
Abstract
NAA10-related syndrome is an extremely rare X-chromosomal disorder, the symptoms of which include intellectual disability (ID), ocular anomalies, or congenital heart diseases, such as hypertrophic cardiomyopathy (HCM). Here, we describe a 4-year-old Japanese male patient who exhibited mild ID, HCM, and specific facial features. A hemizygous mutation (NM_003491.3: c.455_458del, p. Thr152Argfs*6) in exon 7 of NAA10 was detected. We recommend that patients undergo precise medical follow-up considering the characteristics of NAA10-related syndrome.
Similar content being viewed by others

N-alpha-acetylation (N-terminal acetylation, NTA) is one of the most common protein modifications in eukaryotes, and ~80% of the N-termini of human proteins are acetylated1. NAA10 (Xq28) encodes the enzyme N-alpha-acetyltransferase 10 (NAA10), which is the catalytic subunit of the N-terminal acetyltransferase A complex with the protein encoded by NAA15 (4q31.1)2. NTA is essential for the preservation of normal cell function. However, its physiological significance has not been completely elucidated.
NAA10 abnormality in humans was originally known as causing Ogden syndrome (MIM #300855), which is an X-chromosomal inherited disorder. Female patients with Ogden syndrome show mild to severe intellectual disability (ID), and male patients die early in life3. Recently, several male patients with various NAA10 mutations have been reported to survive. These patients exhibit various phenotypes, such as hypertrophic cardiomyopathy (HCM)4, microphthalmia/anophthalmia5, or severe nonsyndromic ID6. These diseases due to NAA10 abnormalities are collectively called NAA10-related syndrome1. We report a Japanese boy carrying a hemizygous NAA10 mutation with HCM and ID but no microphthalmia.
Our patient was a Japanese boy who was the first child of healthy parents. He was delivered by elective cesarean section at 37 weeks of gestation because of a transverse position, and his Apgar score was 8/8 (at 1 and 5 min). His birth weight was 2170 g (small for gestational age). He exhibited genital abnormalities (split scrotum, hypospadias), eyelid drooping, and bilateral overlap of toes at birth. He was diagnosed with congenital heart disease (CHD) by echocardiography. His karyotype was 46,XY.
At 1 month of age, he was referred to our hospital for examination for CHD. Echocardiogram revealed a perimembranous outlet ventricular septal defect (defect: 5.0 mm × 4.5 mm), an atrial septal defect (defect: 3.5 mm × 3.6 mm), and left ventricle (LV) wall thickness (interventricular septum: 5.4 mm; left ventricular posterior wall: 3.7 mm) (Fig. 1a). At the age of 5 months, echocardiogram showed thickening of the LV, especially the interventricular septum. Thus, he was diagnosed with HCM with LV outflow tract obstruction (LVOTO) and trivial-mild mitral regurgitation (MR). After 1 month, the MR and LVOTO worsened, and β-blocker therapy was started.

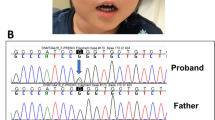
a Echocardiography of the patient at 1 year of age. b NAA10 mutations were identified in the patient and his mother by the Sanger method. Red letters indicate deleted nucleotides. c The genotype of the NAA10 mutation in male patients of the present case and literature. The bold face indicates hypertrophic myopathy, and the dotted line shows microphthalmia or anophthalmia.
At the age of 1 year, he was referred to the Clinical Genetics Department for molecular diagnosis. He showed mild ID, characteristic facial features, including eyelid drooping, exophthalmos, underdeveloped superior crus of antihelix, unilateral hearing loss, bifid scrotum, hypospadias, perodactylia, and CHD. His height at the age of 1 year was 69.6 cm (−2.0 SD), and his body weight was 8.4 kg (body mass index (BMI) 17.3). Routine blood tests and serum amino acid analysis were normal. He started to walk independently at the age of 1 year and 7 months. At the age of 4 years, his height was 90.0 cm (−2.63 SD), and his body weight was 12.2 kg (BMI 15.1). He was able to trot around, but he was unable to speak meaningful words. Brain magnetic resonance at the age of 1 year and 6 months imaging did not show any abnormalities.
To confirm his molecular diagnosis, we analyzed DNA samples derived from his peripheral blood by next-generation sequencing using TruSight One (Illumina, San Diego, CA, USA) after obtaining written informed consent from his parents. All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine (86) and Hyogo Prefectural Kobe Children’s Hospital (28-4) and were in accordance with the ethical standards of the Declaration of Helsinki. We identified a hemizygous four-base deletion (NM_003491.3: c.455_458del, p. Thr152Argfs*6) in exon 7 of NAA10. The same deletion was identified in his asymptomatic mother, although in the heterozygous state (Fig. 1b). Other pathogenic variants, including genes associated with RASopathies, were not identified by TruSight One.
Male patients with NAA10-related syndrome are extremely rare and present various clinical features. Previously, it was considered that boys with NAA10 mutations die early in life2. To our knowledge, 35 male patients from 13 families with 11 types of genetic aberrations in NAA10 have been reported (Table 1). The frequently observed symptoms are as follows: ID, motor developmental delay, growth failure, ophthalmic diseases, skeletal disorders, including scoliosis or digital anomalies, and cardiac disorders. Most NAA10 mutation types in male patients are missense variants, but three families, including the present case, harbor truncated mutations2,7. Patients with NAA10 mutations in further unstructured domains (exons 7 or 8) tend to exhibit microphthalmia or anophthalmia (Fig. 1c). The mutation of the present patient is a frameshift mutation that is identical to that of a boy reported by Cheng3. We presumed that he survived because the mutation is in a region where nonsense-mediated degradation does not occur. However, our patient did not show microphthalmia/anophthalmia. Further investigations are needed to clarify the precise mechanism of microphthalmia in NAA10-related syndrome. Cardiac complications are also observed in male patients with NAA10-related syndrome. However, the genotype–phenotype correlation is not clear. In addition, the exact cause of HCM in the present patient remains unknown.
Female patients with NAA10-related syndrome may display various types of ID6. The mother of the present patient carried an identical NAA10 mutation in the heterozygous state, but she did not exhibit any medical abnormalities. Families with NAA10-related syndrome should be considered during genetic counseling with regard to recurrence in the next child, irrespective of the sex of the child.
In conclusion, we report for the first time a Japanese male patient with NAA10-related syndrome. We recommend that patients undergo precise medical follow-up, particularly for neurodevelopment, cardiac disease, including HCM, ocular abnormalities, and scoliosis. The results of our study are useful for the recognition of NAA10-related syndrome.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2879.
References
Wu, Y. & Lyon, G. J. NAA10-related syndrome. Exp. Mol. Med. 5, 1–10 (2018).
Cheng, H. et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 28, 2900–2919 (2019).
Rope, A. F. et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am. J. Hum. Genet. 89, 28–43 (2011).
Støve, S. I. et al. A novel NAA10 variant with impaired acetyltransferase activity causes developmental delay, intellectual disability, and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 9, 1294–1305 (2018).
Esmailpour, T. et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J. Med. Genet. 51, 185–196 (2014).
Popp, B. et al. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 23, 602–609 (2015).
Johnston, J. J. et al. NAA10 polyadenylation signal variants cause syndromic microphthalmia. J. Med. Genet. 56, 444–452 (2019).
Casey, J. P. et al. NAA10 mutation causing a novel intellectual disability syndrome with long QT due to N-terminal acetyltransferase impairment. Sci. Rep. 5, 16022 (2015).
Saunier, C. et al. Expanding the phenotype associated with NAA10-related N-terminal acetylation deficiency. Hum. Mutat. 37, 755–764 (2016).
Ree, R. et al. A novel NAA10 p.(R83H) variant with impaired acetyltransferase activity identified in two boys with ID and microcephaly. BMC Med. Genet. 20, 101 (2019).
Slavotinek, A., Lee, S. S. & Hamilton, S. P. A. family with X-linked anophthalmia: exclusion of SOX3 as a candidate gene. Am. J. Med. Genet. A 138A, 89–94 (2005).
Acknowledgements
We thank the patient and his family. This work was supported by the Initiative on Rare and Undiagnosed Diseases from the Japan Agency for Medical Research and Development (grant number 0109301h0001) and the Hyogo Science and Technology Association. We also thank Editage (www.editage.jp) and Nature Publishing Group Language Editing for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kazumoto Iijima received grants from Zenyaku Kogyo Co., Ltd.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shishido, A., Morisada, N., Tominaga, K. et al. A Japanese boy with NAA10-related syndrome and hypertrophic cardiomyopathy. Hum Genome Var 7, 23 (2020). https://doi.org/10.1038/s41439-020-00110-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-020-00110-0
This article is cited by
-
Expanding the phenotypic spectrum of NAA10-related neurodevelopmental syndrome and NAA15-related neurodevelopmental syndrome
European Journal of Human Genetics (2023)
