
Abstract
Variants in the retinitis pigmentosa GTPase regulator (RPGR) gene are a major cause of X-linked inherited retinal disorder (IRD). We herein describe the clinical and genetic features of 14 patients from 13 Japanese families harboring RPGR variants in a nationwide cohort. Comprehensive ophthalmological examinations were performed to classify the patients into one of the phenotype subgroups: retinitis pigmentosa (RP) and cone rod dystrophy (CORD). The mean age of onset/at examination was 13.8/38.1 years (range, 0–50/11–72), respectively. The mean visual acuity in the right/left eye was 0.43/0.43 (range, 0.1–1.7/−0.08–1.52) LogMAR unit. Eight patients had RP, and six had CORD. Whole-exome sequencing with target analyses identified 13 RPGR variants in 730 families with IRD, including 8 novel variants. An association between the phenotype subgroup and the position of variants (cutoff of amino acid 950) was revealed. To conclude, the clinical and genetic spectrum of RPGR-associated retinal disorder was first illustrated in a Japanese population, with a high proportion of novel variants. These results suggest the distinct genetic background of RPGR in the Japanese population, in which the genotype–phenotype association was affirmed. This evidence should be helpful monitoring and counseling patients and in selecting patients for future therapeutic trials.
Similar content being viewed by others
Introduction
Inherited retinal disorder (IRD) is a major cause of blindness both in children and working populations in developed countries1. Retinitis pigmentosa (RP) is one of the most prevalent IRDs, and RP represents a heterogeneous group of retinal diseases characterized by progressive bilateral degeneration of rod and cone photoreceptors1,2,3,4,5,6,7,8. The estimated prevalence of RP in European populations is ~1 in 3000–4000 individuals2,3,4,5,6,9. Different patterns of inheritance have been identified in RP and allied disorders, including autosomal dominant (AD), autosomal recessive (AR), X-linked (XL), and mitochondrial inheritance2,3,10.
RP with an X-linked pattern of inheritance (XLRP) accounts for ~10–15% of RP cases, and is associated with the most severe form of the disease3,5,7,8,11. Two major causative genes for XLRP are the retinitis pigmentosa GTPase regulator (RPGR; OMIM; 312610) and RP2 (OMIM; 312600), which accounts for 70–90% and 7–18% of XLRP, respectively9.
Pathogenic variants in the RPGR gene (RP3) were first identified as a cause of XLRP in 199612,13. RPGR contains 19 exons and encodes a 90-kDa protein product localized predominantly to the photoreceptor connecting cilium (CC)12,14. The RPGR protein contains a repeat structure highly similar to the regulator of chromosome condensation 1 (RCC1) at the N-terminus11,12,15. RCC1 plays a crucial role in nucleocytoplasmic transport and regulation of cell-division processing16,17. Later, a novel 3′ terminal exon (well-known as exon open-reading frame 15; ORF15) was identified, which includes a large 3′ terminal exon consisting of exon 15 and extending into part of intron 1518. Biochemical investigations revealed that RPGR-ORF15 is located in the CC, which binds to the axoneme and the basal body19,20. The RPGR protein plays an important role in the transportation of phototransduction components and other outer segment proteins across the CC, although the function of RPGR is not perfectly understood3.
XLRP caused by pathogenic RPGR variants is one of the most severe forms of RP, with early onset of disease, night blindness, myopia, severe generalized rod and cone dysfunction, and progression to legal blindness by the third or fourth decade3,21. Carrier females are mostly asymptomatic or mildly affected with characteristic fundus features and electrophysiological abnormalities22, although the severity of carriers varies.
Pathogenic variants in the RPGR gene were responsible for X-linked cone rod dystrophy (XLCORD) and XL cone dystrophy (XLCOD), in addition to XLRP23,24,25,26,27,28. RPGR-associated retinal disorder (RPGR-RD) accounts for 73% of molecularly confirmed XLCORD cases in a British cohort. RPGR variants identified in XLCORD/XLCOD are frequently located toward the 3′ end of ORF15 in comparison with RPGR variants causing XLRP3,29,30.
XLCORD caused by pathogenic RPGR variants affects males with various onsets ranging from the second to the fourth decade, myopia, generalized cone rod dysfunction (occasionally with rod dysfunction), and diverse rates of progression3. Carrier females are mostly asymptomatic or mildly affected, with varying severity28.
Over 350 disease-associated variants in the RPGR gene have been reported to date in IRD3,15,26,31,32,33,34,35,36,37. A number of studies have been conducted in European populations;3,11,21,27,37,38,39,40,41 however, the characteristics of RPGR-RD in Asian populations remain uncertain due to limited resources8,34,35,36,41. Therefore, large cohort studies are required to understand the RPGR-RD in Asian populations.
The purpose of this study was to characterize the clinical and genetic features of patients and carriers with RPGR-RD in a large nationwide Japanese cohort by clarifying a genotype–phenotype association.
Methods
Participants
The protocol of this study adhered to the tenets of the Declaration of Helsinki, which was approved by the local ethics committee of the participating institutions from Japan (Reference: R18-029). Signed informed consent was obtained from all participants after explanation of the nature and possible consequences of this study.
Patients with a clinical diagnosis of IRD and available genetic data of whole-exome sequencing (WES) were investigated between 2008 and 2018 in the Japan Eye Genetics Consortium (JEGC; http://www.jegc.org/) study42. A total of 1294 subjects from 730 Japanese families registered to the JEGC database were surveyed.
Clinical investigations
Detailed demographic information was obtained, including ethnicity, sex, medical and family history, chief complaints of visual symptoms, and onset of disease. Comprehensive ophthalmological examinations were performed, including measurement of refractive errors, best corrected decimal visual acuity (BCVA) converted to the logarithm of the minimum angle of resolution (LogMAR), fundus photography, fundus autofluorescence (FAF) imaging, spectral-domain optical coherence tomography (SD-OCT), visual field testing, and electrophysiological assessment according to the international standards of the International Society for Clinical Electrophysiology of Vision (ISCEV)43,44.
Phenotype subgroup
For the purpose of this study, phenotype subgroups were defined based on clinical manifestation according to the previous report;45 RP (including rod-cone dystrophy), a progressive retinal dystrophy initially often presenting peripheral atrophy with generalized rod dysfunction greater than cone dysfunction; CORD, a progressive retinal dystrophy initially often presenting with macular atrophy with generalized cone dysfunction greater than rod dysfunction.
RPGR variant detection
Genomic DNA was extracted from all affected subjects and unaffected family members (where available for cosegregation analysis). WES with target analysis of retinal disease-associated genes (RetNet; https://sph.uth.edu/retnet/home.htm) was performed according to previously published methods42. The called variants were filtered by the allele frequency in the general Japanese population (<1%) as listed in the Human Genetic Variation Database (HGVD; http://www.genome.med.kyoto-u.ac.jp/SnpDB/about.htm). Depth and coverage for the target areas were interrogated using the Integrative Genomics Viewer (http://www.broadinstitute.org/igv/). For the purpose of this study, long-read direct sequencing was performed in seven patients with XLRP who were negative for two major XLRP-associated genes (RP2, RPGR) by WES at the National Genetic Reference Laboratory in Manchester, UK, for further screening of RPGR-ORF15 according to the previously published method46. Together with clinical features and pattern of inheritance, disease-causing variants were determined from the detected variants of the retinal disease-associated genes.
In silico molecular genetic analysis
The allele frequencies of all detected RPGR variants were established with the HGVD, Integrative Japanese Genome Variation (iJGVD 3.5k; https://ijgvd.megabank.tohoku.ac.jp/download_3.5kjpn/), 1000 Genomes (http://www.internationalgenome.org/), and the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/). All detected RPGR variants were analyzed with two general and three functional prediction programs; MutationTaster (http://www.mutationtaster.org/), FATHMM (http://fathmm.biocompute.org.uk/9), SIFT (https://www.sift.co.uk/), PROVEAN (http://provean.jcvi.org/index.php), and Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/). All detected RPGR variants were analyzed with evolutionary conservation scores according to the UCSC database (https://genome.ucsc.edu/index.html). Variant classification according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) was conducted for all detected variants47.
Results
Participants
Fourteen affected subjects from 13 Japanese families with a clinical diagnosis of IRD and harboring RPGR variants were ascertained. All 14 affected subjects were registered as a proband (or probands) for each pedigree. Seven females from six families were also registered as carriers.
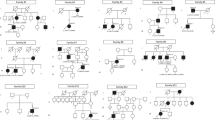
The detailed clinical information of 14 affected subjects (registered as a proband) is presented in Table 1. Pedigrees of 13 families are shown in Fig. 1. All 14 subjects and 7 carriers were originally from Japan, and no mixture of ethnicity was reported.

The solid squares (men) and circles (women) represent the affected patients. Carrier females are represented by circles containing a black spot. Unaffected family members are represented by white icons. The slash symbol indicates deceased individuals. The generation number is shown on the left. The proband of each pedigree is marked by an arrow. Subjects clinically examined and genetically examined are annotated with e + and X
There were two families with definite XL family history (2/13, 15.4%; Families 4, 12; history of multiple affected males in different generations and a female carrier for at least one generation), two families with probable XL history (2/13, 15.4%; Families 9, 10, one affected male and at least a female carrier), five families with possible XL/AD/incomplete AD (5/13, 38.5%; Families 1, 5, 6, 8, 13; a transmission between at least two generations reported or an incomplete transmission anticipated), and three with unknown family history (4/13, 30.8%, Families 2, 3, 7, 11; sporadic).
There were 12 affected males (12/14, 85.7%) and 2 affected females (2/14, 14.2%). For the purpose of this study, the two affected females registered as probands are described as patients, since both had clear visual impairment (8-III:2, 12-III:1). Systemic abnormalities, including hearing loss, were not reported in all patients.
The mean age at the latest examination of 12 affected males and 2 affected females was 38.9 (range, 11–72) and 25.0 years (25, 41), respectively.
Onset, chief complaint, refraction, and visual acuity
The mean age of onset of ten affected males with available records was 14.3 years (range, 0–50). One affected female with available records had onset of disease at the age of 9 (11-III:1).
Night blindness was reported in 4 out of 11 patients with available records (4/11, 36.3%). There were three patients with reduced visual acuity (3-II:1, 4-IV:1, 6-III:4), two with poor visual acuity (2-II:3, 7-II:1), two with photophobia (1-II:2, 5-III:4), one with color vision abnormality (5-III:2), and one with peripheral visual field defect (13-III:3).
The mean refractive error of the right/left eye of ten affected males with available records who had no refractive complication was −3.15/−2.95 diopter (range −8.0–1.0/−7.0–1.0). The mean refractive error of the right/left eye of two affected females with available records was −8.25/−10.25 diopter (−10, −6.5/−13.5, −7.0). One patient had cataracts in both eyes (2-II:3), and one patient underwent refractive surgery for myopia (1-II:2). Five patients had high myopia (5/10, 50.0%; higher than −6.0 diopter).
The mean VA in the right/left eye of ten affected males was 0.49/0.48 LogMAR unit (range 0.1–1.7/−0.08–1.52). The mean VA in the right/left eye of two affected females was 0.13/0.15 (0.1, 0.15/0.15, 0.15) LogMAR unit. Eight patients had relatively favorable VA (8/14, 57.1%; 0.22 LogMAR unit or better in the better eye), five had moderate VA (5/14, 35.7%; between 0.22 and 1.0 LogMAR unit in the better eye), and one had poor VA (1/14, 7.1%; 1.0 LogMAR unit or worse in the better eye).
Retinal images, visual field, and electrophysiological findings
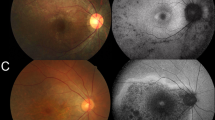
Fundus photographs were available in 12 affected males, and FAF images were obtained in 6 affected males. Representative fundus and FAF images of 12 affected males are presented in Fig. 2.

Fundus photographs and autofluorescence (FAF) images of the right eyes of 12 affected males with RPGR-RD are presented. Central atrophy or parafoveal atrophy was identified in all patients, with tigroid changes (seen in high myopic retina) in six patients (3-II:1, 4-IV:1, 5-III:2, 5-III:4, 6-III:4, and 7-II:1), and bone-spicule pigmentation (found in peripheral retinal atrophy) in five patients (5-III:2, 6-III:4, 7-II:1, 9-II:1, and 13-III:3). Well-marked atrophic changes with a ring of high AF density were found in all patients with available FAF images
Central atrophy or parafoveal atrophy was identified in all 12 patients with available fundus photographs. Tigroid changes (seen in high myopic retina) were observed in six patients (6/12, 50%), and bone-spicule pigmentation (found in peripheral retinal atrophy) was detected in five patients (5/12, 41.7%). Well-marked atrophic changes were demonstrated in FAF images. A ring of high AF density was noted in all six patients with available FAF images.
SD-OCT images were available in 11 affected males. Representative SD-OCT images are shown in Fig. 3. Structural disruption in the photoreceptor layers was observed in all 11 patients. Relatively preserved photoreceptor layers at the fovea were detected in five patients (5/11, 45.5%; 4-IV:1, 7-II:1, 10-III:1, 12-III:1, and 13-III:3).

Spectral-domain optical coherence tomographic images of the right eyes of 11 affected males are presented. Structural disruption in the photoreceptor layers is observed in all patients, with relatively preserved photoreceptor layers at the fovea in five patients (4-IV:1, 7-II:1, 10-III:1, 12-III:1, and 13-III:3)
Visual fields were available in all 12 affected males and 2 affected females. Central scotoma was observed in four patients (4/14, 28.6%), and annular scotoma was detected in three patients (3/14, 21.4%). Concentric visual field defects were found in two patients (2/14, 14.3%), and both central scotomas and concentric defects were observed in one patient (1/14, 7.1%). Two patients had partial paracentral scotoma (2/14, 14.3%), and two had no particular visual field defect (2/14, 14.3%).
Electrophysiological assessment was performed in ten affected males and two affected females. Undetectable responses in both generalized rod and cone systems were observed in five patients (5/12, 41.7%). Severely decreased generalized cone and severely decreased rod responses were demonstrated in one patient (1/12, 8.3%). Severely decreased generalized rod responses and moderately decreased generalized cone responses were identified in one patient (1/12, 8.3%). Severely decreased generalized cone and mildly decreased generalized rod responses were found in three patients (3/12, 25.0%). Moderately decreased generalized cone and generalized rod responses were noted in two patients (2/12, 16.7%).
Phenotype subgroups
Phenotype subgroup classification was performed in all 12 affected males and 2 affected females. There were six males with CORD (6/12, 50%) and six with RP (6/12, 50%). Both affected females were classified into RP.
Clinical information of carrier females
Clinical information of seven carrier females from six families was obtained. The detailed findings are described in Supplementary Table 1. Representative fundus and FAF images are presented in Supplementary Fig. 1. Representative SD-OCT images are shown in Supplementary Fig. 2.
The mean age at the latest examination of seven carriers was 47.1 years (range, 12–78). The mean refractive error of the right/left eye was −7.00/−7.42 diopter (range, −12.5–0.5/−12.0–0.5). The mean BCVA in the right/left eye was 0.14/0.13 LogMAR unit (range, 0.05–0.4/0.0–0.4).
Abnormalities on fundus photographs, FAF, SD-OCT, visual field testing, and electrophysiological assessment were found in five out of seven carriers (5/7, 71.4%). One carrier had reduced visual acuity, and one had night blindness. Retinal atrophy was observed in two carriers, and tapetal reflexes were found in five carriers (5/7, 71.4%). Electrophysiological assessment was available in five carriers, and all five carriers showed decreased responses.
RPGR variants
The variant data of 18 affected, 9 carriers, and 14 unaffected subjects of 13 families are summarized in Table 2.
Thirteen RPGR variants were identified; c.3399delG, p.Pro1134HisfsTer18; c.3308_3309delAT, p.Tyr1103SerfsTer7; c.3178_3179delGA, p.Glu1060ArgfsTer18; c.3104_3105delAG, p.Glu1035GlyfsTer43; c.3092delA, p.Glu1031GlyfsTer58; c.2625dupA, p.Gly876ArgfsTer203; c.2236_2237delGA, p.Glu746ArgfsTer23; c.1693C > T, p.Gln565Ter; c.1070 G > A, p.Gly357Asp; c.832 A > G, p.Thr278Ala; c.628 G > T, p.Glu210Ter; c.679 C > T, p.Gln227Ter; and c.389_390delTT, p.Phe130SerfsTer4. Twelve variants were detected by WES with target analysis of the retinal disease-associated genes. One variant was found by specific direct sequencing for RPGR-ORF15 (p.Gly876ArgfsTer203).
There are eight frameshift, three nonsense, and two missense variants. Five variants have been previously reported18,26,29,32. Eight variants have never been reported, including three frameshift, three nonsense, and two missense variants: p.Pro1134HisfsTer18, p.Glu1035GlyfsTer43, p.Phe130SerfsTer4, p.Gln565Ter, p.Glu210Ter, p.Gln227Ter, p.Gly357Asp, and p.Thr278Ala.
In silico molecular genetic analysis
The detailed results of in silico molecular genetic analyses for the 13 detected RPGR variants are presented in Supplementary Table 2.
Seven frameshift variants were located in ORF15. Six variants, including three nonsense, two missense, and one frameshift variant, were in exons 5, 7, 8, 10, and 14, and all six variants except for one frameshift variant (p. Gly876ArgfsTer203) were located in the RCC1-like domain. The allele frequency for one variant (p. Glu1060ArgfsTer18) in the general population was 0.001134% in the GnomAD database. None of the 13 detected RPGR variants were found in the Japanese general population of the HGVD and iJGVD databases.
General prediction, functional prediction, and conservation were assessed for the 13 variants, and pathogenicity classification according to the ACMG guidelines was pathogenic for eight variants, including six frameshift (p.Glu746ArgfsTer23, p.Glu1031GlyfsTer58, p.Glu1035GlyfsTer43, p.Glu1060ArgfsTer18, p.Tyr1103SerfsTer7, and p.Pro1134HisfsTer18) and two nonsense variants (p.Glu210Ter and p.Gln565Ter), likely pathogenic for three variants including two frameshift (p.Phe130SerfsTer4 and p.Gly876ArgfsTer203) and one nonsense variant (p. Gln227Ter), and uncertain significance for two missense variants (p.Gly357Asp and p.Thr278Ala).
Two missense variants (p. Gly357Asp and p. Thr278Ala) with uncertain significance were found in two probable XL families (Families 9, 10), and no other candidate variants associated with RP were detected in either of these families.
Overall, 13 disease-causing variants in the RPGR gene were ascertained in eight families with RP, and five families with CORD. Together with the clinical features of affected subjects and the model of inheritance in the pedigree, 13 disease-causing variants in the RPGR gene were determined.
Genotype–phenotype association
The locations of variants for two phenotype subgroups were investigated. All variants in eight families with RP were located in exons 1–14 and the 5′ end of ORF15 (< amino acid 950). All variants in five families with CORD were located at the 3′ end of ORF15 (> amino acid 950). A significant genotype–phenotype association between the phenotype subgroup and the position of detected variants was revealed.
Discussion
The clinical and genetic characteristics of RPGR-RD were illustrated in a nationwide cohort of 18 affected and 14 unaffected individuals and 9 carriers from 13 Japanese families with RPGR-RD, detecting 13 variants including 8 novel variants. There were eight families with RP and five families with CORD, which was associated with the position of RPGR variants.
To the best of our knowledge, this study reports the largest cohort of RPGR-RD in the Asian population. RPGR-RD accounts for 66.7% of molecularly confirmed XLRP (12 families; RPGR-8 families, RP2-4 families) in the JEGC cohort. All of molecularly confirmed XLCORD were RPGR-RD (RPGR-5 families). RPGR-RD accounts for 8.1% of 148 families with molecularly confirmed RP in total, and accounts for 4.8% of 105 families with molecularly confirmed CORD and allied disorders. The prevalence of RPGR-RD was similar to that of European cohorts3,48,49, although CACNA1F responsible for XL incomplete congenital night blindness (incomplete type of Miyake’s classification; OMIM: 300071)50,51 was not included as CORD in the JEGC cohort.
Out of 13 detected RPGR variants detected in this study, five variants were located within the RCC1-like domain (5/13, 38.5%), and seven were within the ORF15 domain (7/13, 53.8%). The proportion of ORF15 variants in the present cohort was slightly lower than that of the North American population (66%, reported by Sharon et al.)27.
In this study, five previously reported RPGR variants were identified in five families (Families 2, 3, 5 -CORD; Families 6, 7 -RP). Three of the five previously reported variants in the present cohort were reported in European cases with RP (p. Glu746ArgfsTer23, p.Gly876ArgfsTer203, and p.Glu1031GlyfsTer58)18,26,33. The other two variants were reported in European cases with CORD (p.Glu1060ArgfsTer18 and p.Tyr1103SerfsTer7)29,32. Four of the five families (4/5, 80%) in the present cohort showed a concordant phenotype with previous reports (Families 2, 3, 6, 7). In that study, the clinical effect of these four variants was confirmed in the Japanese population. One family with CORD had a discordant phenotype with a previous report (Family 5, p.Glu1031GlyfsTer58). Given the severely affected retinal findings in two affected males in Family 5, an advanced stage of this phenotype could be described as “RP”.
Eight RPGR variants were reported first in this study, including three frameshift (p.Phe130SerfsTer4, p.Glu1035GlyfsTer43, and p.Pro1134HisfsTer18), three nonsense (p.Glu210Ter, p.Gln227Ter, and p.Gln565Ter), and two missense variants (p.Thr278Ala and p.Gly357Asp). A high proportion of novel variants (8/13, 61.5%) was revealed in the Japanese cohort, which suggests a distinct genetic background for RPGR in the Japanese population compared with the European population.
In silico analysis for eight novel variants predicted pathogenic effects in four variants (p. Glu210Ter, p.Gln565Ter, p.Glu1035GlyfsTer43, and p.Pro1134HisfsTer18), a likely pathogenic effect in two variants (p.Gln227Ter and p.Phe130SerfsTer4), and variants of uncertain significance in two variants (p.Thr278Ala and p.Gly357Asp). These two missense variants (p.Gly357Asp and p.Thr278Ala) were located within the RCC1-like domain, where other missense disease-causing RPGR variants are frequently found37. The pathogenicity of these two missense variants is uncertain, although some association with nucleocytoplasmic transport and regulation of cell-division processing may be anticipated16,17.
A significant association between genotype and phenotype was revealed in this study. Variants located in exons 1–14 and the 5′ end of ORF15 caused RP, and variants at the 3′ end of ORF15 caused CORD. This finding was consistent with previous reports in the European population3,29,30. This fact supports the prediction of the natural history of RPGR-RD in counseling patients. Notably, the mechanism underlying this genotype–phenotype association has not been clarified.
There are limitations to this study. The selection bias related to the disease severity should be inherent, since it is uncommon for genetically affected subjects with good vision to visit clinics/hospitals. This study is a cross-sectional retrospective study; thus, longitudinal natural history studies in a larger cohort could provide more accurate information for disease progression of RPGR-RD. In addition, the molecular mechanisms of disease causation for some novel and previously reported variants are not yet known; therefore, further functional analysis could determine the disease causation of each variant.
In conclusion, the phenotypic and genotypic features of RPGR-RD were documented first in a large cohort of Japanese populations. A broad spectrum of phenotypic and genotypic findings was determined, revealing a considerable genotype–phenotype association. This evidence should be helpful in monitoring and counseling patients and in selecting patients for future therapeutic trials such as gene replacement therapy.
Change history
10 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Liew, G., Michaelides, M. & Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16-64 years), 1999-2000 with 2009-2010. BMJ Open 4, e004015 (2014).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Tee, J. J., Smith, A. J., Hardcastle, A. J. & Michaelides, M. RPGR-associated retinopathy: clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 100, 1022–1027 (2016).
Boughman, J. A., Conneally, P. M. & Nance, W. E. Population genetic studies of retinitis pigmentosa. Am. J. Hum. Genet. 32, 223–235 (1980).
Haim, M. Epidemiology of retinitis pigmentosa in Denmark. Acta. Ophthalmol. Scand. Suppl. 233, 1–34 (2002).
Prokisch, H., Hartig, M., Hellinger, R., Meitinger, T. & Rosenberg, T. A population-based epidemiological and genetic study of X-linked retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 48, 4012–4018 (2007).
You, Q. S. et al. Prevalence of retinitis pigmentosa in North China: the Beijing Eye Public Health Care Project. Acta Ophthalmol. 91, e499–e500 (2013).
Xu, Y. et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 133, 1255–1271 (2014).
Sahel, J. A., Marazova, K. & Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 5, a017111 (2014).
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M. & Bhattacharya, S. S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11, 273–284 (2010).
Lyraki, R., Megaw, R. & Hurd, T. Disease mechanisms of X-linked retinitis pigmentosa due to RP2 and RPGR mutations. Biochem. Soc. Trans. 44, 1235–1244 (2016).
Meindl, A. et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet. 13, 35–42 (1996).
Roepman, R. et al. Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum. Mol. Genet. 5, 1035–1041 (1996).
Hong, D. H. et al. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest. Ophthalmol. Vis. Sci. 44, 2413–2421 (2003).
Shu, X. et al. RPGR mutation analysis and disease: an update. Hum. Mutat. 28, 322–328 (2007).
Bischoff, F. R. & Ponstingl, H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 354, 80–82 (1991).
Hadjebi, O., Casas-Terradellas, E., Garcia-Gonzalo, F. R. & Rosa, J. L. The RCC1 superfamily: from genes, to function, to disease. Biochim. Biophys. Acta. 1783, 1467–1479 (2008).
Vervoort, R. et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 25, 462–466 (2000).
Khanna, H. et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J. Biol. Chem. 280, 33580–33587 (2005).
Shu, X. et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum. Mol. Genet. 14, 1183–1197 (2005).
Tee, J. J. L. et al. Natural history study of retinal structure, progression, and symmetry using ellipzoid zone metrics in RPGR-associated retinopathy. Am. J. Ophthalmol. 198, 111–123 (2019).
Kalitzeos, A. et al. Cellular imaging of the tapetal-like reflex in carriers of Rpgr-associated retinopathy. Retina 39, 570–580 (2019).
Ayyagari, R. et al. X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics 80, 166–171 (2002).
Demirci, F. Y. et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am. J. Hum. Genet. 70, 1049–1053 (2002).
Yang, Z. et al. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum. Mol. Genet. 11, 605–611 (2002).
Bader, I. et al. X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest. Ophthalmol. Vis. Sci. 44, 1458–1463 (2003).
Sharon, D. et al. and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 73, 1131–1146 (2003).
Ebenezer, N. D. et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest. Ophthalmol. Vis. Sci. 46, 1891–1898 (2005).
Branham, K. et al. Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest. Ophthalmol. Vis. Sci. 53, 8232–8237 (2012).
Yang, L. et al. Novel mutations of RPGR in Chinese retinitis pigmentosa patients and the genotype-phenotype correlation. PLoS ONE 9, e85752 (2014).
Fokkema, I. F., den Dunnen, J. T. & Taschner, P. E. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 26, 63–68 (2005).
Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Neidhardt, J. et al. Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol. Vis. 14, 1081–1093 (2008).
Oishi, M. et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest. Ophthalmol. Vis. Sci. 55, 7369–7375 (2014).
Hosono, K. et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci. Rep. 8, 8279 (2018).
Maeda, A. et al. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn J. Ophthalmol. 62, 451–457 (2018).
Bellingrath, J. S. et al. High symmetry of visual acuity and visual fields in RPGR-linked retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 58, 4457–4466 (2017).
Tee, J. J. L., Carroll, J., Webster, A. R. & Michaelides, M. Quantitative analysis of retinal structure using spectral-domain optical coherence tomography in RPGR-associated retinopathy. Am. J. Ophthalmol. 178, 18–26 (2017).
Tee, J. J. L., Kalitzeos, A., Webster, A. R., Peto, T. & Michaelides, M. Quantitative analysis of hyperautofluorescent rings to characterize the natural history and progression in Rpgr-associated retinopathy. Retina 38, 2401–2414 (2018).
Tee, J. J. L. et al. Characterization of visual function, interocular variability and progression using static perimetry-derived metrics in RPGR-associated retinopathy. Invest. Ophthalmol. Vis. Sci. 59, 2422–2436 (2018).
Jin, Z. B., Liu, X. Q., Hayakawa, M., Murakami, A. & Nao-i, N. Mutational analysis of RPGR and RP2 genes in Japanese patients with retinitis pigmentosa: identification of four mutations. Mol. Vis. 12, 1167–1174 (2006).
Fujinami, K. et al. Novel RP1L1 variants and genotype-photoreceptor microstructural phenotype associations in cohort of Japanese patients with occult macular dystrophy. Invest. Ophthalmol. Vis. Sci. 57, 4837–4846 (2016).
Hood, D. C. et al. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 124, 1–13 (2012).
McCulloch, D. L. et al. ISCEV standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 130, 1–12 (2015).
Hull, S. et al. The phenotypic variability of retinal dystrophies associated with mutations in CRX, with report of a novel macular dystrophy phenotype. Invest. Ophthalmol. Vis. Sci. 55, 6934–6944 (2014).
Neidhardt, J. et al. Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 28, 797–807 (2007).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Stone, E. M. et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 124, 1314–1331 (2017).
Gill, J. S., Georgiou, M., Kalitzeos, A., Moore, A. T. & Michaelides M. Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. e-pub ahead of print 24 January 2019; https://doi.org/10.1136/bjophthalmol-2018-313278 (2019).
Miyake, Y., Yagasaki, K., Horiguchi, M., Kawase, Y. & Kanda, T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 104, 1013–1020 (1986).
Nakamura, M., Ito, S., Piao, C. H., Terasaki, H. & Miyake, Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Achc Ophthalmol. 121, 1028–1033 (2003).
Acknowledgements
We are grateful to Dr. Anna Thompson at the National Genetic Reference Laboratory, Manchester, UK, and Dr. Panagiotis Sergouniotis, Manchester University, for their help in genetic screening for RPGR-ORF15 and for clinical genetic data analysis. Grant numbers: Kaoru Fujinami is supported by grants from Grant-in-Aid for Young Scientists (A) of the Ministry of Education, Culture, Sports, Science and Technology, Japan (16H06269), grants from Grant-in-Aid for Scientists to support international collaborative studies of the Ministry of Education, Culture, Sports, Science and Technology, Japan (16KK01930002), grants from National Hospital Organization Network Research Fund, Japan (H30-NHO-Sensory Organs-03), grants from the Foundation Fighting Blindness Alan Laties Career Development Program (CF-CL-0416-0696-UCL), USA, and grants from Health Labour Sciences Research Grant, The Ministry of Health Labour and Welfare, Japan (201711107 A), and grants from Great Britain Sasakawa Foundation Butterfield Awards, UK. Yu Fujinami-Yokokawa is supported by grants from Grant-in-Aid for Young Scientists of the Ministry of Education, Culture, Sports, Science and Technology, Japan (18K16943). Takeshi Iwata is supported by AMED (18ek0109282h0002). Kazushige Tsunoda is supported by the Japan Agency for Medical Research and Development (AMED), the Ministry of Health, Labor and Welfare, Japan (18ek0109282h0002), Grants-in-Aid for Scientific Research, Japan Society for the Promotion of Science, Japan (H26-26462674). Nobuhisa Nao-i is supported by Grants-in-Aid for Scientific Research (C) of the Ministry of Education, Culture, Sports, Science and Technology, Japan (15K-10842). Role of the funder/sponsor: The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the paper; and decision to submit the paper for the publication.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Individual investigators who participate in the sponsored project(s) are not directly compensated by the sponsor but may receive salary or other support from the institution to support their effort on the project(s). Kaoru Fujinami is a paid consultant of Astellas Pharma Inc., Kubota Pharmaceutical Holdings Co., Ltd, Acucela Inc., Novartis AG., and NightstaRx Limited. Kaoru Fujinami reports personal fees from Astellas Pharma Inc., Kubota Pharmaceutical Holdings Co., Ltd., Acucela Inc., Novartis AG., SANTEN Company Limited, the Foundation Fighting Blindness, the Foundation Fighting Blindness Clinical Research Institute, the Japanese Ophthalmology Society, and the Japan Retinitis Pigmentosa Society. The laboratory of Visual Physiology, Division for Vision Research, National Institute of Sensory Organs, National Hospital Organization, Tokyo Medical Center, Tokyo, Japan is supported by grants from Astellas Pharma Inc. (NCT03281005), outside the submitted work.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mawatari, G., Fujinami, K., Liu, X. et al. Clinical and genetic characteristics of 14 patients from 13 Japanese families with RPGR-associated retinal disorder: report of eight novel variants. Hum Genome Var 6, 34 (2019). https://doi.org/10.1038/s41439-019-0065-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-019-0065-7
This article is cited by
-
Application of patient-derived induced pluripotent stem cells and organoids in inherited retinal diseases
Stem Cell Research & Therapy (2023)
