
Abstract
The molecular mechanisms involved in thyroid organogenesis have not been fully elucidated. We report a patient with a de novo germline AKT3 variant, NM_005465.7:c.233A > G, p.(Gln78Arg), who presented with congenital hypothyroidism in addition to typical AKT3-related brain disorders. The report of this patient contributes to delineating the associated yet uncertain endocrine complications of this AKT3 disease-causing variant.
Similar content being viewed by others


Primary congenital hypothyroidism (CH) is the most common form of neonatal endocrine disease. Although recent advances in genetic technologies have identified CH-causing variants in several genes, the molecular mechanisms involved in thyroid organogenesis have not been fully elucidated1.
AKT proteins are a family of serine-threonine kinases that are central molecules of the phosphatidylinositol-3-kinase signaling pathway. They play critical roles in key cellular functions, such as cell cycle progression, proliferation, apoptosis, and metabolism2. AKT3 is expressed at high levels in the brain and endocrine tissues, especially in the thyroid. Recently, the clinical characteristics of AKT3-related brain disorders have been reported3. Somatic (mosaic) activating mutations in AKT3 cause focal or segmental brain malformations, such as focal cortical dysplasia and hemimegalencephaly, whereas germline-activating mutations cause megalencephaly and diffuse bilateral cortical malformations3. Given the major role of AKT3 in cellular signal transduction, it is understandable that mutations in this gene may cause systemic disease. However, endocrine disorders have not been reported, except for hypoglycemia in a few patients3,4. Here, we report a case of a patient with a de novo germline AKT3 variant, NM_005465.7:c.233A > G, p.(Gln78Arg), who presented with CH in addition to typical AKT3-related brain disorders.
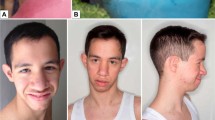
A 39-year-old female was referred to our prenatal diagnosis center at 27 gestational weeks for fetal macrocephaly with cortical maldevelopment, as indicated by transvaginal 3D neurosonographic findings. Fetal magnetic resonance imaging (MRI) revealed that the fetus had asymmetrical megalencephaly with cortical dysplasia (Fig. 1A). Genomic DNA was extracted from amniotic fluid cells and parental lymphocytes using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Trio-based targeted exome sequencing was performed with the TruSight One Expanded Sequencing Panel (6704 genes) according to the manufacturer’s instructions (Illumina Inc., CA, USA). The prepared library was sequenced in 151 bp paired-end mode on an Illumina MiSeq (Illumina Inc., CA, USA). Sequencing reads were aligned and processed as previously reported5. Plausible variants were assessed according to the American College of Medical Genetics (ACMG) guidelines6. We identified a de novo heterozygous variant in AKT3, NM_005465.7:c.233A > G, p.(Gln78Arg), which was confirmed by Sanger sequencing (Fig. 1B). As this variant was not recorded in the public variant database, we concluded that this is a novel variant that is likely pathogenic (PS2, PM2, PP2, and PP3) based on ACMG guidelines. At 35 gestational weeks, 1500 mL amniotic fluid was drained because of polyhydramnios. The patient was delivered by cesarean section at 37 gestational weeks. His birth weight, height, and head circumference were 3529 g (+2.50 standard deviation (SD)), 49.0 cm (+1.24 SD), and 43.5 cm (+8.19 SD), respectively. He had a 1 min Apgar score of 2 and a 5 min Apgar score of 5. Soon after birth, he was intubated because of respiratory distress. The patient was treated with dopamine, dobutamine, and hydrocortisone to maintain blood pressure. His condition was stable, and he was extubated on the fifth day after birth. Sanger sequencing confirmed the presence of a heterozygous AKT3 c.233A > G variant in peripheral blood lymphocytes. MRI of the brain showed asymmetrical megalencephaly with bilateral polymicrogyria (MEG-PMG) (Fig. 2A, B), which are typical presentations of germline-activating AKT3 variants. The newborn screening revealed elevated levels of thyroid-stimulating hormone (TSH), which suggested hypothyroidism. On the 12th day after birth, a thyroid function test was performed. The results showed the elevation of TSH (1841 µIU/mL) and low levels of thyroid hormones (FT4 0.06 ng/dL, FT3 0.45 pg/mL), leading to the diagnosis of primary hypothyroidism. The breadth (cm), depth (cm), and length (cm) of the bilateral thyroid lobes were measured using thyroid ultrasound (right lobe: 0.57, 0.34, 0.92; left lobe: 0.85, 0.19, 1.22) (Fig. 2C). Thyroid volume was calculated using the following formula: thyroid volume (mL) = length (cm) × breadth (cm) × depth (cm) × π/67. The volumes of the right thyroid and left thyroid were 0.093 mL (range (mL): 0.3–1.4) and 0.103 mL (range (mL): 0.4–1.3), respectively. Thyroid scintigraphy using 99mTc pertechnetate did not reveal any functional thyroid tissue (Fig. 2D). Consequently, he was diagnosed with congenital hypothyroidism due to thyroid hypogenesis. L-T4 supplementation was initiated, and his blood levels of thyroid hormone normalized.

A Fetal magnetic resonance imaging (MRI) revealed that the fetus had asymmetrical megalencephaly with cortical dysplasia. B The chromatogram represents a de novo heterozygous c.233A > G (p.Gln78Arg) variant in the AKT3 gene.

A, B MRI of the brain showed asymmetrical megalencephaly with bilateral polymicrogyria. C Thyroid ultrasound examination revealed that both the right and left thyroid lobes were small. D Thyroid scintigraphy by 99mTc pertechnetate showed no functional thyroid gland.
We report the case of a male patient with a novel AKT3 variant, p.Gln78Arg, which localizes to the pleckstrin homology domain. Most previously reported germline AKT3 missense variants causing MEG-PMG are within the pleckstrin homology domain or the catalytic kinase domain and result in elevated kinase activity3. Although functional studies to confirm the pathogenic effect of this variant has not been performed, the presence of typical brain malformations strongly indicates abnormal AKT3 function. He also presented with thyroid dysgenesis. His initial thyroid function test clearly showed primary hypothyroidism, and a thyroid ultrasound examination revealed that his thyroid was small. In addition, thyroid scintigraphy showed no functional thyroid glands. Thus, he was diagnosed with congenital hypothyroidism due to thyroid hypogenesis. To the best of our knowledge, this is the first report to describe a case of an AKT3-related brain disorder associated with thyroid hypogenesis.
The prevalence of thyroid dysgenesis is approximately one in 5000 live births. In Japan, most of these cases have ectopic thyroid (73%), while the rest have thyroid agenesis (12%) and thyroid dysgenesis (15%)8. The pathogenesis of thyroid dysgenesis remains unknown. Thyroid dysgenesis is generally considered sporadic. However, the frequency of familial cases is more than 15 times that of the general population, indicating that genetic factors could be associated with thyroid dysgenesis9. There are some transcription factors involved in thyroid genesis. Hhex is ubiquitously expressed in the foregut endoderm and drives the budding process. Fgf2 and Bmp4, which are derived from the cardiogenic mesoderm, induce thyroid progenitors from the foregut endoderm10. Thyroid progenitors express Nkx2-1 and Pax8 and regulate folliculogenesis. FOXE1 is expressed in thyroid follicular cells and plays an important role in thyroid development. Consequently, variants related to these transcription factors induce thyroid dysgenesis. No likely pathogenic variants of these genes related to thyroid dysgenesis were detected in this patient.
AKT is a serine/threonine-protein kinase that is involved in multiple cellular processes, such as glucose metabolism, apoptosis, cell proliferation, transcription, and cell migration. There are three isoforms of AKT: AKT1, AKT2, and AKT3. AKT3 is a candidate gene for thyroid dysgenesis and dysfunction, as AKT3 is highly expressed in the thyroid gland11. Iwadate et al. showed that NKX2-1 acts on thyroid development via AKT signaling in a mouse model, suggesting the possibility that the phosphorylation of AKT is associated with early stages of thyroid differentiation12. The detailed mechanism by which AKT3 is involved in the development of the thyroid gland is unknown. Reactive oxygen species (ROS) induce apoptosis in embryonic tissue, and a fine balance in ROS generation is required for embryonic and fetal development during the gestational period. In addition, ROS plays a pivotal role in the development and function of the thyroid13. The double knockout of AKT1 and AKT2 in mouse embryonic fibroblasts decreases ROS levels, suggesting that AKT increases ROS production14. Based on the evidence mentioned above, accelerated AKT activity due to AKT3 mutations might increase the production of ROS, resulting in the inhibition of thyroid organogenesis.
In summary, the report of this patient contributes to delineating the associated yet uncertain endocrine complication of this AKT3 disease-causing variant. However, there is the possibility of the incidental occurrence of hypothyroidism due to other causative genes for thyroid genesis that were not covered by the panel sequencing. To elucidate the relationship between AKT3 variants and thyroid dysgenesis, further cases are needed.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3195.
References
Targovnik, H. M., Scheps, K. G. & Rivolta, C. M. Defects in protein folding in congenital hypothyroidism. Mol. Cell Endocrinol. 501, 110638 (2020).
Toker, A. & Marmiroli, S. Signaling specificity in the Akt pathway in biology and disease. Adv. Biol. Regul. 55, 28–38 (2014).
Alcantara, D. et al. Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain 140, 2610–2622 (2017).
Nellist, M. et al. Germline activating AKT3 mutation associated with megalencephaly, polymicrogyria, epilepsy and hypoglycemia. Mol. Genet. Metab. 114, 467–473 (2015).
Pooh, R. K. et al. Fetal megalencephaly with cortical dysplasia at 18 gestational weeks related to paternal UPD mosaicism with PTEN mutation. Genes (Basel). 12, 358 (2021).
Richards, S. et al. ACMG laboratory quality assurance committee. Genet Med. 17, 405–424 (2015).
Perry, R., Hollman, A., Wood, A. & Donaldson, M. Ultrasound of the thyroid gland in the newborn: normative data. Arch. Dis. Child Fetal Neonatal Ed. 87, F209–211 (2002).
Narumi, S., Muroya, K., Asakura, Y., Adachi, M. & Hasegawa, T. Transcription factor mutations and congenital hypothyroidism: systematic genetic screening of a population-based cohort of Japanese patients. J. Clin. Endocrinol. Metab. 95, 1981–1985 (2010).
Castanet, M. et al. Nineteen years of national screening for congenital hypothyroidism: familial cases with thyroid dysgenesis suggest the involvement of genetic factors. J. Clin. Endocrinol. Metab. 86, 2009–2014 (2001).
Nilsson, M. & Fagman, H. Development of the thyroid gland. Development 144, 2123–2140 (2017).
Camargo, R. Y. et al. Histopathological characterization and whole exome sequencing of ectopic thyroid: fetal architecture in a functional ectopic gland from adult patient. Int. J. Endocrinol. 2018, 4682876 (2018).
Iwadate, M., Takizawa, Y., Shirai, Y. T. & Kimura, S. An in vivo model for thyroid regeneration and folliculogenesis. Lab Invest. 98, 1126–1132 (2018).
Li, M. et al. Nicotinamide nucleotide transhydrogenase mutation analysis in Chinese patients with thyroid dysgenesis. Am. J. Med. Genet A. 188, 89–98 (2022).
Nogueira, V. et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 14, 458–470 (2008).
Acknowledgements
We thank Editage (http://www.editage.com) for proofreading, editing, and reviewing this manuscript.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
J.M., T.H., Y.M., K.K., H.M., and T.C. attended to the patient. R.K.P. performed the gene analysis. J.M., R.K.P., and T.C. wrote the manuscript. J.M., T.T., R.K.P., and T.C. gave conceptual advice. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mori, J., Hasegawa, T., Miyamoto, Y. et al. Thyroid hypogenesis is associated with a novel AKT3 germline variant that causes megalencephaly and cortical malformation. Hum Genome Var 9, 18 (2022). https://doi.org/10.1038/s41439-022-00197-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00197-7
