
Abstract
Variants of GRIN1, which encodes GluN1, are associated with developmental delay, epilepsy, and cortical malformation. Here, we report a case of arthrogryposis multiplex congenita with polymicrogyria and infantile encephalopathy caused by a heterozygous variant, c.1949A>C, p.(Asn650Thr) of GRIN1, which could result in the disruption of the third transmembrane domain (M3) of GluN1. This case expands our understanding of the known phenotypes of GRIN1-related neurodevelopmental disorders.
Similar content being viewed by others


N-methyl-D-aspartate receptors (NMDARs) are glutamate-gated ion channels that are essential for synaptic transmission and plasticity in the central nervous system (CNS). They are composed of protein tetramers, comprising two GluN1 subunits and two GluN2 subunits or a mixture of GluN2 and GluN3 subunits1. The GluN1 subunit is encoded by GRIN1, which maps to chromosome 9q34.3. Variations in GRIN1 are associated with NMDAR functional loss or gain that leads to impaired CNS function and results in neurodevelopmental disorders, including developmental delay, epilepsy, muscle tone abnormalities, microcephaly, and cortical malformation (Table 1)2,3,4. For example, heterozygous missense GRIN1 variants have been implicated in neurodevelopmental disorders with hyperkinetic movements and seizures (MIM 614254)3,5. GRIN1 variants have also been linked to infantile encephalopathy with CNS abnormalities6, but they have rarely been associated with arthrogryposis multiplex congenita (AMC). Overall, little is known about the clinical phenotypes associated with GRIN1 variant-related disruption of neuronal development, and expanding the list of known phenotypes is essential for accurate diagnosis and selection of appropriate treatment/management options.
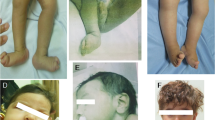
Here, we report the case of a newborn with AMC, polymicrogyria, and infantile-onset epilepsy caused by a novel GRIN1 variant. The proband was found to have perinatal intrauterine growth restriction (IUGR) without oligohydramnios and fetal akinesia upon fetal ultrasound. She was born to healthy nonconsanguineous parents at 39 weeks of gestation, with a birth weight of 2,312 g (−1.8 standard deviation [SD]), a length of 40.4 cm (−4.0 SD), and an occipitofrontal circumference of 29.5 cm (−2.7 SD). After birth, she was transferred to our hospital due to AMC and involuntary movements. On arrival, she exhibited a narrow chest, AMC (symmetrical flexion contractures predominantly in the elbow and knee), club foot, spasticity, and respiratory distress requiring intubation. Brain magnetic resonance imaging (MRI) showed bilateral polymicrogyria in the perisylvian region (Fig. 1). After extubation, she developed epileptic seizures with episodes of desaturation. Electroencephalography revealed focal epileptic activity in the left temporal area. Treatment with diazepam, carbamazepine, and tizanidine was initiated and controlled her symptoms, including seizures and spasticity.

T2-weighted axial (a, b) and coronal (c, d) brain MRI showing bilateral polymicrogyria in the perisylvian and frontal regions.
Informed consent was obtained from the parents in accordance with the Kanagawa Children’s Medical Center Review Board and Ethics Committee. Genetic analysis was performed using the TruSight One Sequencing Panel Kit on the MiSeq platform (Illumina, Inc., San Diego, CA, USA). Exome data alignment and variant calling were performed, and variant annotations were examined7. Targeted sequencing identified a de novo GRIN1 heterozygous variant, NM_007327.3: c.1949A>C, p.(Asn650Thr) (Fig. 2a). The variant has not been previously reported in the general population (The Genome Aggregation Database (https://gnomad.broadinstitute.org/)). The CADD score (27.7) indicated that the variant was deleterious, and Provean (Deleterious), SIFT (Damaging), and PolyPhen-2 (Damaging) analyses predicted different types of pathogenicity. According to the American College of Medical Genetics and Genomics guidelines, the variant is pathogenic (PS2_mod + PM1 + PM2 + PM5 + PP2 + PP3)8.

a Sanger sequencing chromatograms of a de novo heterozygous GRIN1 variant, c.1949A>C, p.(Asn650Thr). b The variant found in this study is shown in the structural domains of GRIN1, including the ligand-binding site domain (S2) and the transmembrane domain (M3). The variation is located in the M3 domain. Blue triangles indicate other reported GRIN1 variants with polymicrogyria.
The Asn650Thr variant results in a mutation in the third transmembrane domain (M3) of GluN1. The M3 domain is a highly conserved region among NMDARs and has a high impact on their functionality9. The M3 and the adjacent second ligand-binding (S2) domains are hot spots of GRIN1 variations associated with polymicrogyria (Fig. 2b)4. In fact, the Asn650Thr variant caused a change at the same amino acid position in the M3 domain that has previously been reported in a variant causing polymicrogyria (Asn650Ile)4.
AMC can be caused by various conditions, mainly genetic diseases. All causes have fetal akinesia in common. The causes of fetal akinesia may be CNS, connective tissue, or skeletal abnormalities10. Malformations, such as arthrogryposis, club foot, and micrognathia, have been reported to occur in ~30% of patients with bilateral perisylvian polymicrogyria11. We hypothesize that the AMC in our patient was likely caused by a complex mechanism secondary to the neurodevelopmental deficit caused by polymicrogyria, which is an abnormality of cortical formation during early development12. Thus, the AMC in our patient may have been caused by a variety of different pathways, including functional disruption of NMDARs.
GRIN1 encodes GluN1, which is expressed as a part of the NMDA receptor complex on the neuronal surface during embryonic brain development, critically influencing NMDAR function after birth13. Our findings indicate that alterations of GRIN1, especially those impacting the M3 and S2 domains, can have serious impacts on the early development and function of the CNS. Further investigation of the relationship between AMC and polymicrogyria could clarify the neurodevelopmental mechanism underlying this phenotype, especially during fetal growth.
Puduri et al. suggested that polymicrogyria with AMC may have an underlying genetic background based on a large research database of polymicrogyria14. They showed that typical CNS involvement and lower motor neuron defects were often observed in patients with polymicrogyria and AMC. BICD2 and PI4KA are the causative genes underlying the complex phenotype, including AMC and polymicrogyria15,16. These genes are also responsible for other neurological features involved in peripheral nervous pathology or cerebellar hypoplasia. Our patient could move her upper and lower limbs without a decreased deep tendon reflex; however, a nerve conduction study was not performed. The brain MRI did not show cerebellar hypoplasia (Fig. 1). In line with these results, polymicrogyria with AMC is likely to be a heterogeneous disorder, and further genetic analyses are required to elucidate the complex underlying mechanisms.
In conclusion, we identified a novel GRIN1 variant responsible for AMC and cortical abnormalities in a newborn. To our knowledge, this is the first report of severe neurodevelopmental disorders associated with AMC linked to a GRIN1 variant. Our findings expand the known phenotypes in the spectrum of GRIN1-related neurodevelopmental disorders, which is essential for accurate diagnosis and development of specific therapeutic strategies.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2900.
References
Paoletti, P., Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400 (2013).
Platzer, K. & Lemke, J. R. GRIN1-Related Neurodevelopmental Disorder. (eds Adam, M. P., Ardinger, H. H., Pagon, R. A., Wallace, S. E., Bean, L. J. H., Stephens, K. et al.) (GeneReviews((R)), Seattle, 2019).
Lemke, J. R. et al. Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86, 2171–2178 (2016).
Fry, A. E. et al. De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 141, 698–712 (2018).
Hamdan, F. F. et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 88, 306–316 (2011).
Ohba, C. et al. GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia 56, 841–848 (2015).
Murakami, H., Enomoto, Y., Tsurusaki, Y., Sugio, Y. & Kurosawa, K. A female patient with X-linked Ohdo syndrome of the Maat-Kievit-Brunner phenotype caused by a novel variant of MED12. Congenit. Anom. 60, 91–93 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17, 405–424 (2015).
Leventer, R. J. et al. Clinical and imaging features of cortical malformations in childhood. Neurology 53, 715–722 (1999).
Hall, J. G. Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 57, 464–472 (2014).
Jansen, A. & Andermann, E. Genetics of the polymicrogyria syndromes. J. Med. Genet. 42, 369–378 (2005).
Yuan, H., Erreger, K., Dravid, S. M. & Traynelis, S. F. Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. J. Biol. Chem. 280, 29708–29716 (2005).
Chen, W. et al. Grin1 mutation associated with intellectual disability alters NMDA receptor trafficking and function. J. Hum. Genet. 62, 589–597 (2017).
Poduri, A. et al. The syndrome of perisylvian polymicrogyria with congenital arthrogryposis. Brain Dev. 32, 550–555 (2010).
Peeters, K. et al. Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am. J. Hum. Genet. 92, 955–964 (2013).
Pagnamenta, A. T. et al. Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum. Mol. Genet. 24, 3732–3741 (2015).
Acknowledgements
We would like to thank the patient’s family members for granting permission to conduct and publish this study. This research was supported by a grant-in-aid from the Ministry of Health, Labor and Welfare, Japan, JSPS KAKENHI 20K08270 (K.K.); The Initiative on Rare and Undiagnosed Diseases (IRUD) 18ek0109301, BIRTHDAY 1-2 19gk0110038 from Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishimura, N., Kumaki, T., Murakami, H. et al. Arthrogryposis multiplex congenita with polymicrogyria and infantile encephalopathy caused by a novel GRIN1 variant. Hum Genome Var 7, 29 (2020). https://doi.org/10.1038/s41439-020-00116-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-020-00116-8
