
Abstract
Intellectual developmental disorder with dysmorphic facies, speech delay, and T-cell abnormalities (MIM # 618092) is a congenital disorder derived from pathogenic variants of the B-cell leukemia/lymphoma 11B gene (BCL11B). Several variants have been reported to date. Here, through comprehensive genomic analysis, a novel BCL11B truncation variant, NM_138576.4(BCL11B_v001): c.2439_2452dup [p.(His818Argfs*31)], was identified in a Japanese male patient with developmental delay, distinctive features, and early craniosynostosis.
Similar content being viewed by others


B-cell leukemia/lymphoma 11B (BCL11B; MIM* 606558) is a lineage-specific, Kruppel-like Cys2-His2 zinc-finger-containing transcriptional regulator that acts bifunctionally as a repressor or activator1. In 2016, the first case of human BCL11B alteration was reported to have severe combined immunodeficiency (SCID)2; the patient also showed intellectual impairment, spastic quadriplegia, and seizures. To date, twenty-seven patients with BCL11B alterations have been reported3. Recently, we identified a novel BCL11B truncation variant in a patient with developmental delay, distinctive features, and early craniosynostosis.
The patient was a 5-year-old Japanese boy who had been delivered via cesarean section (because of breech presentation) with birth weight, birth length, and occipitofrontal circumference (OFC) of 2680 g (10th–25th percentile), 47.8 cm (25th–50th percentile), and 33.5 cm (mean), respectively. The pregnancy was uncomplicated, and there was no asphyxia at birth. Newborn screening for metabolic disorders was negative. There was no history of a compromised infection. His parents were nonconsanguineous, and there was no family history of congenital anomalies. He had sucking difficulties due to his small mouth, although his body weight gain was normal. He achieved head control at 4 months, although he could not maintain his trunk in the prone position and could not roll over at 6 months. His mother brought him to our hospital with suspected gross motor movement delay at 6 months.
At 6 months of age, his body weight, length, and OFC were 7645 g (25th–50th percentile), 68.8 cm (50th–75th percentile), and 43.0 cm (25th–50th percentile), respectively. He showed distinctive features, including prominent forehead, arched eyebrows, midface hypoplasia, thin upper lip, small mouth, long philtrum, retrognathia, and low-set ears (Fig. 1a–d). No abnormal findings were observed in the chest or abdomen. Neurological examination revealed hypotonia with scarf signs. There were no abnormal data in routine blood examinations, including liver function, thyroid function, lactate/pyruvate, and amino acid profiles. Metabolomic analysis of urine also showed normal results. Conventional chromosomal G-banding revealed a normal male karyotype (46,XY). Skull 3D-computed tomography (CT) images at 10 months revealed partial early fusion of the sagittal and lambda sutures (Fig. 1e). Surgical cranioplasty was performed twice, at 12 and 18 months. Brain magnetic resonance imaging (MRI) at 11 months showed a slightly reduced volume of the cerebrum but no abnormal signals (Fig. 1f, g).

Facial appearance of the patient at 6 months of age (a, b) and 5 years of age (c, d), suggesting deformity of the skull, forehead protrusion, arched eyebrows, flat nose base, thin upper lip, small mouth, long philtrum, retrognathia and low-set ears. e 3-dimensional CT image of the skull at 10 months of age shows partial early fusion of the sagittal and lambda sutures (arrows). T1-(f) and T2-(g)-weighted brain MRI at 11 months of age showed no apparent abnormalities.
His motor development was delayed, with rolling over occurring at 8 months, sitting without support at 10 months, crawling at 11 months, and walking alone at 28 months. In addition, his language development was severely delayed, with no speaking or mimicking. Furthermore, he exhibited autistic behaviors at three years of age. He started baby food at 5 months of age, although he had difficulty feeding using a spoon due to sensitivity around the mouth. Thus, he consumed baby food dissolved with formula using a baby bottle. He was allergic to eggs, milk, wheat, and soy. He started oral rehabilitation and was gradually able to eat food with a spoon at 3 years of age. He had no history of recurrent infection, and his laboratory data, including immunoglobin; CD4/CD8 ratio; and CD3, CD10, CD19, and CD20 profiles, were normal.
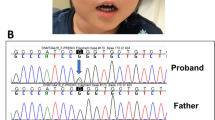
For precise diagnosis, this patient was enrolled in the research project “Initiative on Rare and Undiagnosed Disorders (IRUD)”, which was performed in accordance with the Declaration of Helsinki and approved by the ethics committee of our institution4. After obtaining informed consent from the family, blood samples were collected from the patient and his parents. Genomic DNA was extracted from the peripheral blood samples following a standard protocol, and exome sequencing was performed as described previously using trio samples, including parental samples, at 3 years and 11 months of age5. The results showed a de novo heterozygous variant of NM_138576.4(BCL11B_v001):c.2439_2452dup [p.(His818Argfs*31)]. Standard Sanger sequencing confirmed this finding (Fig. 2a). The identified variant has not been reported previously and is not registered in any database. The clinical information of this patient is summarized in Table 1 together with previous reports2,3,6,7,8,9,10,11,12,13,14,15.

a Sanger sequencing indicates overlapping electropherograms due to the 14-bp duplication. b Locations of the BCL11B variants are depicted on the primary structure (modified data from Prasad et al., 2020). The positions of the previously reported BCL11B variants are shown in black. The variant reported in this study is shown in red (in the 4th zinc-finger motif). Most of the variants are located on exon 4. Circles, rhombuses, triangles, and rectangles indicate missense, truncation, nonsense, and splicing mutations, respectively. NH3 N-terminus, COOH C-terminus.
BCL11B plays an essential role in the development of the nervous, immune, and cardiovascular systems and is also involved in skin, dental, and cranial development16. In murine models, biallelic loss of Bcl11b leads to defects in the development of the central nervous system17,18, epidermis19, and teeth3,20, as well as in the development and maintenance of the T-cell lineage21, resulting in perinatal lethality. The role of Bcl11b in craniofacial skeleton formation has also been reported in murine models22,23; however, it is less common in humans.
The first reported case of a BCL11B variant, p.N441K, exhibited SCID associated with neurological features2. The identified variant is located in the zinc-finger C2H2-type domain. Thus, a dominant-negative effect on the DNA-binding structural interface was suggested. A subsequent report included 27 additional patients with BCL11B alterations, including our patient (Table 1). In total, 21 variants have been reported, including frameshift variants (9), missense variants (9), nonsense variants (2), and a splicing variant (1) (Fig. 2b). Patients harboring missense variants may show the most severe clinical findings, which also supports the mechanism of dominant-negative effects. However, variants leading to protein truncation (frameshift and nonsense) were shared by more than half of patients. Truncation variants are predicted to activate nonsense-mediated mRNA decay, resulting in haploinsufficiency. However, 10 truncation variants are located in the last exon (exon 4) and are thus predicted to escape nonsense-mediated mRNA decay, probably resulting in a protein with loss of C-terminal DNA-binding zinc-finger domains3.
BCL11B is located on human chromosome 14p32.2 and consists of four exons, and it encodes five zinc-finger C2H2-type domains. Three of them were located in the C-terminal region of the last exon (Fig. 2b). BCL11B has an amino acid length of 894, and over half of the amino acids are encoded by exon 4 (Fig. 2b). Therefore, most truncation variants are predicted to cause the loss of the last C-terminal DNA-binding zinc-finger domains.
In this study, we identified a unique 14-bp tandem duplication in exon 4, which was predicted to be involved in protein truncation, as discussed above. This patient showed severe developmental delay, autistic features, and distinctive findings of craniosynostosis, which was repaired by cranioplasty in early infancy.
Craniosynostosis is a disorder of skull formation caused by premature ossification of cranial sutures, occurring in ~1 in 2250 births6. Although the first two reports of BCL11B alterations showed no findings of craniosynostosis, five patients have been reported to show craniosynostosis in association with BCL11B alterations, suggesting a relatively less common manifestation in humans6,11.
Genetic alterations have been identified in approximately a quarter of craniosynostosis cases. The most frequent activating mutations are in genes coding for FGF receptors24,25, as well as for other genes, including the twist basic helix-loop-helix transcription factor 1 gene (TWIST1) and the ephrin-B2 gene (EFNB). Owing to advances in comprehensive genetic testing, several BCL11B alterations have recently been reported in children with craniodiaphyseal abnormalities. Bcl11b regulates FGF-dependent signaling pathways. Analysis of Bcl11b-deficient mice and neural crest-specific inactivation (Bcl11b ncc–/–) mice revealed its role in craniofacial development and maintenance of suture patency22,23. Furthermore, Bcl11b is highly expressed in the mesenchyme of the murine craniofacial skeleton during embryonic development22,23.
The variants of the five reported cases of craniosynostosis were missense variants (p.Arg3Ser, p.Gly667Glu, p.Gly582Ser, p.Pro673Arg, and p.Pro422Leu), and four of the five variants were located in exon 46,11. Gaillard et al.11 hypothesized that these missense variants may either have a dominant-negative effect or induce new target genes, thereby causing a more severe phenotype in patients with these missense variants. These findings indicate that truncation variants usually cause loss of function, but the variant identified in this study may have a dominant-negative effect.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3255.
References
Satterwhite, E. et al. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood 98, 3413–3420 (2001).
Punwani, D. et al. Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N. Engl. J. Med. 375, 2165–2176 (2016).
Lessel, D. et al. BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain 141, 2299–2311 (2018).
Takahashi, Y. et al. Six years’ accomplishment of the Initiative on rare and undiagnosed diseases: nationwide project in Japan to discover causes, mechanisms, and cures. J. Hum. Genet. 67, 505–513 (2022).
Yamamoto-Shimojima, K. et al. Elucidation of the pathogenic mechanism and potential treatment strategy for a female patient with spastic paraplegia derived from a single-nucleotide deletion in PLP1. J. Hum. Genet. 64, 665–671 (2019).
Goos, J. A. C. et al. A de novo substitution in BCL11B leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum. Mol. Genet. 28, 2501–2513 (2019).
Qiao, F. et al. A de novo heterozygous frameshift mutation identified in BCL11B causes neurodevelopmental disorder by whole exome sequencing. Mol. Genet. Genom. Med. 7, e897 (2019).
Homma, T. K. et al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J. Pediatr. 215, 192–198 (2019).
Prasad, M. et al. BCL11B-related disorder in two canadian children: expanding the clinical phenotype. Eur. J. Med. Genet. 63, 104007 (2020).
Yang, S. et al. Mutant BCL11B in a patient with a neurodevelopmental disorder and T-cell abnormalities. Front. Pediatr. 8, 544894 (2020).
Gaillard, L. et al. Case report and review of the literature: congenital diaphragmatic hernia and craniosynostosis, a coincidence or common cause? Front. Pediatr. 9, 772800 (2021).
Lu, H. Y. et al. A novel germline heterozygous BCL11B variant causing severe atopic disease and immune dysregulation. Front. Immunol. 12, 788278 (2021).
Alfei, E. et al. Progressive clinical and neuroradiological findings in a child with BCL11B missense mutation: expanding the phenotypic spectrum of related disorder. Neuropediatrics 53, 283–286 (2021).
Harrer, P. et al. A de novo BCL11B variant case manifesting with dystonic movement disorder regarding the article “BCL11B-related disorder in two canadian children: expanding the clinical phenotype (Prasad et al., 2020). Eur. J. Med. Genet. 65, 104635 (2022).
Che, F. et al. Identification of two novel variants of the BCL11B gene in two Chinese pedigrees associated with neurodevelopmental disorders. Front. Mol. Neurosci. 15, 927357 (2022).
Lennon, M. J., Jones, S. P., Lovelace, M. D., Guillemin, G. J. & Brew, B. J. Bcl11b-A critical neurodevelopmental transcription factor-roles in health and disease. Front. Cell Neurosci. 11, 89 (2017).
Arlotta, P. et al. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45, 207–221 (2005).
Simon, R. et al. A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. Embo J. 31, 2922–2936 (2012).
Golonzhka, O. et al. Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and permeability barrier formation. J. Invest. Dermatol. 129, 1459–1470 (2009).
Golonzhka, O. et al. Ctip2/Bcl11b controls ameloblast formation during mammalian odontogenesis. Proc. Natl Acad. Sci. USA 106, 4278–4283 (2009).
Wakabayashi, Y. et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat. Immunol. 4, 533–539 (2003).
Kyrylkova, K., Iwaniec, U. T., Philbrick, K. A. & Leid, M. BCL11B regulates sutural patency in the mouse craniofacial skeleton. Dev. Biol. 415, 251–260 (2016).
Holmes, G., van Bakel, H., Zhou, X., Losic, B. & Jabs, E. W. BCL11B expression in intramembranous osteogenesis during murine craniofacial suture development. Gene Expr. Patterns 17, 16–25 (2015).
Cunningham, M. L., Seto, M. L., Ratisoontorn, C., Heike, C. L. & Hing, A. V. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod. Craniofac. Res. 10, 67–81 (2007).
Purushothaman, R., Cox, T. C., Maga, A. M. & Cunningham, M. L. Facial suture synostosis of newborn Fgfr1(P250R/+) and Fgfr2(S252W/+) mouse models of Pfeiffer and Apert syndromes. Birth Defects Res A Clin. Mol. Teratol. 91, 603–609 (2011).
Acknowledgements
We appreciate the cooperation of the patient and his parents for this study. This work was supported by the Initiative on Rare and Undiagnosed Diseases (grant number 22ek0109549(h0002)) from the Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Contributions
T.Yama. designed this study. K.E. organized the study and drafted the manuscript. O.M., T.Yana., K.C., Y.Ai., and Y.H. contributed to acquisition of clinical information. K.S.Y., M.N., Y.I., Y.M., and Y.As. conducted data analysis. S.N. and T.Yama. critically reviewed the manuscript. All authors contributed to the analysis and interpretation of data. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
The consent to use the photos of the child was obtained from the child’s guardian. The photos are never used for anything other than medical purposes.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eto, K., Machida, O., Yanagishita, T. et al. Novel BCL11B truncation variant in a patient with developmental delay, distinctive features, and early craniosynostosis. Hum Genome Var 9, 43 (2022). https://doi.org/10.1038/s41439-022-00220-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00220-x
