
Abstract
We report the first case of classic lipoid congenital adrenal hyperplasia and combined pituitary hormone deficiency. We identified pathogenic variants in the STAR gene: a novel variant of c.126_127delCCinsG, namely, p.Thr44Profs*2 and an already reported variant of c.634C>T, namely, p.Gln212*. The association with combined pituitary hormone deficiency might be just a coincidence.
Similar content being viewed by others
Lipoid congenital adrenal hyperplasia (LCAH; OMIM 201710) is an autosomal recessive disease caused by biallelic pathogenic variants in the STAR gene1. STAR is expressed in steroidogenic cells in the adrenals and gonads, facilitating transport of cholesterol from the outer to inner mitochondrial membrane, where the first step in steroidogenesis commences. LCAH is endocrinologically characterized by impairment of all classes of steroidogenesis in adrenals and gonads. LCAH is composed of the classic type and nonclassic type2. Patients with classic LCAH develop early infantile-onset primary adrenal insufficiency. 46,XY patients show complete female external genitalia, and 46,XX patients achieve thelarche and menarche spontaneously, although menstrual cycles are often anovulatory and menopause occurs early. Patients with nonclassic LCAH develop relatively late-onset primary adrenal insufficiency without any episode of salt loss. 46,XY patients showed complete male external genitalia, and 46,XX patients can achieve spontaneous pregnancy and delivery3,4.
Combined pituitary hormone deficiency (CPHD) is a congenital condition that causes deficiencies in more than two hormones produced by the pituitary gland. Approximately 30 genes have been identified as responsible for CPHD5. We report, for the first time, a Japanese female with classic LCAH and CPHD. We identified a novel variant of c.126_127delCCinsG, namely, p.Thr44Profs*2 and an already reported mutant of c.634C>T, p.Gln212* in the STAR gene.
A Japanese female had frequent vomiting 1 week after birth and generalized seizures at the age of 3 months. Reportedly, she was clinically diagnosed with primary adrenal insufficiency, although her medical record was discarded. Glucocorticoid and mineralocorticoid supplementation therapy was started. At 2 years of age, she had generalized skin pigmentation. Her karyotype was 46,XX. Her endocrinological data are summarized in Table 1 and were compatible with primary adrenal insufficiency. At the age of 12 years, she presented with menarche, with no subsequent menstruation. At the first visit to our hospital at the age of 35 years, her height was 147 cm (−2.0 SD), and her body weight was 95 kg (+5.4 SD). She had taken daily doses of hydrocortisone 15 mg, fludrocortisone 0.1 mg, and dexamethasone 0.5 mg. On examination, her blood pressure was 135/85 mmHg. Her breast and pubic hair were Tanner I. The intelligence quotient score was 45. She had right low-frequency sensorineural hearing loss between 20 and 40 dB. Abdominal plain CT revealed a slightly atrophic uterus and seemingly normal ovaries. Pituitary magnetic resonance imaging revealed that her pituitary was slightly smaller than that of a female of reproductive age. The endocrinological baseline data are presented in Table 1. The gonadotropin-releasing hormone stimulation test showed that the basal/peak LH and FSH values were 3.61/9.20 mIU/mL and 7.76/11.20 mIU/mL, respectively. The growth hormone-releasing peptide-2 stimulation test and arginine stimulation test showed peak growth hormone (GH) values of 3.31 and 0.12 ng/mL, respectively. These data indicated that she had CPHD, namely, hypogonadotropic hypogonadism (HH) and adult GH deficiency (GHD).
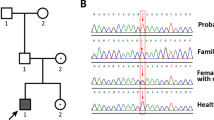
After obtaining informed consent from the patient and her sister and with the approval of the Institutional Review Board of Keio University School of Medicine, genomic DNA was extracted from leukocytes of the patient. We PCR-amplified and sequenced the STAR gene. The primer sequences and PCR conditions were provided previously6. We identified a novel variant of c.126_127delCCinsG, namely, p.Thr44Profs*2, and an already reported mutant of c.634C>T, p.Gln212* (ref. 7) (Fig. 1). c.126_127delCCinsG was absent from databases, including the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/), Human Genetic Variation Database (HGVD; http://www.hgvd.genome.med.kyoto-u.ac.jp/), Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). Parental genetic analysis was refused. We also analyzed major or possible causative genes for CPHD and/or HH, namely, AXL, CCDC141, CHD7, DMXL2, DUSP6, FEZF1, FGF8, FGF17, FGFR1, FLRT3, FSHB, GH1, GHR, GHRH, GHRHR, GHSR, GLI2, GNRH1, GNRHR, GPR161, HESX1, HS6ST1, IGF1, IGF1R, IL17RD, KAL1, KISS1, KISS1R, LHB, LHX3, LHX4, LEP, LEPR, MKRN3, NELF, OTUD4, OTX2, PAX6, PCSK1, PGM1, PNPLA6, POLR3A, POLR3B, POU1F1, PROK2, PROKR2, PROP1, RNF216, SEMA3A, SEMA7A, SIX3, SIX6, SOX2, SOX10, SPRY4, STAT5B, STUB1, TAC3, TACR3, TBX19, and TUBB3, using next-generation sequencing and the MiSeq instrument (Illumina, San Diego, CA, USA), according to the SureSelect protocol (Agilent Technologies, Santa Clara, CA, USA)8. No pathogenic variant among the tested genes was identified.

The left panel shows c.126_127delCCinsG in the proband as well as the wild type (A). The right panel shows c.634C>T in the proband (B).
The diagnosis of classic LCAH in this patient is convincing due to the clinical history of genetic variants in the STAR gene. Notably, one of them was novel: c.126_127delCCinsG. This variant must be pathogenic because it was not reported in the variant database and caused a stop codon, theoretically leading to nonsense-mediated mRNA decay in vivo. As parental genetic analysis was refused, we cannot prove compound heterozygosity.
She had an atypical clinical course, manifesting as LCAH. First, she had CPHD composed of HH and adult GHD. To the best of our knowledge, no case of LCAH and CPHD has been reported4,5. CPHD cannot be explained by STAR gene variants, as the STAR protein is not expressed in the pituitary gland (https://www.proteinatlas.org/ENSG00000147465-STAR/tissue). The onset of CPHD may be around pubertal age, judging from her height (147 cm) without any growth-promoting treatment and the presence of menarche. At present, the cause of CPHD is unknown, as no variant was identified in major or possible causative genes for CPHD and/or HH. Potential causes of CPHD include a monogenic disorder, which has not been established as a disease entity, secondary sequelae due to hypoglycemia by early-onset adrenal insufficiency, and/or others. The association between classic LCAH and CPHD might be just a coincidence. Second, she had mental delay. This might be secondary sequelae due to hypoglycemia. Third, she had right low-frequency sensorineural hearing loss, again, probably due to secondary sequelae due to hypoglycemia.
In conclusion, we identified a novel STAR variation in a patient with classic LCAH and CPHD.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at: https://doi.org/10.6084/m9.figshare.hgv.2963, https://doi.org/10.6084/m9.figshare.hgv.2966.
References
Bose, H. S., Sugawara, T., Strauss, J. F. III, Miller, W. L., Consortium ICLAH: International Congenital Lipoid Adrenal Hyperplasia Consortium. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N. Engl. J. Med. 335, 1870–1878 (1996).
Baker, B. Y. et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J. Clin. Endocrinol. Metab. 91, 4781–4785 (2006).
Hatabu, N. et al. Pubertal development and pregnancy outcomes in 46,XX patients with nonclassic lipoid congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 104, 1866–1870 (2019).
Ishii, T. et al. Clinical features of 57 patients with lipoid congenital adrenal hyperplasia: criteria for nonclassic form revisited. J. Clin. Endocrinol. Metab. 105, dgaa557 (2020).
Fang, Q. et al. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr. Rev. 37, 636–675 (2016).
Ishii, T. et al. Pubertal and adult testicular functions in nonclassic lipoid congenital adrenal hyperplasia: a case series and review. J. Endocr. Soc. 3, 1367–1374 (2019).
Nakae, J. et al. Analysis of the steroidogenic acute regulatory protein (StAR) gene in Japanese patients with congenital lipoid adrenal hyperplasia. Hum. Mol. Genet. 6, 571–576 (1997).
Hamada, J. et al. A novel SOX10 variant in a Japanese girl with Waardenburg syndrome type 4C and Kallmann syndrome. Hum. Genome Var. 7, 30 (2020).
Acknowledgements
This work was supported by a Grant-in-Aid from the Ministry of Health, Labor and Welfare of Japan (Nanjiseisikkanseisakukenkyujigyo (20FC1020)).
Author information
Authors and Affiliations
Contributions
M. Higa, A.Z., A.T., N.M., T.M., T.T., M.S., and H.M. managed the patients and prepared the manuscript. M. Honda and T.H. performed the molecular analyses. T.H. supported the manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Tomonobu Hasegawa has the following financial relationships to disclose: research funding from Novo Nordisk Pharma Ltd. and JCR Pharmaceuticals Co., Ltd.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Higa, M., Zaha, A., Takushi, A. et al. Novel STAR gene variant in a patient with classic lipoid congenital adrenal hyperplasia and combined pituitary hormone deficiency. Hum Genome Var 8, 6 (2021). https://doi.org/10.1038/s41439-021-00138-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-021-00138-w
