
Abstract
Mobile element insertions (MEIs) contribute to genomic diversity, but they can be responsible for human disease in some cases. Initial clinical testing (BRCA1, BRCA2 and PALB2) in a 40-year-old female with unilateral breast cancer did not detect any pathogenic variants. Subsequent reanalysis for MEIs detected a novel likely pathogenic insertion of the retrotransposon element (RE) c.7894_7895insSVA in BRCA2. This case highlights the importance of bioinformatic pipeline optimization for the detection of MEIs in genes associated with hereditary cancer, as early detection can significantly impact clinical management.
Similar content being viewed by others

Hereditary breast and ovarian cancer (HBOC) syndrome, caused by monoallelic germline pathogenic variants (PVs) in BRCA1 (MIM# 604370) and BRCA2 (MIM# 600185), is predominantly associated with an increased risk of breast and ovarian cancer in females1. Prompt detection of BRCA1/2 PVs in women allows for surveillance and/or risk-reducing (RR) strategies, which have been demonstrated to reduce cancer-related morbidity and mortality1. Furthermore, knowledge of a woman’s BRCA1/2 status following a diagnosis of cancer can inform surgical decision-making, such as pursuing RR contralateral mastectomy (RRCM) or RR bilateral salpingo-oophorectomy (RRBSO), and may guide therapeutic decisions, such as platinum-based neoadjuvant chemotherapy or poly ADP-ribose polymerase inhibitors2,3. Variant detection is also important for clarifying risk for family members4. While advances in sequencing technology and analysis have improved the detection of BRCA1/2 PVs, testing is uninformative for a number of breast and ovarian cancer cases with high clinical suspicion5,6. Some of these cases may be explained by PVs in other cancer predisposition genes7. However, some of these individuals may carry PVs in the tested genes that were not detected by sequencing technologies at the time4.
Mobile element insertions (MEIs) are one of these classes of variants. They occur when transposable elements (TEs) “jump” to alternate locations in the genome through DNA transposases or reverse transcription8,9,10. There are many subsets of TEs, but in humans, long interspersed element-1s function as retrotransposons. They are copied into RNA and reinserted into the DNA by a reverse transcriptase enzyme9. They are capable of copying not only their own sequence but also other elements such as Alu elements, short interspersed element-variable number tandem repeat-Alus (SINE-VNTR-Alus or SVAs), and U6s10. While most TEs move throughout the genome without adverse consequences, they are occasionally inserted into critical regions, leading to disruption of transcription, splicing, or translation8,10.
Pathogenic MEIs have been demonstrated to cause a number of genetic conditions, including HBOC and other cancer predisposition syndromes11. The prevalence of pathogenic MEIs among disease-causing variants is estimated to be between 0.16 and 0.3%8; however, this is likely underestimated due to challenges with detection. Historic PCR amplification of sequencing targets might underrepresent or miss alleles containing these insertions due to size (under-amplification) or allele dropout. With improved coverage, capture-based next-generation sequencing allows for the detection of large copy number variants and rearrangements, such as TEs. However, detection requires data analysis optimization and functional confirmation12.
Here we report a case involving the identification of a previously undetected pathogenic MEI in BRCA2 using an updated variant calling method in a patient with early-onset breast cancer.
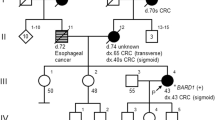
A 40-year-old Emirati female presented with unilateral breast cancer. The initial biopsy revealed invasive ductal carcinoma (IDC), and it was estrogen (ER) and progesterone receptor (PR) positive and human epidermal growth factor receptor 2 (HER2) negative. Although there was no reported family history of HBOC-associated cancers (Fig. 1), she met National Comprehensive Cancer Network (NCCN) guidelines for BRCA1/2 testing2. Expedited testing for BRCA1, BRCA2, and PALB2 was ordered to inform surgical decisions. She expressed a preference for bilateral mastectomy should a PV be detected.

The proband is the only child to her parents; however, she has three half-siblings through her mother and numerous aunts, uncles, and cousins on both sides of the family. While this suggests that the MEI is de novo in the proband, it is possible that some members of the family may have the variant and have simply not developed cancer due to age-related penetrance. Her grandparents are reported to have died of noncancer-related causes in their 60s and 70s, and it is possible that they died before cancer would have developed. Only sequencing of family members could confirm the de novo status of the BRCA2 MEI; however, samples were not available.
Testing revealed two variants that were classified by the testing laboratory as being of uncertain significance (VUS): BRCA2 (NM_000059.3) c.6842G>A (p.Gly2281Glu) and PALB2 (NM_024675.3) c.3464C>G (p.Ser1155Cys). Using ACMG variant classification guidelines, both variants had moderate evidence for pathogenicity (PM2) given their low frequency in population databases, such as GnomAD. In silico predictions provided benign supporting (BP4) evidence for the BRCA2 and PALB2 variants13. Thus current evidence is insufficient to determine the role of these two variants in disease. The patient consequently proceeded with unilateral mastectomy for the affected breast. The final histology was stage IIIA (T3 N1 M0) grade 3 IDC ER+/PR+/HER2−. Cascade testing was unavailable for her relatives, and first-degree relatives were considered to be at moderate risk for breast cancer given her diagnosis.
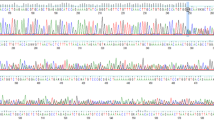
Eight months later, her sequence was reanalyzed using an updated bioinformatic pipeline with increased sensitivity for TE detection. This pipeline development, named Mobster, was built for MEI detection and run with default parameters with post hoc filtering requiring at least 15 supporting reads14. An ~2000 base pair SVA insertion (c.7666_7667insSVA) was detected in BRCA2, which is a retrotransposon composite element with a portion of SINEs, VNTRs, and backward Alu repeats. This SVA appears to be 5′ truncated and is missing the CCCTCT repeats. The presence of a 17-bp target site duplication, L1 endonuclease recognition sequence, and poly-A tail together indicate that this SVA insertion occurred by L1-mediated target-primed reverse transcription (Fig. 2). This novel variant appears to disrupt the BRCA2 DNA-binding domain15 and is classified as likely pathogenic13. The variant was confirmed by Sanger sequencing.

a An IGV image from the proband. Colored bases indicate a mismatch relative to the reference genome. Strings of colored bases in a row indicate clipped reads at the SVA insertion breakpoint. The sequence on the bottom represents the reference genome. b Clipped reads at the insertion breakpoint were used to infer the structure of the SVA. A consensus sequence was built from both the right- and left-clipped read clusters. The right-clipped consensus represents the 5′ sequence of the SVA and demonstrates some 5′ truncation relative to an SVA-E sequence. The left-clipped consensus represents the poly-A tail of the sequence. The box labeled “presumed 3′ SVA” is not sequenced and not drawn to scale. If there are no additional rearrangements within the SVA sequence, then the nonsequenced portion is likely ~2kb long. A primer designed based on the SINE-R region of known SVA-E elements was used to amplify the 3′ insertion breakpoint.
This variant confirmed a diagnosis of HBOC, which had significant implications for risk management. Assuming that this variant conferred a similar risk to other BRCA2 PVs, the patient’s contralateral breast cancer risk was approximately 35 and 53% at 10 and 20 years, respectively1. The patient was recommended to undergo annual mammograms and magnetic resonance imaging for her remaining right breast and may consider RRCM. Of significance, she was now considered to be at increased ovarian cancer risk (~17%; 95% confidence interval)1, and RRBSO was recommended by age 45 years2. She subsequently underwent total hysterectomy (for endometriosis) and RRBSO and opted for annual breast surveillance. Cascade testing was recommended for her at-risk relatives, although it was possible that the MEI was de novo in view of absent cancer family history (Fig. 1). Her children have a 50% chance of inheriting the variant and should consider testing as adults.
This case highlights the causative role MEIs can play in hereditary cancer syndromes. The overall prevalence of MEIs is likely underestimated and may be more common in some genes than in others11. One large cohort study found that RE insertions accounted for a larger proportion of PVs in BRCA2 than in BRCA1 and APC11. They postulated that this may be attributable to the larger size of BRCA2, as well as being a possible “hot spot” for oncogenic insertions11. The specific detected TE in this case (SVA) is larger and rarer than other Alu elements and is thought to comprise ~0.2% of the genome11. It is important to note, however, that SVAs also make up a disproportionate number of new MEIs given that this is an active family with active source elements. These SVAs have been reported in a number of other genes, but to date, only insertions in a few other genes, such as PMS2 and CASP8, have been associated with increased cancer risk. This is the first report of an SVA in BRCA2. Optimized analysis specifically for TEs will improve our understanding of the prevalence of these variants and help us better assess which genes should be more stringently evaluated for TEs in clinical testing.
It is also important to test for MEIs in all patients with a clinical suspicion of hereditary cancer syndromes, regardless of family history. Previous studies of TEs in human disease have focused on family history and founder variants11,16. MEIs may present as de novo variants and should be considered if PVs are not initially detected by standard sequencing and deletion/duplication analysis. It is important to inquire whether the laboratory performing testing has the capability to detect MEIs. Thorough evaluation for MEIs in regions exhibiting evidence of insertion should be considered a routine part of clinical testing.
This is the first report of an MEI in a person of Arabic descent. There appears to be some correlation between ancestry and the likelihood of detecting a germline TE. The percentage of TE-based PVs may vary by ancestry, as Africans had a much higher proportion of TEs when compared to Europeans11,16. Founder variants have been previously described in a number of populations, including African and Latin/Caribbean populations, and most notably, the Portuguese BRCA2 c.156_157insAlu variant17.
This case demonstrates the contributory role of MEIs in cancer predisposition syndromes and contributes to the body of literature implicating them as a mechanism for disease. Improvements in bioinformatic analysis with increased sensitivity for MEI detection are important for clinical decision-making. While MEIs may be more prevalent in certain genes or ethnicities5,11, they should be considered in all individuals with suspected cancer syndromes. Patients should be counseled that reanalysis of genetic sequencing may impact their results and future clinical management. There is a need for better guidelines for patients, clinicians, and laboratories on sequencing reanalysis and follow-up for VUSs and uninformative results18.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2888.
References
Kuchenbaecker, K. B. et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416 (2017).
Pilarski, R. et al. FORCE: Facing Our Risk of Cancer Empowered NCCN Guidelines Version 3.2019 Genetic/Familial High-Risk Assessment: Breast and Ovarian. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf (2019).
Hartmann, L. C. & Lindor, N. M. The role of risk-reducing surgery in hereditary breast and ovarian cancer. N. Engl. J. Med. 374, 454–468 (2016).
Machado, P. M. et al. Screening for a BRCA2 rearrangement in high-risk breast/ovarian cancer families: evidence for a founder effect and analysis of the associated phenotypes. J. Clin. Oncol. 25, 2027–2034 (2007).
Sluiter, M. D. & Van Rensburg, E. J. Large genomic rearrangements of the BRCA1 and BRCA2 genes: review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res. Treat. 125, 325–349 (2011).
Beck, A. C. et al. Rate of BRCA mutation in patients tested under NCCN genetic testing criteria. Am. J. Surg. 219, 145–149 (2020).
Kurian, A. W. et al. Genetic testing and results in a population-based cohort of breast cancer patients and ovarian cancer patients. J. Clin. Oncol. 37, 1305–1315 (2019).
Hancks, D. C. & Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mob. DNA 7, 9 (2016).
Hancks, D. C. & Kazazian, H. H. Active human retrotransposons: variation and disease. http://www.repeatmasker.org/ (2012). Cited 31 Jul 2018.
Kazazian, H. H. & Moran, J. V. Mobile DNA in health and disease. N. Engl. J. Med. 377, 361–370 (2017).
Qian, Y. et al. Identification of pathogenic retrotransposon insertions in cancer predisposition genes. Cancer Genet. 216–217, 159–169 (2017).
Tang, Z. et al. Human transposon insertion profiling: analysis, visualization and identification of somatic LINE-1 insertions in ovarian cancer. Proc. Natl Acad. Sci. USA 114, E733–E740 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Thung, D. T. et al. Mobster: accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 15, 488 (2014).
Yang, H. J. BRCA2 function in DNA binding and recombination from a BRCA2–DSS1-ssDNA structure. Science 297, 1837–1848 (2002).
Hancks, D. C. & Kazazian, H. SVA retrotransposons: evolution and genetic instability. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2945828/pdf/nihms199583.pdf (2010). Cited 26 Jul 2018.
Peixoto, A. et al. The role of targeted BRCA1/BRCA2 mutation analysis in hereditary breast/ovarian cancer families of Portuguese ancestry. Clin. Genet. 88, 41–48 (2015).
Vears, D. F., Sénécal, K. & Borry, P. Genetic health professionals’ experiences with initiating reanalysis of genomic sequence data. Fam. Cancer https://doi.org/10.1007/s10689-020-00172-7 (2020).
Acknowledgements
The authors thank the patient and her family for their participation in this study. We also thank GeneDx for contributing to the description of the genetic findings.
Author information
Authors and Affiliations
Contributions
R.D., V.T., S.-T.L., E.C., T.S., and J.N. clinically characterized the subject and collected the samples. L.Y., R.T., and W.B.-F. analyzed the genetic test results. N.D. wrote the manuscript. All authors critically revised and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
J.N. received funding from AstraZeneca for breast and ovarian cancer research unrelated to the current study. L.Y., R.T., and W.B.-F. are salaried employees of GeneDx/OPKO Health. The remaining authors declare no competing interests.
Patient consent
Patient consent was obtained under IRB number 2011/826/B.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deuitch, N., Li, ST., Courtney, E. et al. Early-onset breast cancer in a woman with a germline mobile element insertion resulting in BRCA2 disruption: a case report. Hum Genome Var 7, 24 (2020). https://doi.org/10.1038/s41439-020-00111-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-020-00111-z
This article is cited by
-
Retrotransposon insertion as a novel mutational cause of spinal muscular atrophy
Human Genetics (2023)
