
Abstract
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is inherited in an autosomal dominant manner and is characterized by a combination of developmental abnormalities and a predisposition to tumor formation. Hedgehog receptor Patched 1 (PTCH1) has been identified as the mutated gene in NBCCS. We identified the PTCH1_c.3298_3299insAAG_p.1099_1100insE mutation in the transmembrane region, which comprises a sterol transporter whose abnormal function is reportedly related to pathogenicity.
Similar content being viewed by others


Nevoid basal cell carcinoma syndrome (NBCCS, OMIM: 109400), also known as Gorlin syndrome, was first reported by Gorlin and Goltz in 19601. NBCCS is an autosomal dominant inherited disease characterized by bifid ribs and palmar pits, as well as a predisposition to various tumors, including basal cell carcinoma (BCC), medulloblastoma, ovarioma, cardiac fibroma, odontogenic keratocyst, and skin patch2,3,4. At birth, patients with NBCCS typically exhibit macrocephaly or rib anomalies. As the patients' age, palmar and plantar pits become evident. At the age of approximately 10 years, a odontogenic keratocyst, formerly known as a jaw cyst, is observed as the first notable symptom in the diagnosis of NBCCS. At the age of approximately 20 years, BCC can develop at any location on the body, particularly on the eyelid. However, in Japanese patients with NBCCS, the incidence of BCC is significantly low, whereas the frequency of odontogenic keratocysts is relatively high3,5.
The gene responsible for causing NBCCS is the human homologue of the Drosophila patched gene Patched 1 (PTCH1)6. PTCH1, a Hedgehog (HH) receptor, is located on chromosome 9q22.3, consists of 23 exons, and encodes a 1447-amino-acid integral membrane protein with 12 transmembrane (TM) regions, five of which form a sterol-sensing domain and two extracellular loops at which the N-terminal domain of the HH ligand binds7,8. The PTCH1 protein functions to inhibit the transmembrane protein Smoothened (SMO). Once extracellular HH ligands bind to the PTCH1 receptor, PTCH1 releases SMO inhibition, allowing SMO to participate in downstream signaling and activate GLI transcription factors. HH signaling plays an essential role during embryogenesis and maintains stem cell populations in certain adult tissues9,10. Unliganded PTCH1 inhibits HH signaling; this repression is released when HH ligands bind to PTCH17,8. In this study, we investigated PTCH1 germline mutations in Japanese familial NBCCS.
Patients were diagnosed with NBCCS in the Department of Oral and Maxillofacial Surgery, Kawasaki Medical University Hospital, and Hiroshima University Hospital based on both clinical and genetic analyses. Each patient in the family with NBCCS was designated F1, F2, F3, F4, F5, F6, and F7. The pedigree is shown in Fig. 1a. F1–F5 had odontogenic keratocysts. F4 and F5 had undergone surgery for removal of odontogenic keratocysts several times at the Kawasaki Medical University Hospital. F1–F7 exhibited palmar and plantar pits, F3 and F4 showed kyphoscoliosis and calcification of the falx cerebri, and F4 had BCC, skin patch, and hairy skin patch. F6 and F7 had a congenital deficiency of the second premolar. Based on interviews with the patients, the husband of F5 also had a deficiency of the second premolar, and his mother had congenital deficiency of several incisors. The main phenotypes of F1–F7 are shown in Fig. 1b.

a Pedigree of familial NBCCS: NBCCS was inherited in four generations. b Phenotypes of familial NBCCS. JC: Jaw Cysts, P: Pits, RM: Rib Malformation, KS: Kyphoscoliosis, BS: Bridging of Sella, CFC: Calcification of Falx Cerebri (c) Genotypes of familial NBCCS.
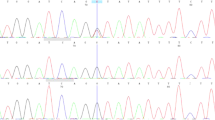
We detected irregular band shifts in exon 19 in F1–F5 through PCR-SSCP (data not shown). Direct sequencing revealed the c.3298_3299insAAG mutation in exon 19 of PTCH1 in F1–F7 (Fig. 2). To screen for other pathogenic mutations affecting the HH signaling pathway, we performed NGS with the MiSeq sequencer using the TruSight One panel for familial NBCCS and identified the following mutations: PTCH1_c.1665T>C_p.N555N (exon 12), _c.2560+9G>C (intron 15), _c.2887+21G>A (intron 17), _c.3298_3299insAAG_p.1099_1100insE (exon 19), and _c.3944C>T_p.P1315L (exon 23). Among the detected mutations, only PTCH1_c.3298_3299insAAG_p.1099_1100insE was specifically shared in our cases of familial NBCCS (F1–F7) and was not shared in WT1–WT4 (Fig. 1c). In contrast, there were no other pathogenic mutations in HH signaling-related molecules, such as PTCH2, SHH, SMO, SUFU, GLI1, GLI2, and GLI3.

The results of direct sequencing of PTCH1 exon 19 are shown. Though an insertion was not detected in WT, an AAG insertion was detected between coding sequences 3298 and 3299 in F4–F7.
The detected mutation, PTCH1_c.3298_3299insAAG_p.1099_1100insE, was located in the 10th transmembrane region and mapped closely to sterol-binding sites. Recently, through structure-guided mutational analysis, Gong et al. revealed that the interaction between Shh-N and PTCH1 is steroid-dependent11. Moreover, PTCH1_c.3298_3299insAAG was not found in Togo Var, Exome Variant Server, dbSNP, dbVar, or ClinVar, confirming that the mutation is novel. In addition, PTCH1_c.3298_3299insAAG was predicted as disease causing by Mutation Taster2 and as deleterious by PROVEAN software. Thus, we predicted that the specifically shared mutation, PTCH1_c.3298_3299insAAG_p.1099_1100insE, would be responsible for the pathogenesis of NBCCS11,12.
Of note, c.1665T>C (rs1805155), located in a sterol-sensing domain, is synonymous and the most common SNP in PTCH1. Clinical significance is benign in ClinVar. To our knowledge, there is no prior report about the pathogenicity of this SNP. c.2560+9G>C and c.2887+21G>A are intronic SNPs. Although c.2887+21G>A is not reported, c.2560+9G>C (rs2066829) is registered as an intronic variant and benign in ClinVar. It is unknown whether the PTCH1 polymorphisms located in introns cause a functional change. However, intronic polymorphisms have been demonstrated in association with other complex diseases, including the association of IRF6 with cleft lip/palate13,14. c.3944C>T (rs357564) results in an amino acid change in the topological domain of the C-terminus of PTCH1. Though the interpretation is benign in ClinVar, some researchers have concluded that, in combination with oral contraceptive use, c.3944C>T-carrier in PTCH1 is associated with an increased risk of breast cancer15. Because PTCH1 has a sterol-sensing domain, long-term exogenous hormone use is reportedly related to breast cancer risk. Although the above three SNPs were also detected in wild-type PTCH1 in this family, it was recently suggested that SNPs in key genes involved in the HH signaling pathway are associated with susceptibility to odontogenic cystic lesions16.
Here, we reported the detection of a novel mutation, PTCH1_c.3298_3299insAAG_p.1099_1100insE, in Japanese familial NBCCS. Therefore, we conclude that PTCH1_ c.3298_3299insAAG is the “likely pathogenic” mutation of NBCCS.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2927.
References
Gorlin, R. J. & Goltz, R. W. Multiple nevoidbasal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N. Engl. J. Med. 262, 908–912 (1960).
Fujii, K. et al. Mutations in the human homologue of Drosophila patched in Japanese nevoid basal cell carcinoma syndrome patients. Hum. Mutat. 21, 451–452 (2003).
Fujii, K. & Miyashita, T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr. Int. 56, 667–674 (2014).
Wilson, L. C., Ajayi-Obe, E., Bernhard, B. & Maas, S. M. Patched mutations and hairy skin patches: a new sign in Gorlin syndrome. Am. J. Med. Genet. A 140, 2625–2630 (2006).
Johnson, R. L. et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671 (1996).
Marigo, V., Davey, R. A., Zuo, Y., Cunningham, J. M. & Tabin, C. J. Biochemical evidence that patched is the Hedgehog receptor. Nature 384, 176–179 (1996).
Stone, D. M. et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134 (1996).
Kato, C. et al. Nevoid basal cell carcinoma syndrome caused by splicing mutations in the PTCH1 gene. Fam. Cancer 16, 131–138 (2017).
McMahon, A. P., Ingham, P. W. & Tabin, C. J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 53, 1–114 (2003).
Varjosalo, M. & Taipale, J. Hedgehog: functions and mechanisms. Genes Dev. 22, 2454–2472 (2008).
Gong, X. et al. Structural basis for the recognition of Sonic Hedgehog by human Patched 1. Science 361, eaas8935 (2018).
Kowatsch, C., Woolley, R. E., Kinnebrew, M., Rohatgi, R. & Siebold, C. Structures of vertebrate Patched and Smoothened reveal intimate links between cholesterol and Hedgehog signalling. Curr. Opin. Struct. Biol. 57, 204–214 (2019).
Zucchero, T. M. et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N. Engl. J. Med 351, 769–780 (2004).
Letra, A. et al. Follow-up association studies of chromosome region 9q and nonsyndromic cleft lip/palate. Am. J. Med. Genet. A 152A, 1701–1710 (2010).
Chang-Claude, J. et al. The patched polymorphism Pro1315Leu (C3944T) may modulate the association between use of oral contraceptives and breast cancer risk. Int. J. Cancer 103, 779–783 (2003).
Magic, M. et al. Hedgehog signaling pathway and vitamin D receptor gene variants as potential risk factors in odontogenic cystic lesions. Clin. Oral. Investig. 23, 2675–2684 (2019).
Acknowledgements
The authors would like to thank all individuals who participated in this study. This work was carried out in part at the Natural Science Center for Basic Research and Development, Hiroshima University. This research was financially supported by the Grant-in-Aid for Scientific Research (B) to T.O. (Grant number: 18H03000), Grant-in-Aid for Young Scientists to Y.N. (20K18725), Grant-in-Aid for Young Scientists to A.H. (19K191980A), and Grant-in-Aid for Scientific Research (C) to S.T. (18K097440A) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Ethics Committee of Human Genome/Gene Analysis Research at Hiroshima University (approval number: hi-72). All participants provided informed consent prior to their inclusion in this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nakase, Y., Hamada, A., Kitamura, N. et al. Novel PTCH1 mutations in Japanese familial nevoid basal cell carcinoma syndrome. Hum Genome Var 7, 38 (2020). https://doi.org/10.1038/s41439-020-00126-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-020-00126-6
This article is cited by
-
Establishment of induced pluripotent stem cells derived from patients and healthy siblings of a nevoid basal cell carcinoma syndrome family
In Vitro Cellular & Developmental Biology - Animal (2023)
-
Multiple orthokeratinized odontogenic cysts: clinical, pathological, and genetic characteristics
Diagnostic Pathology (2022)
