
Abstract
The rapid spread of a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to an ongoing pandemic of coronavirus disease 2019 (COVID-19). Recently, angiotensin-converting enzyme 2 (ACE2) has been shown to be a functional receptor for SARS-CoV-2 to enter host target cells. Given that angiotensin receptor blockers (ARBs) and an ACE inhibitor (ACEI) upregulated ACE2 expression in animal studies, the concern might arise regarding whether ARBs and ACEIs would increase the morbidity and mortality of COVID-19. On the other hand, animal data suggested a potential protective effect of ARBs against COVID-19 pneumonia because an ARB prevented the aggravation of acute lung injury in mice infected with SARS-CoV, which is closely related to SARS-CoV-2. Importantly, however, there is no clinical or experimental evidence supporting that ARBs and ACEIs either augment the susceptibility to SARS-CoV-2 or aggravate the severity and outcomes of COVID-19 at present. Until further data are available, it is recommended that ARB and ACEI medications be continued for the treatment of patients with cardiovascular disease and hypertension, especially those at high risk, according to guideline-directed medical therapy based on the currently available evidence.
Similar content being viewed by others
Introduction
A novel coronavirus, named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was discovered in December 2019 in Wuhan, China, and an ongoing pandemic of coronavirus disease 2019 (COVID-19) has been spreading around the world as of early April 2020. The clinical spectrum of COVID-19 ranges from asymptomatic upper respiratory infection to critically ill pneumonia associated with acute respiratory distress syndrome (ARDS) [1,2,3]. In initial reports from China, the prevalence of older age, hypertension, diabetes mellitus, and cardiovascular disease (CVD) was high in COVID-19 patients, and individuals with these comorbidities tended to have greater case fatality rates [3, 4]. In addition, it has been shown that angiotensin-converting enzyme 2 (ACE2) is a functional receptor for SARS-CoV-2 infection [5,6,7]. Given that experimental studies suggested that angiotensin receptor blockers (ARBs) and an ACE inhibitor (ACEI) could increase ACE2 expression in cardiovascular and renal systems [8,9,10,11,12,13,14,15,16,17,18,19,20], concerns may have been raised regarding whether ARBs and ACEIs would augment the infection of SARS-CoV-2 and the severity of COVID-19 in hypertension and CVD patients receiving these drugs [21]. In contrast, ARBs have potential benefits in the prevention and treatment of lung injury caused by COVID-19 [22]. These conflicting views appear to arise based on the results of experimental studies, especially those of severe acute respiratory syndrome coronavirus (SARS-CoV), because direct clinical data on COVID-19 are lacking at present. SARS-CoV caused the SARS epidemic in China in 2002–2003, and its virological characteristics are closely related to those of SARS-CoV-2 [23, 24]. Here, available investigations on the association of the renin–angiotensin system (RAS), particularly ACE2 and angiotensin II, with SARS-CoV-induced lung injury and the latest information on COVID-19 were reviewed as of early April 2020, which would provide insight into COVID-19 and the direction of future research on COVID-19.
ACE2 as a receptor for SARS-CoV and SARS-CoV-2
ACE2 belongs to the membrane-bound carboxydipeptidase family and is widely distributed in the human body, including the heart, kidney, small intestine, and, to a lesser extent, the lung. Lung ACE2 expression is concentrated mainly in type II alveolar cells and macrophages and modestly in bronchial and tracheal epithelial cells [25]. ACE2 degrades angiotensin II to generate angiotensin 1-7, which activates the mas oncogene receptor that negatively regulates a variety of angiotensin II actions mediated by angiotensin II type 1 receptor (AT1R) [26]. Therefore, it is thought that the ACE2/angiotensin 1-7/mas receptor axis has counteracting effects against the excessively activated ACE/angiotensin II/AT1R axis, as seen in hypertension, cardiac hypertrophy, heart failure, and other CVDs [26].
On the other hand, human ACE2 is an established functional receptor by which SARS-CoV enters host target cells (Fig. 1) [23, 24]. The transmembrane spike glycoprotein (S protein) of SARS-CoV binds to the cellular membrane ACE2; SARS-CoV then attaches to the target cells, followed by SARS-CoV-S protein priming by cellular surface proteases, such as transmembrane protease serine 2 (TMPRSS2), allowing the fusion of viral and cellular membranes and resulting in SARS-CoV entry and replication in the target cells [6]. Moreover, ACE2 knockout greatly reduces viral infection and replication in mice after experimental SARS-CoV infection [27]. Thus, it is suggested that the binding of the SARS-CoV-S protein to ACE2 is crucial for SARS-CoV infection.

Possible scheme of the association of ACE2, angiotensin II, and AT1R and acute lung injury of SARS and COVID-19. Ang II, angiotensin II; ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; ACEI, angiotensin-converting enzyme inhibitor; AT1R, angiotensin II type-1 receptor; ARB, angiotensin II type-1 receptor blocker; SARS-CoV, severe acute respiratory syndrome coronavirus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; S-protein, spike-glycoprotein
Recently, it has been shown that the SARS-CoV-2-S protein shares nearly 80% amino acid identity with the SARS-CoV-S protein, and the binding affinity of the SARS-CoV-2-S protein to human ACE2 is similar to that of the SARS-CoV-S protein [5,6,7]. Anti-human ACE2 antibodies or antisera can inhibit SARS-CoV-2-S protein-mediated entry into cultured cells in vitro [5,6,7]. Accordingly, SARS-CoV-2 utilizes ACE2 for target cell entry as well. However, it is still not clear whether ACE2 is the sole receptor for SARS-CoV-2 infection because CD209L (a glycoprotein of C-type lectin also called L-SIGN) is identified as a possible secondary receptor for SARS-CoV in cultured Chinese hamster ovary cells [28].
ACE2 and acute lung injury
Experimental SARS-CoV infection induces acute respiratory failure and lung parenchymal injury characterized by alveolar wall thickening, pulmonary vascular hyperpermeability, and inflammatory cell infiltration in mice [27]. After SARS-CoV infection in mice, lung ACE2 protein levels are greatly reduced, while ACE levels are not changed [27]. These findings are consistent with the previous observation that coronaviruses specifically downregulate ACE2 expression in host cells, depending on virus replication [29]. In addition, SARS-CoV-induced acute lung injury is remarkably attenuated in ACE2 knockout mice compared with wild-type mice [27]. Lung angiotensin II levels increase in wild-type mice after SARS-CoV infection [27]. Acid aspiration mice, an ARDS model, show functional and pathological changes mimicking SARS-CoV-induced lung injury [30]. The lung angiotensin II levels increase after acid aspiration, and SARS-CoV-S protein treatment elicits a further angiotensin II increase [27]. Interestingly, exogenous recombinant ACE2 treatment rescues acid aspiration-induced acute lung injury [30]. Taking this evidence together, the following hypothesis has been raised for the mechanism underlying SARS-CoV-induced lung injury: SARS-CoV infection downregulates lung ACE2 and in turn, shifts the balance toward the dominance of the ACE/angiotensin II/AT1R system over the ACE2/angiotensin 1-7/mas receptor system in the lung. As a result, noncompeting angiotensin II accumulation occurs, resulting in acute lung injury through AT1R activation [27].
It should be noted that a similar mechanism is proposed for the severe lung injury caused by the avian influenza A virus H5N1, which has spread worldwide in humans with a high mortality rate. After H5N1 virus infection in mice, lung ACE2 expression is downregulated, and serum angiotensin II levels increase [31]. Acute lung injury is augmented by ACE2 knockout in H5N1-infected mice, while the administration of recombinant human ACE2 ameliorates H5N1-induced lung injury in mice [31]. Moreover, in other mouse models of acute lung injury/ARDS, such as acid aspiration, sepsis, and drug-induced lung fibrosis, it has been shown that ACE2 expression is downregulated in response to noxious stimuli and that acute lung injury is aggravated by ACE2 knockout and rescued by exogenous ACE2 administration or AT1R knockout [30, 32]. Accordingly, it is suggested that the angiotensin II/AT1R-induced aggravation resulting from ACE2 downregulation and the protective effects of ACE2 are not specific to lung injury in SARS but rather may be common to acute lung injury/ARDS induced by various viral infections and lung diseases.
Effects of RAS inhibitors on ACE2 and SARS-CoV-induced lung injury
Several kinds of ARBs (e.g., olmesartan, telmisartan, losartan, and azilsartan) have been shown to increase the mRNA or protein levels of ACE2 in animal models of heart diseases (hypertensive hypertrophy, autoimmune myocarditis, dilated cardiomyopathy, myocardial infarction, and diabetic cardiomyopathy) [8,9,10,11,12,13,14] and chronic kidney disease (hypertensive nephropathy and diabetic nephropathy) [15, 16], as well as normal rat heart and renal vasculature [17, 18] and hypertensive rat aorta (but not the carotid artery) [19, 20]. ACEI does not inhibit ACE2 as a pharmacological property. In normal rats, lisinopril has been shown to modestly increase cardiac ACE2 mRNA levels while not changing cardiac ACE2 activity [17]. In the myocardial infarction model, valsartan, ramipril, and their combination have no effects on cardiac ACE2 expression [33]. It should be noted that the doses of ARBs and ACEIs used in these animal studies were much greater than those in clinical practice so that the observed effects of these drugs on ACE2 expression and activity could not be extrapolated to clinical situations in humans. Importantly, no clinical data exist regarding the effects of ARBs and ACEIs on human tissue ACE2 expression or activity in vivo. Although there are several clinical studies investigating the changes in plasma or urine levels of the soluble form of ACE2 in hypertension and CVD patients receiving ARBs or ACEIs, the soluble form of ACE2 in the plasma and urine is not a reliable indicator of the activity of tissue ACE2, namely, membrane-bound ACE2 [22]. Importantly, there are no data on the effects of ARBs and ACEIs on lung ACE2 expression either in animal models or humans. It is still unknown whether the changes in ACE2 levels (for example, ARB-induced upregulation) actually facilitate greater engagement and entry of SARS-CoV and other viruses, even in animal models.
On the other hand, it has been shown that pretreatment with losartan, an ARB, ameliorates acute pulmonary edema and lung injury in SARS-CoV-S protein-treated mice after acid aspiration (Fig. 2) [27]. Taken together with the lung angiotensin II level elevation [27], this evidence suggested that even after considering the speculation that viral load would be enhanced by ACE2 upregulation, the net effects of ARBs would be favorable to prevent SARS-CoV-induced acute lung injury in this model. In addition, there is another possibility that the ARB pretreatment-induced baseline ACE2 increase prior to SARS-CoV infection would result in higher ACE2 levels that remained after SARS-CoV-induced downregulation and consequently conferred protection against lung injury in this model [22, 34]. However, in interpreting the results of this study, it should be noted that the effects of losartan have been observed merely in mice treated with SARS-CoV-S protein in addition to acid aspiration but not in mice treated with SARS-CoV-S protein alone. Additionally, data on lung ACE2 levels before and after treatment with SARS-CoV-S protein and acid aspiration are lacking in mice pretreated with or without losartan.
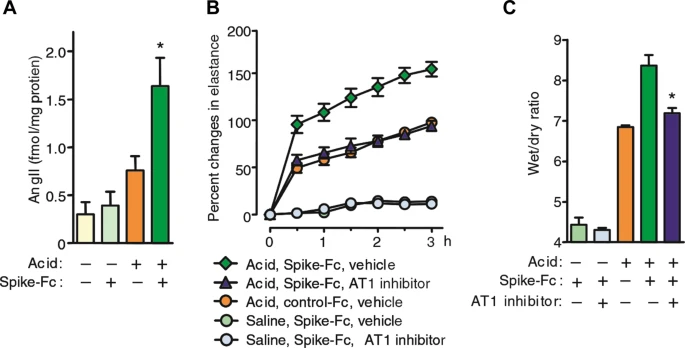
SARS-CoV spike mediates lung injury through modulation of angiotensin II in acid aspiration mice. a Lung angiotensin II levels in SARS-CoV-S protein (spike-Fc protein)- or control Fc protein-treated mice after acid or saline aspiration. *P < 0.05 vs. control-treated mice after acid aspiration. b Effects of losartan (15 mg/kg) (AT1 inhibitor) on lung elastance measurements in acid plus spike-Fc protein-treated mice (n = 4–6 per group). P < 0.05 comparing losartan-treated spike-Fc–challenged mice with vehicle-treated spike-Fc–challenged mice. c Effects of losartan (15 mg/kg) on lung edema (wet/dry weight ratio) in acid plus spike-Fc protein-treated mice (n = 4–6 per group). P < 0.05 vs. vehicle-treated wild-type mice after acid injury. AT1, angiotensin II type-1 receptor. Cited from ref. [27]
Current evidence in COVID-19
Earlier studies reported that the case fatality rate was high in COVID-19 patients with comorbidities such as older age, hypertension, diabetes mellitus, CVD, chronic pulmonary disease, and malignancy [3, 4]. Because patients with hypertension and CVD are likely treated with ACEIs or ARBs, the concern is raised regarding whether the use of ACEIs and ARBs may aggravate the morbidity and mortality of COVID-19 [21]. However, it was possible that the observed phenomenon would result from reverse causality because older patients are at the highest risk for COVID-19 and concurrently tend to have multiple comorbidities, including hypertension and CVD, and because adjustments for age and other possible confounding factors were not performed in such earlier studies [22, 35, 36]. Indeed, in a recent retrospective, multicenter cohort study enrolling 191 confirmed COVID-19 patients in Wuhan, China, multivariable regression analysis revealed that independent risk factors for in-hospital death are older age, higher Sequential Organ Failure Assessment score, and higher D-dimer levels at the early stage of COVID-19, although hypertension, diabetes, and coronary artery disease were not included [37].
A small case study reported that plasma angiotensin II levels were markedly elevated and linearly associated with viral load and lung injury severity in COVID-19 pneumonia patients [38]. Given that a remarkable elevation of circulating levels was also documented in various kinds of cytokines (IL-6, IL-10, TNF-alpha, etc.), it is unknown whether these findings of angiotensin II are the cause or result of a systemic cytokine storm in COVID-19 patients [39]. In the context of the effects of ARBs and AECIs, a retrospective, single-center analysis enrolling 112 COVID-19 patients showed that the use of ARBs and ACEIs had no effects on the morbidity and mortality of COVID-19 patients with CVD [40]. Currently, a case-control study is underway to clarify the effects of ACEIs and ARBs on the severity of COVID-19 in Italy (NCT04318418). Most importantly, at present, there is no large-scale study with convincing evidence to determine whether ARBs and ACEIs play a neutral, beneficial, or harmful role in the susceptibility to SARS-CoV-2 and the severity and outcomes of COVID-19 patients [22, 34,35,36, 41].
Potential therapies targeting ACE2 and angiotensin II
In addition to vaccine development and antiviral agents, the modulation of the RAS, especially by ACE2 and angiotensin II, is highlighted as a potential therapeutic target for COVID-19. Based on an animal study suggesting the beneficial net effects of ARB in SARS-CoV-infected acute lung injury mice [27], multicenter, double-blinded placebo-controlled, randomized trials (RCTs) are currently being conducted to investigate the effects of losartan on mortality and hospital admission in COVID-19 patients requiring hospital admission (NCT04312009) and not requiring hospital admission (NCT04311177), respectively.
Recombinant ACE2 is another candidate to prevent COVID-19 pneumonia. As described above, the efficacy of exogenous recombinant ACE2 has been shown in animal models of acute lung injury/ARDS [30, 31]. It is also possible that the circulating soluble form of ACE2 may act as a decoy receptor that binds to SARS-CoV-2 and competitively inhibits virus entry to host target cells mediated by membrane-bound ACE2. To investigate the efficacy of human recombinant ACE2 in the treatment of COVID-19 patients, a randomized, controlled, pilot clinical study was planned (NCT04287686). However, this study was withdrawn on March 17, 2020.
TMPRSS2 is a membrane protease for ACE2 priming, which is a crucial step for the fusion of SARS-CoV-2 and target cell membranes and the resultant viral entry into the cells [6]. Nafamostat mesylate and camostat mesylate are TMPRSS2 inhibitors that have been used for the treatment of acute pancreatitis in Japan. Currently, a clinical trial evaluating the efficacy of nafamostat and camostat for treating COVID-19 is underway in Japan (https://www.u-tokyo.ac.jp/focus/en/articles/z0508_00083.html).
Risk limits of RAS inhibitor therapy in the COVID-19 pandemic
The concern regarding whether ARBs and ACEIs may have harmful effects on the morbidity and mortality of COVID-19 patients is based on the speculation that these drugs would upregulate ACE2, a receptor for SARS-CoV-2, which would increase the viral load and then augment lung injury. However, the evidence of ACE2 upregulation is limited only in animal studies using the relatively high (namely, pharmacological) doses of several ARBs and one ACEI. Additionally, it remains unclear whether the ARB/ACE-induced ACE2 increase actually enhances the susceptibility to SARS-CoV-2 or SAR-CoV. Importantly, no clinical studies exist supporting that ARBs and ACEIs augment susceptibility to infection and worsen (or improve) all-cause and cardiovascular outcomes in not only COVID-19 patients but also SARS patients at present. Therefore, the hypothesis underlying the concern would not be readily extrapolated to humans, particularly to COVID-19 patients.
In patients with heart failure, prior myocardial infarction, other CVDs, and cerebrovascular disease, the benefit of ARBs and ACEIs has been established in the improvement of quality of life and survival outcomes, while the discontinuation leads to new onset or exacerbation of heart failure, CVD, and stroke and the worsening of the prognosis [42]. Thus, RAS inhibitors should be continued in high-risk patients who have received guideline-directed medical therapy (GDMT). In general, switching from one to another class of antihypertensive agents does not infrequently result in destabilization of blood pressure control and requires frequent visits for drug titration to achieve appropriate and stable blood pressure control and to avoid adverse effects. Blood pressure destabilization increases the incidence of heart failure, stroke, and other CVDs. In addition, frequent visits to the clinic or hospital expose patients to a greater risk of infection in a COVID-19 pandemic situation. Accordingly, even in stably controlled hypertension patients without compelling indications, it is thought that not only simple discontinuation but also switching from ARB or ACEI to other antihypertensive agents should be avoided unless physicians make a decision based on specific considerations.
Cardiovascular comorbidities are common in COVID-19 patients, and such patients are at great risk for morbidity and mortality [1,2,3,4]. Myocardial damage ranging from silent troponin T elevation to fulminant myocarditis has been documented in COVID-19 patients [35]. In confirmed or suspected COVID-19 patients, ACEI and ARB medications are recommended for the treatment of heart failure and other CVD according to GDMT based on the available evidence at this time [35].
In conclusion, at present, the use of ARBs and AECIs should not be withheld in established standard therapy for not only high-risk patients such as those with heart failure, prior myocardial infarction, other CVD, and cerebrovascular disease but also patients with hypertension in stable condition according to the recent statements released by the European Society of Hypertension, the European Society of Cardiology, and the American Heart Association/Heart Failure Society of America/American College of Cardiology [43,44,45]. The evidence on COVID-19 is being accumulated each day. Large-scale clinical studies with convincing evidence, not only RCTs but also real-world big data analyses as well as experimental studies, are awaited to solve the question of whether ARBs and ACEIs have neutral, favorable, or harmful effects on the susceptibility to SARS-CoV-2 and the severity and outcomes of COVID-19.
Finally, we should learn from the lessons of COVID-19 without being bound by common sense in the ante-COVID-19 era. In addition, we should reconstruct a novel strategy for the treatment of hypertension and CVD in the post-COVID-19 era.
References
Han Q, Lin Q, Jin S, You L. Recent insights into 2019-nCoV: a brief but comprehensive review. J Infect. 2020;80(4):373–7. https://doi.org/10.1016/j.jinf.2020.02.010.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020; https://doi.org/10.1056/NEJMoa2002032.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China. JAMA. 2020; https://doi.org/10.1001/jama.2020.2648.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–3. https://doi.org/10.1038/s41586-020-2012-7.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80. https://doi.org/10.1016/j.cell.2020.02.052.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–92. https://doi.org/10.1016/j.cell.2020.02.058.
Agata J, Ura N, Yoshida H, Shinshi Y, Sasaki H, Hyakkoku M, et al. Olmesartan is an angiotensin II receptor blocker with an inhibitory effect on angiotensin-converting enzyme. Hypertens Res. 2006;29:865–74.
Wang X, Ye Y, Gong H, Wu J, Yuan J, Wang S, et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1-7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol. 2016;97:180–90.
Sukumaran V, Veeraveedu PT, Gurusamy N, Lakshmanan AP, Yamaguchi K, Ma M, et al. Olmesartan attenuates the development of heart failure after experimental autoimmune myocarditis in rats through the modulation of ANG 1-7 mas receptor. Mol Cell Endocrinol. 2012;351:208–19.
Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, Ma M, et al. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int J Biol Sci. 2011;7:1077–92.
Sukumaran V, Veeraveedu PT, Lakshmanan AP, Gurusamy N, Yamaguchi K, Ma M, et al. Olmesartan medoxomil treatment potently improves cardiac myosininduced dilated cardiomyopathy via the modulation of ACE-2 and ANG 1-7 Mas receptor. Free Radic Res. 2012;46:850–60.
Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–6.
Sukumaran V, Tsuchimochi H, Tatsumi E, Shirai M, Pearson JT. Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1-7/Mas receptor cascade. Biochem Pharm. 2017;144:90–9.
Iwanami J, Mogi M, Tsukuda K, Wang XL, Nakaoka H, Ohshima K, et al. Role of angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis in the hypotensive effect of azilsartan. Hypertens Res. 2014;37:616–20.
Lakshmanan AP, Thandavarayan RA, Watanabe K, Sari FR, Meilei H, Giridharan VV, et al. Modulation of AT-1R/MAPK cascade by an olmesartan treatment attenuates diabetic nephropathy in streptozotocin-induced diabetic mice. Mol Cell Endocrinol. 2012;348:104–11.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–10.
Soler MJ, Ye M, Wysocki J, William J, Lloveras J, Batlle D. Localization of ACE2 in the renal vasculature: amplification by angiotensin II type 1 receptor blockade using telmisartan. Am J Physiol Ren Physiol. 2009;296:F398–405.
Zhong JC, Ye JY, Jin HY, Yu X, Yu HM, Zhu DL, et al. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul Pept. 2011;166:90–7.
Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H1013–9.
Sommerstein R, Gråni C. Rapid response: preventing a Covid-19 pandemic: ACE inhibitors as a potential risk factor for fatal Covid-19. BMJ. 2020;368:m810 https://doi.org/10.1136/bmj.m810.
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin–angiotensin–aldosterone system Iinhibitors in patients with Covid-19. N Engl J Med. 2020;382:1653–9. https://doi.org/10.1056/NEJMsr2005760.
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–3.
Turner AJ, Hiscox JA, Hooper NM. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharm Sci. 2004;25:291–4.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev. 2018;98:505–53.
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. 2005;11:875–9.
Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci USA. 2004;101:15748–53.
Dijkman R, Jebbink MF, Deijs M, Milewska A, Pyrc K, Buelow E, et al. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. J Gen Virol. 2012;93:1924–9.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6.
Zou Z, Yan Y, Shu Y, Gao R, Sun Y, Li X, et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. 2014;5:3594.
Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L178–85.
Burchill LJ, Velkoska E, Dean RG, Griggs K, Patel SK, Burrell LM. Combination renin–angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci. 2012;123:649–58.
Kuster GM, Pfister O, Burkard T, Zhou Q, Twerenbold R, Haaf P, et al. SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19?. Eur Heart J. 2020; https://doi.org/10.1093/eurheartj/ehaa235.
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. Coronavirus disease 2019 (COVID-19) and cardiovascular disease. Circulation. 2020; https://doi.org/10.1161/CIRCULATIONAHA.120.046941.
Danser AHJ, Epstein M, Batlle D. Renin-angiotensin system blockers and the COVID-19 pandemic: at present there is no evidence to abandon renin-angiotensin system blockers. Hypertension. 2020; https://doi.org/10.1161/HYPERTENSIONAHA.120.15082.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–74.
Pedersen SF, Ho YC. SARS-CoV-2: a storm is raging. J Clin Investig. 2020; https://doi.org/10.1172/JCI137647.
Peng YD, Meng K, Guan HQ. Clinical characteristics and outcomes of 112 cardiovascular disease patients infected by 2019-nCoV. Zhonghua Xin Xue Guan Bing Za Zhi. 2020;48:E004.
Patel AB, Verma A. COVID-19 and angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. What is the evidence?. JAMA. 2020; https://doi.org/10.1001/jama.2020.4812.
Umemura S, Arima H, Arima S, Asayama K, Dohi Y, Hirooka Y, et al. The Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2019). Hypertens Res. 2019;42:1235–481.
ESC Council on Hypertension. Position statement of the ESC Council on hypertension on ACE-inhibitors and angiotensin receptor blockers. 2020. https://www.escardio.org/Councils/Council-on-Hypertension-(CHT)/News/position-statement-of-the-esc-council-on-hypertension-on-ace-inhibitors-and-ang.
European Society of Hypertension. Statement of the European Society of Hypertension (ESH) on hypertension, renin angiotensin system blockers and COVID-19. 2020. https://www.eshonline.org/spotlights/esh-statement-on-covid-19/.
Statement from the American Heart Association, the Heart Failure Society of America, and the American College of Cardiology. Patients taking ACE-i and ARBs who contract COVID-19 should continue treatment, unless otherwise advised by their physician. 2020. https://newsroom.heart.org/news/patients-taking-ace-i-and-arbs-who-contract-covid-19-should-continue-treatment-unless-otherwise-advised-by-their-physician.
Funding
This work was supported in part by a JSPS KAKENHI Grant (19K08503).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kai, H., Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—lessons from available evidence and insights into COVID-19. Hypertens Res 43, 648–654 (2020). https://doi.org/10.1038/s41440-020-0455-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-020-0455-8
Keywords
This article is cited by
-
SARS-CoV-2 spike protein-ACE2 interaction increases carbohydrate sulfotransferases and reduces N-acetylgalactosamine-4-sulfatase by p38 MAPK
Signal Transduction and Targeted Therapy (2024)
-
Challenges of mesenchymal stem cells in the clinical treatment of COVID-19
Cell and Tissue Research (2024)
-
The prognostic significance of insulin resistance in COVID-19: a review
Journal of Diabetes & Metabolic Disorders (2024)
-
Modulation of pulmonary immune function by inhaled cannabis products and consequences for lung disease
Respiratory Research (2023)
-
Insights into organoid-based modeling of COVID-19 pathology
Virology Journal (2023)
