
Abstract
Leigh syndrome is the most genetically heterogenous phenotype of mitochondrial disease. We describe a patient with Leigh syndrome whose diagnosis had not been confirmed because of normal metabolic screening results at the initial presentation. Whole-exome sequencing identified pathogenic variants in NARS2, the gene encoding a mitochondrial asparaginyl-tRNA synthetase. One of the biallelic variants was novel. This highlights the essential role of genetic testing for a definite diagnosis of Leigh syndrome.
Similar content being viewed by others


Leigh syndrome (MIM 516060) is the most common childhood mitochondrial disorder and has an estimated prevalence of 1 per 40,000 live births1. This neurodegenerative disorder is genetically heterogeneous, and approximately 100 causative genes have been identified in either the mitochondrial or the nuclear genomes to date2. Increased lactate is an important biochemical marker in the diagnosis of patients with suspicion of mitochondrial disorders. Here, we report a case of a 24-year-old female with Leigh syndrome whose diagnosis had not been confirmed because she had normal blood and cerebrospinal fluid (CSF) lactate concentrations at presentation. Whole-exome sequencing (WES) analysis identified biallelic pathogenic variants in NARS2 (MIM 612803). The gene is located on 11q14.1 and encodes mitochondrial asparaginyl-tRNA synthetase 2. The enzyme catalyzes the ligation of asparagine to tRNA molecules, so it plays a critical role in protein biosynthesis3. To date, 18 different NARS2 disease-causing variants have been described in 22 affected patients4,5,6,7,8,9,10,11,12,13. This report aimed to better understand the phenotypic variability of NARS2-associated disease and the potential diagnostic pitfall.
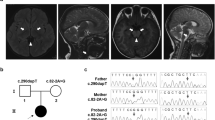
The patient, now aged 24 years, was the first child of healthy, nonconsanguineous Japanese parents, and her sister was healthy. The patient was born at 40 weeks gestation after an uneventful pregnancy. Her birth weight, length, and head circumference were 2700 g (−0.9 SD), 48 cm (−0.8 SD), and 30 cm (−2.6 SD), respectively. No signs of perinatal distress were observed. Her psychomotor development was normal until one year of age; she acquired head control, sat without support, and stood with assistance by three months, seven months, and by 12 months of age, respectively. She suffered from a urinary tract infection at one year and one month of age and developed generalized tonic and clonic seizures. Thereafter, she exhibited developmental regression, which she lost her ability to stand and maintain a sitting position by one year and five months. Brain magnetic resonance imaging (MRI) at one year and seven months demonstrated symmetrical faint high signal lesions in the putamen (Fig. 1a). The patient developed generalized tonic and myoclonic seizures at one year and eight months of age. Electroencephalography showed generalized spike-waves. The antiepileptic drug valproic acid (VPA) was administered, but she had a cluster of tonic seizures. At the age of two years and three months, she was referred to our hospital. Neurological examination revealed muscle hypotonia and a profound delay in psychomotor development. She also showed involuntary movements, such as chorea and intermittent opisthotonos posturing. The auditory brainstem response test revealed severe bilateral auditory impairment. Laboratory examinations revealed normal lactate levels in both the blood and CSF (12 mg/dl and 10 mg/dl, respectively). Follow-up MRIs revealed symmetrical high signal intensity lesions involving the putamen and periaqueductal gray matter, which are characteristic of Leigh syndrome (Fig. 1). However, single-voxel proton magnetic resonance spectroscopy (MRS) obtained from the abnormal regions in the basal ganglia did not show a lactate peak (Supplementary Fig. 1). At the age of three years, she developed VPA-induced Fanconi syndrome that was detected incidentally with findings such as hypophosphatemia, metabolic acidosis with a normal anion gap, glycosuria, and generalized hyperaminoaciduria. However, these findings were normalized after VPA administration was stopped. During infections, the patient was repeatedly found to have liver dysfunction (aspartate aminotransferase, up to 469 U/L; and alanine aminotransferase, up to 278 U/L), which was coincident with increased lactate and pyruvate levels in blood (lactate, up to 46.0 mg/dl; pyruvate, up to 2.1 mg/dl). In the following years, the patient exhibited a slowly progressive clinical course. Her psychomotor development was severely delayed without head control, eye pursuit, or the use of meaningful words. Because of repeated aspiration pneumonia owing to feeding and swallowing difficulties, she was fed via a gastrostomy tube and required tracheostomy tube placement.

Note the bilateral symmetric high signal lesions involving the dorsal putamen (arrows, a–c) and diffuse progressive cerebral atrophy at the age of 1.7 (a, d), 2.3 (b, e), and 3.3 (c, f) years old. Brain stem lesions become apparent at the age of 3.3 years (arrows, f).
To reveal the underlying genetic etiology of Leigh syndrome, WES analysis was conducted for the patient and her asymptomatic parents after written informed consent was obtained from her parents. The study protocol was approved by the Committee for Ethical Issues at Asahikawa Medical University (approval number 17112). WES analysis revealed that the patient harbored compound heterozygous variants in NARS2, with a paternally inherited NM_024678:c.731 C > G (p.Ala244Gly) variant and a maternally inherited NM_024678:c.556 A > G (p.Asn186Asp) variant. These variants were confirmed by Sanger sequencing (Fig. 2a). Both variants occurred at the catalytic domain of NARS2 (Fig. 2b, c). The paternal variant, p.Ala244Gly, has been reported in a patient with Leigh syndrome8. This variant was predicted to be “probably damaging” with a score of 0.983 by PolyPhen-2 and “deleterious” with a score of 0.001 by SIFT. The maternal variant, p.Asn186Asp, was not registered in the Genome Aggregation Database (http://gnomad.broadinstitute.org) or Human Genome Mutation Database (http://www.hgmd.cf.ac.uk/). The variant was predicted to be “probably damaging” with a score of 0.999 by PolyPhen-2 and “deleterious” with a score of 0.001 by SIFT. Based on the American College of Medical Genetics and Genomics standards and guidelines, these variants were classified as likely pathogenic.

a Partial sequence chromatograms for NARS2 in the patient and her parents. The patient harbors the biallelic heterozygous variants, with a paternally inherited NM_024678:c.731 C > G (p.Ala244Gly) variant and a maternally inherited NM_024678:c.556 A > G (p.Asn186Asp) variant. b Schematic illustration of NARS2 at the DNA level. c Schematic illustration of NARS2 at the protein level. Both variants occur at the catalytic domain. Amino acid residues were numbered according to the GenBank reference sequence (NP_078954).
Diagnosis of Leigh syndrome is made based on the neuropathological or neuroradiological findings of bilateral symmetrical lesions within the brainstem and basal ganglia structures. These lesions must be accompanied by elevated levels of lactate in the blood or CSF, indicating abnormal energy metabolism1. Lactic acidosis is a hallmark of all mitochondrial diseases, but is neither invariably present nor necessarily severe14. Among the previously reported 22 cases with NARS2-associated disease, at least seven patients showed normal lactate levels at presentation4,5,7,10,12,13. Our case also showed normal lactate levels in both the blood and CSF, but elevated levels in catabolic states such as vomiting, diarrhea, and fever. Recently, several serum biomarkers such as growth differentiation factor 15 and fibroblast growth factor 21 have been reported as diagnostic indicators of mitochondrial diseases; however, a lack of validated biomarkers for diagnosing mitochondrial diseases has not been resolved15. A definite diagnosis is only possible with genetic confirmation. Early diagnosis is crucial for optimizing care; avoidance of the drugs (e.g., VPA) metabolized by the mitochondria is recommended. VPA can contribute to mitochondrial dysfunction in proximal renal tubular cells and may cause Fanconi syndrome in patients with mitochondrial diseases16. Recognition of these clinical characteristics may facilitate the early diagnosis and proper treatment of patients with Leigh syndrome, improve their long-term outcomes, and help adapt appropriate genetic counseling.
Data availability
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3160, https://doi.org/10.6084/m9.figshare.hgv.3163.
References
Rahman, S. et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol. 39, 343–351 (1996).
Rahman, J., Noronha, A., Thiele, I. & Rahman, S. Leigh map: a novel computational diagnostic resource for mitochondrial disease. Ann. Neurol. 81, 9–16 (2017).
Rajendran, V., Kalita, P., Shukla, H., Kumar, A. & Tripathi, T. Aminoacyl-tRNA synthetases: structure, function, and drug discovery. Int J. Biol. Macromol. 111, 400–414 (2018).
Simon, M. et al. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 11, e1005097 (2015).
Vanlander, A. V. et al. Two siblings with homozygous pathogenic splice-site variant in mitochondrial asparaginyl-tRNA synthetase (NARS2). Hum. Mutat. 36, 222–231 (2015).
Sofou, K. et al. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genom. Med. 3, 59–68 (2015).
Mizuguchi, T. et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J. Hum. Genet. 62, 525–529 (2017).
Lee, J. S. et al. Genetic heterogeneity in Leigh syndrome: highlighting treatable and novel genetic causes. Clin. Genet. 97, 586–594 (2020).
Palombo, F. et al. Autozygosity-driven genetic diagnosis in consanguineous families from Italy and the Greater Middle East. Hum. Genet. 139, 1429–1441 (2020).
Seaver, L. H. et al. Lethal NARS2-related disorder associated with rapidly progressive intractable epilepsy and global brain atrophy. Pediatr. Neurol. 89, 26–30 (2018).
Sofou, K., Kollberg, G., Hedberg-Oldfors, C. & Oldfors, A. The phenotypic variability and natural history of NARS2 associated disease. Eur. J. Paediatr. Neurol. 31, 31–37 (2021).
Vafaee-Shahi, M. et al. Novel phenotype and genotype spectrum of NARS2 and literature review of previous mutations. Ir. J. Med. Sci. 2021.
Štěrbová, K. et al. Novel variants in the NARS2 gene as a cause of infantile-onset severe epilepsy leading to fatal refractory status epilepticus: case study and literature review. Neurogenetics 22, 359–364 (2021).
DiMauro, S., Schon, E. A., Carelli, V. & Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 9, 429–444 (2013).
Davis, R. L., Liang, C. & Sue, C. M. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 86, 2010–2015 (2016).
Niaudet, P. & Rotig, A. The kidney in mitochondrial cytopathies. Kidney Int. 51, 1000–1007 (1997).
Acknowledgements
This work was supported in part by a grant from the Initiative on Rare and Undiagnosed Diseases in Pediatrics (IRUD-P) project of the Japan Agency for Medical Research and Development (AMED). We thank the participants and their families for their cooperation in this study. We would like to thank Editage (www.editage.com) for the English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tanaka, R., Takeguchi, R., Kuroda, M. et al. Novel NARS2 variant causing leigh syndrome with normal lactate levels. Hum Genome Var 9, 12 (2022). https://doi.org/10.1038/s41439-022-00191-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00191-z
This article is cited by
-
Novel NARS2 variants in a patient with early-onset status epilepticus: case study and literature review
BMC Pediatrics (2024)
-
Valproic acid
Reactions Weekly (2022)

{kind=link}