mearns M, Velkoska E, Burrell LM. Angiotensin converting enzyme 2 activity and human atrial fibrillation: increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace. 2017;19:1280–7.
Roberts MA, Velkoska E, Ierino FL, Burrell LM. Angiotensinconverting enzyme 2 activity in patients with chronic kidney disease. Nephrol Dial Transplant. 2013;28:2287–94.
Anguiano L, Riera M, Pascual J, Valdivielso JM, Barrios C, Betriu A, et al. Circulating angiotensin converting enzyme 2 activity as a biomarker of silent atherosclerosis in patients with chronic kidney disease. Atherosclerosis. 2016;253:135–43.
Mogi M, Kawajiri M, Tsukuda K, Matsumoto S, Yamada T, Horiuchi M. Serum levels of renin-angiotensin system components in acute stroke patients. Geriatr Gerontol Int. 2014;14:793–8.
Sama IE, Ravera A, Santema BT, van Goor H, Ter Maaten JM, Cleland JGF, et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur Heart J. 2020;41:1810–7.
Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181:905–913.
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020. https://doi.org/10.1016/j.thromres.2020.04.013.
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020;191:9–14.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77:1–9.
Xiang-Hua Y, Le-Min W, Ai-Bin L, Zhu G, Riquan L, Xu-You Z, et al. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am J Respir Crit Care Med. 2010;182:436–7.
Tsai LK, Hsieh ST, Chang YC. Neurological manifestations in severe acute respiratory syndrome. Acta Neurol Taiwan. 2005;14:113–9.
Umapathi T, Kor AC, Venketasubramanian N, Lim CC, Pang BC, Yeo TT, et al. Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J Neurol. 2004;251:1227–31.
Avula A, Nalleballe K, Narula N, Sapozhnikov S, Dandu V, Toom S, et al. COVID-19 presenting as stroke. Brain Behav Immun. 2020;87:115–9.
Gonzalez-Pinto T, Luna-Rodriguez A, Moreno-Estebanez A, Agirre-Beitia G, Rodriguez-Antiguedad A, Ruiz-Lopez M Emergency room neurology in times of COVID-19: malignant ischaemic stroke and SARS-CoV-2 infection. Eur J Neurol. 2020. https://doi.org/10.1111/ene.14286.
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382:e60.
Tunc A, Unlubas Y, Alemdar M, Akyuz E. Coexistence of COVID-19 and acute ischemic stroke report of four cases. J Clin Neurosci. 2020;77:227–9.
Valderrama EV, Humbert K, Lord A, Frontera J, Yaghi S. Severe acute respiratory syndrome coronavirus 2 infection and ischemic stroke. Stroke. 2020;51:e124–e127.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–9.
Zhang L, Yan X, Fan Q, Liu H, Liu X, Liu Z, et al. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J Thromb Haemost. 2020;18:1324–9.
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Muller MCA, et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14888.
Ren B, Yan F, Deng Z, Zhang S, Xiao L, Wu M, et al. Extremely high incidence of lower extremity deep venous thrombosis in 48 patients with severe COVID-19in Wuhan. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.120.047407.
Paranjpe I, Fuster V, Lala A, Russak A, Glicksberg BS, Levin MA, et al. Association of treatment dose anticoagulation with inhospital survival among hospitalized patients with COVID-19. J Am Coll Cardiol. 2020. https://doi.org/10.1016/j.jacc.2020.05.001.
Yang Y, Tang H. Aberrant coagulation causes a hyperinflammatory response in severe influenza pneumonia. Cell Mol Immunol. 2016;13:432–42.
Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86.
Choudhury A, Mukherjee S In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020. https://doi.org/10.1002/jmv.25987.
Chakraborty C, Sharma AR, Bhattacharya M, Sharma G, Lee SS, Agoramoorthy G Consider TLR5 for new therapeutic development against COVID-19. J Med Virol. 2020. https://doi.org/10.1002/jmv.25997.
Moreno-Eutimio MA, Lopez-Macias C, Pastelin-Palacios R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARSCoV, and MERS-CoV genomes. Microbes Infect. 2020;22:226–9.
Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83.
Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. 2016;13:3–10.
Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014;58:515–23.
Visseren FL, Bouwman JJ, Bouter KP, Diepersloot RJ, de Groot PH, Erkelens DW. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb Haemost. 2000;84:319–24.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2015432.
Ma D, Chen CB, Jhanji V, Xu C, Yuan XL, Liang JJ, et al. Expression of SARS-CoV-2 receptor ACE2 and TMPRSS2 in human primary conjunctival and pterygium cell lines and in mouse cornea. Eye (Lond). 2020;34:1212–9.
Aimes RT, Zijlstra A, Hooper JD, Ogbourne SM, Sit ML, Fuchs S, et al. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb Haemost. 2003;89:561–72.
Probst CK, Montesi SB, Medoff BD, Shea BS, Knipe RS. Vascular permeability in the fibrotic lung. Eur Respir J. 2020. https://doi.org/10.1183/13993003.00100-2019.
Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–40.
Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020;395:1771–8.
Yim D, Curtis N, Cheung M, Burgner D. Update on Kawasaki disease: epidemiology, aetiology and pathogenesis. J Paediatr Child Health. 2013;49:704–8.
Shulman ST, Rowley AH. Kawasaki disease: insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol. 2015;11:475–82.
Chang LY, Lu CY, Shao PL, Lee PI, Lin MT, Fan TY, et al. Viral infections associated with Kawasaki disease. J Formos Med Assoc. 2014;113:148–54.
Nigro G, Zerbini M, Krzysztofiak A, Gentilomi G, Porcaro MA, Mango T, et al. Active or recent parvovirus B19 infection in children with Kawasaki disease. Lancet. 1994;343:1260–1.
Nakamura A, Ikeda K, Hamaoka K. Aetiological significance of infectious stimuli in Kawasaki disease. Front Pediatr. 2019;7:244.
Arentz M, Yim E, Klaff L, Lokhandwala S, Riedo FX, Chong M, et al. Characteristics and outcomes of 21 critically Ill patients with COVID-19 in Washington State. JAMA. 2020;323:1612–4.
Bangalore S, Sharma A, Slotwiner A, Yatskar L, Harari R, Shah B, et al. ST-segment elevation in patients with Covid-19 - a case series. N Engl J Med. 2020;382:2478–80.
Fried JA, Ramasubbu K, Bhatt R, Topkara VK, Clerkin KJ, Horn E, et al. The variety of cardiovascular presentations of COVID-19. Circulation. 2020;141:1930–6.
Kim IC, Kim JY, Kim HA, Han S. COVID-19-related myocarditis in a 21-year-old female patient. Eur Heart J. 2020;41:1859.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1017.
Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.0950.
Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1096.
Doyen D, Moceri P, Ducreux D, Dellamonica J. Myocarditis in a patient with COVID-19: a cause of raised troponin and ECG changes. Lancet. 2020;395:1516.
Nguyen JL, Yang W, Ito K, Matte TD, Shaman J, Kinney PL. Seasonal influenza infections and cardiovascular disease mortality. JAMA Cardiol. 2016;1:274–81.
Harari R, Bangalore S, Chang E, Shah B. COVID-19 complicated by acute myocardial infarction with extensive thrombus burden and cardiogenic shock. Catheter Cardiovasc Interv. 2020. https://doi.org/10.1002/ccd.28992.
Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–704.
Prabhu SD. Cytokine-induced modulation of cardiac function. Circulation Res. 2004;95:1140–53.
Minhas AS, Scheel P, Garibaldi B, Liu G, Horton M, Jennings M, et al. Takotsubo syndrome in the setting of COVID-19 infection. JACC Case Rep. 2020. https://doi.org/10.1016/j.jaccas.2020.04.023.
Wichmann D, Sperhake JP, Lutgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Internal Med. 2020. https://doi.org/10.7326/M20-2003.
Tavazzi G, Pellegrini C, Maurelli M, Belliato M, Sciutti F, Bottazzi A, et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020;22:911–5.
Nicin L, Abplanalp WT, Mellentin H, Kattih B, Tombor L, John D, et al. Cell type-specific expression of the putative SARSCoV- 2 receptor ACE2 in human hearts. Eur Heart J. 2020;41:1804–6.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–13.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382:1708–20.
Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, et al. Acute kidney injury in patients hospitalized with Covid-19. Kidney Int. 2020;98:209–18.
Goldfarb DS, Benstein JA, Zhdanova O, Hammer E, Block CA, Caplin NJ, et al. Impending shortages of kidney replacement therapy for COVID-19 patients. Clin J Am Soc Nephrol. 2020;15:880–2.
Chu KH, Tsang WK, Tang CS, Lam MF, Lai FM, To KF, et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005;67:698–705.
Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020;97:829–38.
Pei G, Zhang Z, Peng J, Liu L, Zhang C, Yu C, et al. Renal involvement and early prognosis in patients with COVID-19 pneumonia. J Am Soc Nephrol. 2020;31:1157–65.
Larsen CP, Bourne TD, Wilson JD, Saqqa O, Sharshir MA. Collapsing glomerulopathy in a patient with coronavirus disease 2019 (COVID-19). Kidney Int Rep. 2020;5:935–9.
Peleg Y, Kudose S, D’Agati V, Siddall E, Ahmad S, Kisselev S, et al. Acute kidney injury due to collapsing glomerulopathy following COVID-19 infection. Kidney Int Rep. 2020;5:940–5.
Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98:219–27.
Farkash EA, Wilson AM, Jentzen JM. Ultrastructural evidence for direct renal infection with SARS-CoV-2. J Am Soc Nephrol. 2020. https://doi.org/10.1681/ASN.2020040432.
Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. 2020. https://doi.org/10.1056/NEJMc2011400.
Batlle D, Soler MJ, Sparks MA, Hiremath S, South AM, Welling PA, et al. Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J Am Soc Nephrol. 2020. https://doi.org/10.1681/ASN.2020040419.
Mizuiri S, Hemmi H, Arita M, Ohashi Y, Tanaka Y, Miyagi M, et al. Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am J Kidney Dis. 2008;51:613–23.
Williams VR, Scholey JW. Angiotensin-converting enzyme 2 and renal disease. Curr Opin Nephrol Hypertens. 2018;27:35–41.
Xiong F, Tang H, Liu L, Tu C, Tian JB, Lei CT, et al. Clinical characteristics of and medical interventions for COVID-19 in hemodialysis patients in Wuhan, China. J Am Soc Nephrol. 2020. https://doi.org/10.1681/ASN.2020030354.
de Lusignan S, Dorward J, Correa A, Jones N, Akinyemi O, Amirthalingam G, et al. Risk factors for SARS-CoV-2 among patients in the Oxford Royal College of General Practitioners Research and Surveillance Centre primary care network: a crosssectional study. Lancet Infect Dis. 2020;S1473-3099:30371–6.
Alberici F, Delbarba E, Manenti C, Econimo L, Valerio F, Pola A, et al. A report from the Brescia Renal COVID Task Force on the clinical characteristics and short-term outcome ofhemodialysis patients with SARS-CoV-2 infection. Kidney Int. 2020;98:20–6.
Valeri AM, Robbins-Juarez SY, Stevens JS, Ahn W, Rao MK, Radhakrishnan J, et al. Presentation and outcomes of patients with ESKD and COVID-19. J Am Soc Nephrol. 2020. https://doi.org/10.1681/ASN.2020040470.
Basile C, Combe C, Pizzarelli F, Covic A, Davenport A, Kanbay M, et al. Recommendations for the prevention, mitigation and containment of the emerging SARS-CoV-2 (COVID-19) pandemic in haemodialysis centres. Nephrol Dial Transplant. 2020;35:737–41.
Betjes MG. Immune cell dysfunction and inflammation in endstage renal disease. Nat Rev Nephrol. 2013;9:255–65.
Kliger AS, Silberzweig J. Mitigating risk of COVID-19 in dialysis facilities. Clin J Am Soc Nephrol. 2020;15:707–9.
Li J, Xu G. Lessons from the experience in Wuhan to reduce risk of COVID-19 infection in patients undergoing long-term hemodialysis. Clin J Am Soc Nephrol. 2020;15:717–9.
Watnick S, McNamara E. On the frontline of the COVID-19 outbreak: keeping patients on long-term dialysis safe. Clin J Am Soc Nephrol. 2020;15:710–3.
Sommerstein R, Kochen MM, Messerli FH, Grani C. Coronavirus disease 2019 (COVID-19): do angiotensin-converting enzyme inhibitors/angiotensin receptor blockers have a biphasic effect? J Am Heart Assoc. 2020;9:e016509. https://doi.org/10.1161/JAHA.120.016509.
Kreutz R, Algharably EAE, Azizi M, Dobrowolski P, Guzik T, Januszewicz A, et al. Hypertension, the renin-angiotensin system, and the risk of lower respiratory tract infections and lung injury: implications for COVID-19. Cardiovasc Res. 2020. https://doi.org/10.1093/cvr/cvaa097.
Kickbusch I, Leung G. Response to the emerging novel coronavirus outbreak. BMJ. 2020;368:m406.
Gurwitz D Angiotensin receptor blockers as tentative SARSCoV- 2 therapeutics. Drug Dev Res. 2020. https://doi.org/10.1002/ddr.21656.
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID-19 and cardiovascular disease. Circulation. 2020;141:1648–55.
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020;382:1653–9.
Danser AHJ, Epstein M, Batlle D. Renin-angiotensin system blockers and the COVID-19 pandemic: at present there is no evidence to abandon renin-angiotensin system blockers. Hypertension. 2020;75:1382–5.
Yang G, Tan Z, Zhou L, Yang M, Peng L, Liu J, et al. Effects Of ARBs and ACEIs on virus infection, inflammatory status and clinical outcomes in COVID-19 patients with hypertension: a single center retrospective study. Hypertension. 2020;76:51–8.
de Abajo FJ, Rodriguez-Martin S, Lerma V, Mejia-Abril G, Aguilar M, Garcia-Luque A, et al. Use of renin-angiotensinaldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: a case-population study. Lancet. 2020;395:1705–14.
Li J, Wang X, Chen J, Zhang H, Deng A. Association of reninangiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) infection in Wuhan. China. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1624.
Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circulation Res. 2020;126:1671–81.
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Reninangiotensin- aldosterone system blockers and the risk of Covid-19. N Engl J Med. 2020;382:2431–40.
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med. 2020;382:2441–48.
Guo X, Zhu Y, Hong Y. Decreased mortality of COVID-19 with renin-angiotensin-aldosterone system inhibitors therapy in patients with hypertension: a meta-analysis. Hypertension. 2020. https://doi.org/10.1161/HYPERTENSIONAHA.120.15572.
Sugimoto T, Mizuno A, Kishi T, Ito N, Matsumoto C, Fukuda M, et al. Coronavirus disease 2019 (COVID-19) information for cardiologists- systematic literature review and additional analysis. Circ J. 2020;84:1039–43.
Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res. 2020;43:648–54.
International Society of Hypertension. A statement from the International Society of Hypertension on COVID-19. https://ish-world.com/news/a/A-statement-from-the-International-Society-of-Hypertension-on-COVID-19/. Accessed 7 June 2020.
European Society of Hypertension. Statement of the European Society of Hypertension (ESH) on hypertension, renin angiotensin system blockers and COVID-19. https://www.eshonline.org/spotlights/esh-statement-on-covid-19/. Accessed 7 June 2020.
ESC Council on Hypertension. Position statement of the ESC Council on hypertension on ACE-inhibitors and angiotensin receptor blockers. https://www.escardio.org/Councils/Councilon-Hypertension-(CHT)/News/position-statement-of-theesccouncil-on-hypertension-on-ace-inhibitors-and-ang. Accessed 7 June 2020.
Statement from the American Heart Association tHFSoAatACoC. Patients taking ACE-i and ARBs who contract COVID-19 should continue treatment, unless otherwise advised by their physician. https://newsroom.heart.org/news/patients-taking-acei-and-arbs-who-contract-covid-19-should-continue-treatmentunless-otherwise-advised-by-their-physician. Accessed 7 June 2020.
Japanese Society of Hypertension. Information of COVID-19. https://www.ipnsh.jp/corona.html. Accessed 7 June 2020.
Kishi T, Hirano T, Mizuno A, Hashimoto Y, Matsumoto C, Fukuda M, et al. Joint declaration on COVID-19 by the Japan stroke and Japanese Circulation Societies. Circ Rep. https://doi.org/10.1253/circrep.CR-20-0040.
Japanese Circulation Society. Information of COVID-19 in cardiology. https://www.j-circ.or.jp/covid-19/. Accessed 7 June 2020.
Higashi Y, Kihara Y, Noma K. Endothelial dysfunction and hypertension in aging. Hypertens Res. 2012;35:1039–47.
Kario K. Disaster hypertension - its characteristics, mechanism, and management. Circ J. 2012;76:553–62.
Hoshide S, Nishizawa M, Okawara Y, Harada N, Kunii O, Shimpo M, et al. Salt intake and risk of disaster hypertension among evacuees in a shelter after the Great East Japan Earthquake. Hypertension. 2019;74:564–71.
Kario K, Nishizawa M, Hoshide S, Shimpo M, Ishibashi Y, Kunii O, et al. Development of a disaster cardiovascular prevention network. Lancet. 2011;378:1125–7.
Nishizawa M, Hoshide S, Okawara Y, Matsuo T, Kario K. Strict blood pressure control achieved using an ICT-based home blood pressure monitoring system in a catastrophically damaged area after a disaster. J Clin Hypertens. 2017;19:26–29.
Udell JA, Zawi R, Bhatt DL, Keshtkar-Jahromi M, Gaughran F, Phrommintikul A, et al. Association between influenza vaccination and cardiovascular outcomes in high-risk patients: a meta-analysis. JAMA. 2013;310:1711–20.
Acknowledgements
We are grateful to Dr. Hiromi Rakugi at Osaka University, Dr. Naoki Kashihara at Kawasaki Medical School, Dr. Masashi Mukoyama at Kumamoto University, Dr. Hisashi Kai at Kurume University Medical Center, Dr. Tomohiro Katsuya at Katsuya Clinic, Dr. Tatsuo Shimosawa at International University of Health and Welfare, Dr. Koichi Node at Saga University, Dr. Katsuyuki Miura at Shiga University of Medical Science, and Dr. Kimika Arakawa at National Hospital Organization Kyushu Medical Center for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shibata, S., Arima, H., Asayama, K. et al. Hypertension and related diseases in the era of COVID-19: a report from the Japanese Society of Hypertension Task Force on COVID-19. Hypertens Res 43, 1028–1046 (2020). https://doi.org/10.1038/s41440-020-0515-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-020-0515-0
Keywords
This article is cited by
-
Association between blood pressure and COVID-19 severity
Hypertension Research (2024)
-
Emerging topics on basic research in hypertension: interorgan communication and the need for interresearcher collaboration
Hypertension Research (2023)
-
What impacts do the new ESH 2023 guidelines have on the management of hypertension in Japan?
Hypertension Research (2023)
-
Long COVID and hypertension-related disorders: a report from the Japanese Society of Hypertension Project Team on COVID-19
Hypertension Research (2023)
-
Hypertension management before and under the COVID-19 pandemic: lessons and future directions
Hypertension Research (2023)
